
Abstract
The legality of wood products often depends on their origin, creating a need for forensic tools that verify claims of provenance for wood products. The neotropical tree species Cedrela odorata (Spanish cedar) is economically valuable for its wood and faces threats of overexploitation. We developed a 140 SNP assay for geographic localization of C. odorata specimens. Target capture and short-read sequencing of 46 C. odorata specimens allowed us to identify 140 spatially informative SNPs that differentiate C. odorata specimens by latitude, temperature, and precipitation. We assessed the broad applicability of these SNPs on 356 specimens from eight Cedrela species, three tissue types, and a range of DNA mass inputs. Origin prediction error was evaluated with discrete and continuous spatial assignment methods focusing on C. odorata specimens. Discrete classification with random forests readily differentiated specimens originating in Central America versus South America (5.8% error), while uncertainty increased as specimens were divided into smaller regions. Continuous spatial prediction with SPASIBA showed a median prediction error of 188.7 km. Our results demonstrate that array SNPs and resulting genotypes accurately validate C. odorata geographic origin at the continental scale and show promise for country-level verification, but that finer-scale assignment likely requires denser spatial sampling. Our study underscores the important role of herbaria for developing genomic resources, and joins a growing list of studies that highlight the role of genomic tools for conservation of threatened species.
Similar content being viewed by others


Introduction
Biodiversity loss is of global concern, and is due in part to deforestation and high consumer demand for wood and wood products (Nellemann 2012; Elias 2012; van Zonneveld et al. 2018). Forests of Central and South America (or “neotropical” forests) face the largest threat because they support the most terrestrial biodiversity, with an estimated 16,000 tree species contained within the Amazon rainforest alone (Pennington et al. 2015; Pennington and Lavin 2016; Dick and Pennington 2019). Thirty-five to 72% of wood sourced in the Amazon is thought to be acquired from illegal logging (Saunders and Reeve 2014), and illegal logging accounts for 50–90% of forestry activities across tropical forests globally (Hoare 2015; Sheikh et al. 2019). Laws are in place to protect economically valuable tree species from overexploitation and promote sustainable practices (e.g., U.S. Lacey Act [2008]; European Union timber regulation [2010]; Australian Illegal Logging Prohibition Act [2012]; Japanese Clean Wood Act [2017]), but these remain difficult to enforce because of the sheer scale of illegal logging, and the challenge of identifying protected species and their countries of origin, especially after wood is transported from the site of harvest, processed, and enters commercial markets (Dormontt et al. 2015, 2020; Wiedenhoeft et al. 2019).
Illegal logging affects many tree species, but highly valuable—often rare and endangered—species are common targets. Spanish cedar (Cedrela odorata L.; Meliaceae) and congeners are among the most valuable neotropical hardwoods, making them particularly vulnerable to illegal harvesting. In 2001, C. odorata was listed under the protections of the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES) Appendix III requiring validated documentation of species identity and source for both export and import documentation, protecting populations in Bolivia, Brazil, Colombia, Guatemala, and Peru (Ferriss 2014), and in 2019, C. odorata and all species in the genus Cedrela were elevated to CITES Appendix II, due to the similarity of sawn logs and processed wood across species (Gasson 2011). However, CITES protection does not entirely eliminate illegal harvesting. An investigation focused on logging of Spanish cedar (C. odorata) and mahogany (Swietenia macrophylla King) in Peru found 112 CITES export records of illegal wood listing origins that had been fabricated (Urrunaga et al. 2012). Urrunaga et al. (2012) provided evidence that illegal logging is systematic, common, and environmentally and economically damaging in Peru. In 2017, a snapshot of the scale of illegal logging in Peru was provided by the same team of investigators; a cargo ship from Peru, the Yaca Kallpa, was seized and contained nearly 10,000 m3 of illegal wood (Conniff 2017; Bargent 2017). Despite the anticipated increase in global scrutiny for Cedrela wood and wood products regardless of geographic origin, tools for predicting the origin of C. odorata wood remain valuable to process seizures predating CITES Appendix II listing, and could be valuable for verifying supply chains and identifying responsible parties.
Forensic tools that can accurately predict the geographic origin of wood have potential to assist the enforcement of trade restrictions for protected species. Genetic approaches have been used to determine the origin and trade routes of protected species, including elephant ivory (Wasser et al. 2004, 2018), sturgeon and paddlefish caviar (Doukakis et al. 2012; Ogden et al. 2013), tigers (Linacre and Tobe 2008; Gupta et al. 2011; Kitpipit et al. 2012), birds (Abe et al. 2012; Coghlan et al. 2012; White et al. 2012), fishes (Zarraonaindia et al. 2012; Clemento et al. 2014), and plants (Ogden et al. 2008; Degen et al. 2013; Blanc-Jolivet et al. 2018; Paredes-Villanueva et al. 2019). Anatomical, chemical, genetic, and isotopic methods have all been applied to address questions of taxonomy and origin of wood (Dormontt et al. 2015); however, a single method does not typically address questions of both taxonomy and source. Cedrela odorata and closely allied species will likely require multiple techniques for validation of taxonomy and origin because its wood lacks the anatomical features required for discrimination among species (Gasson 2011), and variation in wood chemistry does not vary in a manner that is geographically predictive (Paredes-Villanueva et al. 2018). While methods for DNA extraction and recovery from wood are improving (Dumolin-Lapègue et al. 1999; Asif and Cannon 2005; Rachmayanti et al. 2006; Tnah et al. 2012; Jiao et al. 2012, 2018; Yu et al. 2017; Dormontt et al. 2020), genetic markers of short length, such as single nucleotide polymorphisms (SNPs), are increasingly being used in wildlife forensics because they are suitable for low concentration, degraded DNA extracts (Ogden et al. 2009). Here, we evaluate the power of SNPs to resolve geographic origin of C. odorata across much of its range in Central America and western South America.
Materials and methods
Development of the SNP genotyping array for this study first involved the sequencing of a design panel of C. odorata specimens. From SNP variants detected in a design panel of specimens, we selected 140 candidate SNPs for the genotyping array on the basis of geographic and environmental differentiation. DNAs from a screening panel of 376 specimens were genotyped with these 140 array SNPs, and we were able to assess genotyping efficiency for DNAs derived from eight Cedrela species, three tissue types, a range of mass inputs. We evaluated discrete and continuous spatial origin prediction methods on a group of specimens from the screening panel, representing C. odorata and closely allied taxa (referred to as C. odorata sensu lato). Methods for each of these steps are described below.
Design panel sequencing
We used hybridization-based target capture and massively-parallel sequencing (Cronn et al. 2012; Heyduk et al. 2016) to identify SNPs from a panel of 46 C. odorata herbarium specimens (Appendix 1; Table S1; Fig. S1) from Peru and surrounding countries. Hybridization capture probes were designed based on gene models of a C. odorata individual originating in Mexico as described in Finch et al. (2019). Sequencing yield and depth were assessed using methods previously described (Finch 2018; Finch et al. 2019) and included in the Supporting Information for this article (Appendix 1; Table S2) (Finch 2019a, b). One Peruvian specimen (C. odorata 300; Table S1) was selected as the nuclear reference, and captured sequence reads were assembled de novo using SPAdes (v. 3.6.1; Bankevich et al. 2012). These enriched nuclear contigs were filtered to ensure that assembled sequences contained the target probe sequences, and did not contain sequences with identity to the C. odorata chloroplast genome (Finch et al. 2019) or mitochondrial genes (Kuravadi et al. 2015) (Appendix 2; Table S3).
Sequence reads from the 46 specimens were aligned to the C. odorata 300 reference using BWA-MEM (v. 0.7.17; Li and Durbin 2010; Li 2013), and sequence alignments were used as inputs for probabilistic variant calling using SAMtools (v. 1.10; Li et al. 2009) and the Genome Analysis Toolkit (GATK) (v. 3.7; McKenna et al. 2010). Best-practice guidelines for GATK variant calling were used, including indel region realignment, and high-stringency variant filtering for coverage, mapping quality, and variant position. Initial SNP filtering was performed with VCFtools (v. 0.1.17; Danecek et al. 2011) to remove insertion/deletion variants, sites with greater than 85% missing data, sites with more than two alleles, and sites with a minor allele frequency (MAF) lower than 5%. By applying these filtering parameters, we sought to provide a set of ‘high confidence’ SNPs for further analysis and eventual candidate selection for inclusion on a genotyping array.
Candidate SNP selection and SNP assay development
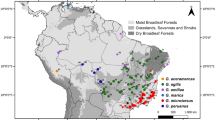
We identified SNPs showing the highest differentiation in allele frequencies (FST) (Weir and Cockerham 1984) for groups based on latitude (LAT; decimal degrees), mean annual temperature (MAT; °C × 10), and annual precipitation (AP; mm). In this way, FST was used to measure the partitioning of allelic variance in alternative groupings, not to make specific statements or inferences of population genetic parameters (e.g., probability of identity by descent, panmixis, or migration). Specimens were divided into a northern and southern LAT group based on a gap in the sampling distribution at 7.5° S latitude (Fig. 1a). Specimens were grouped into low, moderate, or high MAT (low < 20 °C; 20° < moderate < 25 °C; high > 25 °C; Fig. 1b) and AP (low < 2000 mm; 2000 < moderate < 3000 mm; high > 3000 mm; Fig. 1c) categories based on their tercile rank for these climate variables (Table S1) at their geographic source based on the WorldClim 2 dataset (Fick and Hijmans 2017) using R (see Table S7 for R packages and citations; R. Core Team et al. 2013; Finch 2019a, b). These measures exhibit low pairwise correlations (LAT × AP, r2 = 0.29; LAT × MAT, r2 = 0.01; AP × MAT, r2 = 0.14; Fig. S2), so genetic associations with these gradients are likely to include SNPs that respond to different neutral and selective forces.

SNP candidate selection via identification of highly differentiated SNPs (high relative FST) among C. odorata specimens (n = 46) based on: a Latitudinal (LAT) groups, b Mean Annual Temperature (MAT) groups, and c Annual Precipitation (AP) groups. Bar plots show the distribution of values and the number of counts in d LAT, e MAT, and f AP groups. g The distribution of FST for 140 SNPs selected for genotyping array development (gray), superimposed with the individual distributions of FST for SNPs selected for LAT (green), MAT (gold), and AP (blue). h The proportion of 140 SNPs selected for genotyping array development that were differentiated based on LAT (green), MAT (gold), and AP (blue). Map labels show country codes: COL (Colombia), ECU (Ecuador), PER (Peru), BOL (Bolivia), and BRA (Brazil)
LAT, MAT, and AP groups were then treated as ‘populations’ for calculating FST on a per-marker basis with VCFtools resulting in three lists of SNPs associated with these groups. To develop a SNP assay based on these loci, we sorted each SNP-FST list in descending order, concatenated the 150 top FST SNPs from each group into a single list, and filtered redundant SNPs to one SNP per contig of reference sequence. If two or more non-identical SNPs appeared on the same contig, we selected the SNP position with the highest FST. We further filtered the list to retain SNPs with a MAF ≥ 20% to avoid SNPs that were nearly-fixed across much of the geographic range. This resulted in 152 high FST and high MAF SNPs, none of which showed evidence of linkage disequilibrium (r ≤ 0.3). Contig names and SNP positions were converted into BED format, and BEDtools (v. 2.25.0; Quinlan and Hall 2010) was used to extract a multi-fasta file containing positions ± 100 bp flanking each SNP. The multi-fasta was used in primer design and multiplexing for a MassARRAY iPLEX® assay (Agena Bioscience, Inc., San Diego, CA, USA), using the MassARRAY Assay Design Suite software (Agena Bioscience, Inc.). From this list of sequences, we identified 140 SNP loci that could be evaluated in 4 multiplex groups. Resulting mass spectra were scored using Typer Viewer (v. 4.0.24.17; Agena Bioscience). All steps of SNP analysis (assay design, oligonucleotide synthesis, amplification, mass spectrometry, and SNP calling) were performed by NeoGen Genomics Inc. (Lincoln, NE, USA).
SNP assay screening
Samples screened for our selected SNPs derived from three sources (Table S4; Fig. S3): (i) 234 herbarium specimens from the Missouri Botanical Garden Herbarium (MO), (ii) 36 field collections by collaborators at the Universidad Nacional Agraria La Molina (Lima, Peru), Universidad Cayetano Heredia (Lima, Peru), and Servicio Nacional Forestal y de Fauna Silvestre (SERFOR; Lima, Peru), and (iii) 86 field collections by collaborators at the Instituto de Investigaciones de la Amazonía Peruana (IIAP; Iquitos, Peru), von Thüenen Institute of Forest Genetics (VTI; Braunschweig, Germany), and Universidad Autónoma Gabriel René Moreno (Santa Cruz, Bolivia). DNA was extracted using a modified CTAB procedure (AG-Biotech LLC, Monterey, CA, USA) from herbarium-derived dry leaf tissue from MO (150–200 mg from fragment packets). Leaf- and cambium-derived DNA from La Molina/Cayetano/SERFOR were extracted from fresh tissue using a modified CTAB protocol (Healey et al. 2014). These samples yielded < 100 ng of total DNA, so we used whole-genome amplification (Genomiphi™ V2 Amplification Kit, GE Healthcare, Chicago, IL, USA) to amplify the DNA using manufacturer’s instructions. DNA from the remaining 86 samples was derived from fresh material dried in silica gel according to Dumolin et al. (1995) at VTI or IIAP. All samples were quantified via fluorometry (Qubit, ThermoFisher Scientific, Waltham, MA, USA).
MassARRAY SNP genotyping was performed on 376 samples representing 356 unique specimens and 20 technical replicates. Included in our assay were representatives of C. odorata (n = 196), C. fissilis Vell. (n = 62), C. angustifolia DC (n = 28), C. montana Mortiz ex Turcz. (n = 22), C. nebulosa T. D. Penn. & Daza (n = 17), C. saltensis M. A. Zapater & del Castillo (n = 10), C. longipetiolulata Harms (n = 2), C. weberbaueri Harms (n = 2), and Cedrela field-collected samples that were not identified to species (n = 17; Table S4; Fig. S3). Replicated DNAs from two C. odorata individuals (C. odorata 83, C. odorata 291; Table S4) were examined across a range of DNA mass inputs (50, 75, 100, 200, and 300 ng). Our target DNA mass was 300 ng (recommended by NeoGen Genomics Inc.), although some samples had as little as 50 ng. In total, 330 DNAs derived from leaf tissue (primarily herbarium specimens) and 26 derived from cambium.
Assay assessment
To evaluate the broader use of the SNP array, SNP call rates (the proportion of successful diploid SNP genotyping calls at 140 diploid loci; Table S4) were determined for all samples, species, tissue types, and input concentrations. For population comparisons involving replicated samples (C. odorata 83, C odorata 291), we chose replicates with the highest call rates (50 ng dilutions in both cases). Loci were removed from all analyses if genotyping call rates were < 0.75 across specimens to strike a balance between missing information and sample retention. R packages adegenet (Jombart and Ahmed 2011), poppr (Kamvar et al. 2014), and hierfstat (Goudet 2005) were used to calculate observed heterozygosity, MAF, and FST for each SNP. FST was calculated on a per-marker basis treating species as populations, and by comparing northern and southern LAT groups (described above) as populations for C. odorata based on herbarium labels or field identification.
The R package adegenet was used to evaluate genetic structure among screening panel specimens with Discriminant Analysis of Principal Components (DAPC) (Jombart and Ahmed 2011), an ordination method based on genetic distances. We used DAPC clusters to define a reference database of specimens representing C. odorata and closely allied species (referred to as C. odorata sensu lato; n = 190; Appendix 3).
Discrete spatial classification with random forests
We classified C. odorata into discrete regional groups based on SNPs using random forests, a classification method that provides a consensus classification based on ‘a forest’ of many classification trees (Breiman 2001). Since our screened specimens represented dispersed samples and not necessarily populations, we designed classification tests to determine the classification accuracy obtained with our specimens and the SNP array at three geographic scales: (i) ‘range-wide’ with categories “Central America” and “South America,” (ii) ‘target countries’ with categories “Ecuador,” “Peru,” and “Bolivia,” and (iii) ‘narrow regional,’ within our target countries, with categories “NW,” “NE,” “SW,” and “SE.” Narrow regional groups were selected to represent an area approximately the size of Peruvian departments, and to maintain approximately equal sample sizes within groups.
For this analysis, we used our reference database of C. odorata s. l. and loci showing a call rate ≥ 0.75 with genotypes coded as categorical variables ‘0’ (genotype homozygous for the reference allele), ‘1’ (heterozygous), or ‘2’ (homozygous for the alternate allele). Since random forests is not tolerant of missing data, we used the R package synbreed (Wimmer et al. 2012) to impute allelic data on a per-locus basis from sampled genotypes under the assumption of Hardy–Weinberg equilibrium.
We generated a random forest of 500 classification trees for each classification question allowing each tree to have as many branches as loci (mtry = number of loci minus 1). In each case, predictor variables for classification were the loci and region of origin was the grouping variable for each specimen. To avoid biasing misclassifications, sample sizes for each regional class in the grouping variable were held constant, with the sample size determined by the class with the smallest number of specimens (Table 1) (Sun et al. 2009). Since classification error varies in random forests due to random sampling (i.e., random starting specimen and random starting locus), we calculated the mean of the median error across 5000 random forests (2,500,000 total classification trees), and evaluated the range and distribution of errors. Observed classification error was compared to random expectations by randomizing the classes by the grouping variable. This method was used to understand the baseline random classification accuracy for random forest tests with different numbers of classes (Finch et al. 2017).
Continuous spatial assignment with SPASIBA
Spatial classification methods have been developed to provide continuous estimates of origin based on an interpolated surface of allele frequencies. The R package SPASIBA (Guillot et al. 2016) uses a training set of georeferenced genotypes to predict the highest probability origin for test genotypes. SPASIBA estimates the spatial auto-covariance of allele frequencies assuming that covariance diminishes with increased geographic distance (i.e., isolation-by-distance). Allele frequencies for individuals geographically proximate to those included in the training set are estimated under the assumption of a population in Hardy–Weinberg equilibrium, and loci are assumed to be in linkage equilibrium. Predicted origins for “unknowns” can be estimated for areas where no training genotypes exist.
We used the same data in SPASIBA analysis as was used in random forest analysis above with the exception that we limited the geographic scope of our analysis to specimens from the target countries (Ecuador, Peru, Bolivia) and additional samples from Brazil and Bolivia below − 17.5° S latitude (n = 148). We used two cross-validation methods to assess the performance of SPASIBA. The first method was a k-fold cross-validation (k-fold CV); we modeled spatial auto-covariance of allele frequencies by randomly selecting 90% of C. odorata specimens (n = 133) as a training set, and used the remaining 10% (n = 15) as validation samples to determine the error of predicted origins. K-fold CV follows recommendations to assess model performance, avoid overfitting, and increase computational efficiency (Lever et al. 2016). In practical use, however, a legal inquiry involving wood would likely employ all reference specimens to identify the origin of confiscated material. To assess the predictive accuracy of SPASIBA via this framework, we performed a leave-one-out cross-validation (LOOCV) by predicting the origin of each specimen based on composite allele frequencies from all other available specimens (n = 147). By including both cross-validation techniques, we: (i) gain an understanding of the range of prediction errors for a single individual as a function of different training and validation sets, (ii) provide a conservative estimate of prediction error that is less prone to overfitting, and (iii) obtain the optimal predictive accuracy for each specimen in our dataset. For both cross-validation methods, we defined prediction error as the distance between the known and predicted origins. Spatial predictions were made in decimal degrees; these were converted to Haversine distances (in kilometers) with the R package geosphere (Hijmans 2016).
For the k-fold CV analysis, we tested the spatial resolution of predicted origins with data sets containing missing data and imputed data separately (imputation as described above) because SPASIBA is tolerant of missing data. Since the selection of training samples influences the accuracy of continuous geographic assignment (Guillot et al. 2016), we repeated the SPASIBA analysis 100 times with each data set (missing data; missing data imputed) to evaluate the distribution of prediction error. We compared the k-fold CV results to our random forest analysis by filtering the k-fold CV results to show prediction error for the specimens used for the target country and narrow regional analyses. As with random forests, observed classification error was compared to random expectations by randomizing genotypes of the geo-referenced training data set, and repeating predictions for the validation samples.
With the LOOCV analysis, we were interested in knowing whether the optimal predicted origin and assignment errors were related to sample density. To evaluate this, we computed the mean pairwise geographic distance to the ten nearest neighbors for each sample, and assessed the relationship between the mean ‘nearest-neighbor distance’ and prediction error for each sample (Appendix 4).
All maps were drawn using the base map shapefiles from the World Borders Dataset (https://thematicmapping.org/).
Results
SNP assay development
Target capture and short read sequencing of the design panel of C. odorata specimens (n = 46; Appendix 1) resulted in 4.4 × 108 paired sequence reads (9.0 × 1010 bp total) with a mean individual sequence yield of 9.6 × 106 paired reads (range: 6.3 × 105–4.7 × 107). The sequence yield for the ‘reduced representation’ nuclear reference for C. odorata 300 was 1.4 × 107 paired reads (2.8 × 109 bp; Table S2). The C. odorata 300 de novo assembly (Appendix 2) yielded 9,139 assembled contigs with a mean length of 982.5 bp (range 156–4,053 bp; sum of length = 9.0 × 106 bp; Table S3), and was used for read mapping and variant calling. On average, 3.6 × 106 reads mapped to each target reference contig (range 1.8 × 105–1.7 × 107 reads; Table S2) for an average depth of 53.7X per target (range 1.1X–443.6X; Table S2). Estimated mean individual depth ranged from 5.9X to 277.3X (Table S2). Our initial vcf contained 1.6 × 106 sequence variants before filtering to remove insertion/deletion variants (5.9% of total variants), multi-allelic variants (7.8%), SNPs with greater than 85% missing information (46.1%), and SNPs with a MAF less than 5% (31.1%). The resulting, filtered sequence matrix of C. odorata specimens from target countries (n = 46; Table S1; Fig. 1a–c) included 144,083 SNPs, and was used as the basis for evaluating allelic associations with geographic and climatic variation, and for developing a SNP assay for spatial assignment.
Figure 1 shows the geographic distribution of samples for each grouping (LAT, MAT, and AP) used to identify spatially informative SNPs via FST (Fig. 1a–c), as well as their predicted values (Fig. 1d–f). SNPs selected for AP showed the strongest allelic differentiation relative to SNPs selected for LAT and MAT, with a mean FST of 0.42 (interquartile range: 0.41—0.45; Fig. 1g). SNPs based on MAT showed a similar median FST of 0.44 (interquartile range 0.43–0.46; Fig. 1g) with a higher mean FST (0.46) and a higher maximum FST (0.62). Surprisingly, LAT SNPs showed lower differentiation, with a median FST of 0.23 (interquartile range 0.22–0.26 Fig. 1g). Our reduced SNP assay included 61 SNPs from the AP list, 53 SNPs from the MAT list, and 26 SNPs from the LAT list (Fig. 1h). These SNPs were converted to an Agena MassARRAY assay, and together array SNPs from the LAT, MAT, and AP lists showed a mean FST of 0.41 and per-SNP FST values ranged from 0.21 to 0.62.
SNP assay results
Across all samples and SNPs screened by MassARRAY, we observed 22.3% missing data (23,708 uncalled alleles out of 106,400 potential alleles), with specimens showing call rates (CR) of 0.00 to 0.96 across 140 SNPs. Three factors influenced CR: (i) DNA concentration, as input DNA mass above 50 ng resulted in decreasing CR (Fig. S4); (ii) the use of herbarium leaves, which showed a lower CR on average than other sources (Fig. S5); and (iii) the inclusion of multiple species, as DNAs derived from C. angustifolia and C. montana showing substantially lower mean CR than other species (CR = 0.62 and 0.71, respectively; Fig. S6). We discarded loci showing CR’s < 0.75 (i.e., 25 additional loci) to mitigate the impact of missing data. This yielded a Cedrela dataset with 352 individuals (four individuals discarded as failures), 99 loci, and 1.32% missing data. This dataset showed a mean observed heterozygosity of 0.05 (range 0–0.37) and a mean MAF of 0.26 (range 0.02–1). Treating species as populations, we calculated a median FST of 0.20 (range 0.01–0.56). Filtering for only C. odorata from South America (defined by herbarium labels and field identifications) produced a dataset with 135 specimens that showed a mean call rate of 0.81 (range 0.1–0.95). This C. odorata dataset included 99 loci and 1.01% missing data, and yielded a mean observed heterozygosity of 0.11 (range 0–0.44), a mean MAF of 0.34 (range 0.04–1), and a per-SNP median FST of 0.01 (range − 0.02 to 0.25) for geographic groups similar to those shown in Fig. 1a.
DAPC identified nine clusters (Fig. S7a). Based on label and field identification, a reference database for C. odorata sensu lato was defined by genetic information with ‘exclusive’ C. odorata (DAPC clusters 1, 2, 4, 7) and closely allied taxa (5, 8; Fig. S7b; Finch 2019b; Finch et al. in preparation), and we used these specimens to test discrete and continuous spatial assignment methods (Appendix 3; Fig. S7). Clusters 9 and 6 were excluded because they were largely composed of specimens identified as C. angustifolia and C. montana or C. fissilis, respectively (Appendix 3; Fig. S7b). Cluster 3 was also excluded because it showed the lowest mean CR (0.34 compared to CR > 0.7 for all other clusters). The resulting dataset included 190 C. odorata s. l. from Central and South America used for random forest classification, and 148 C. odorata s. l. for SPASIBA analyses focused on South America. After defining the C. odorata s. l. dataset and removing individuals with high levels of missing information, we were able to retain a larger number of loci for random forest classification (116 SNPs) and SPASIBA continuous assignment (118 SNPs).
Random forest classification
Range-wide
This analysis included 190 C. odorata s. l. specimens from Central and South America and 116 SNP loci. Each individual was assigned to one of two regional classes (Table 1; Fig. 2a). The estimated mean of the median classification error of 5.8% for observed data was much lower than the estimated mean of the median classification error from 5,000 randomizations (51.1%; Table 1, Fig. 2g).

Summary of random forest classification analyses and resulting geographic classification errors. Maps show the geographic range of specimens categorized into regional classes for: a range-wide, b target country, and c narrow regional classification. Boxplots show the classification error (%) estimates by class for: d range-wide, e target country, and f narrow regional classes. Density plots show the distribution of classification error estimated for: g range-wide, h target country, and i narrow regional classification. Light gray distributions indicate error for observed genotypes, and dark gray distributions indicate error for genotypes after randomizing regional class identifiers in the grouping variable. Maps labels show country codes: NIC (Nicaragua), CR (Costa Rica), PAN (Panama), COL (Colombia), VEN (Venezuela), ECU (Ecuador), PER (Peru), BOL (Bolivia), and BRA (Brazil)
Target countries
This analysis included 141 C. odorata s. l. specimens from three countries (Fig. 2b) and 116 SNP loci. The estimated mean classification error of 34.3% for observed data was lower than the estimated mean classification error from 5,000 randomizations (68.4%; Table 1, Fig. 2h).
Narrow regional
This analysis included 129 C. odorata s. l. specimens from four narrow regions approximately 3.8 × 105 km2 in size (Fig. 2c) and 116 SNP loci. The estimated mean classification error of 34.7% was lower than the mean classification error of 76.9% estimated from 5000 randomizations (Table 1; Fig. 2i).
Classification errors for the range-wide comparison were almost equally distributed between groups, with mean errors of 4.42% for samples from Central America and 7.05% for samples from South America (Fig. 2d). Finer-scale classification tests showed classification asymmetry, where spatial assignment error was not equal across classes. For example, classification errors for the target country comparison showed that specimens from Ecuador had lower classification error (30.7%) than either Bolivia or Peru (35.1% and 37.0%, respectively; Fig. 2e). Similarly, specimens from the NE class had a substantially lower classification error (21.3%), than specimens from the NW, SW or SE classes (39.5%, 39.1%, and 39.0% respectively; Fig. 2f).
SPASIBA assignment
We investigated the accuracy of continuous assignments 148 C. odorata s. l. specimens with 118 SNPs, SPASIBA, and two cross-validation methods (sample distribution shown in Fig. S9). The median deviation between the known sampling location and predicted origin for the k-fold CV was 259.6 km (25%ile = 96.1 km; 75%ile = 820.3 km; Table 2; Fig. 3a) with a maximum error of 2540.8 km. Median k-fold CV prediction error with observed data was lower than the estimated median error from randomized genotypes (median 904.1 km; 25%ile = 494.6 km; 75%ile = 1408.7 km; maximum = 3,033.8 km; Table 2). We evaluated the estimation error in the imputed dataset used for random forest analysis to determine whether imputation of missing data influences prediction errors, and found that imputation had little influence on prediction error (imputed error = 268.4 km; unimputed error = 252.5 km; Table S5; Fig. S8).

Distributions of prediction errors (in km) for C. odorata s. l. specimens used for continuous spatial assignment with SPASIBA: a prediction error for South American specimens across 200 k-fold CV replicates (imputed and unimputed combined) with observed genotypes (light gray) and 148 LOOCV replicates with observed unimputed data (blue), b prediction error for specimens from target country groups, and c prediction error for specimens from narrow regional groups. Red dashed lines indicate the estimated mean prediction error for randomized genotypes (Table 2)
The k-fold CV results also showed that specimens from the Peru (Fig. 2b) showed a lower median prediction error (169.8 km) than Ecuador (598.0 km) and Bolivia (341.4 km; Table 2; Fig. 3b), a pattern that appears different from random forest classification results for countries (Fig. 2e). Similarly, specimens from the NE narrow region (Fig. 2c) showed a lower median prediction error (133.3 km) than the other narrow regional groups (NW = 598.1 km; SW = 135.0 km; SE = 424.0 km; Table 2; Fig. 3b); this pattern appears similar to our findings from random forest classification of these regional classes (Fig. 2f).
The median prediction error was lower under the LOOCV framework than the k-fold CV (188.7 km; 25%ile = 92.2 km; 75%ile = 754.3 km; Table 2; Fig. 3a). This was a 27.3% decrease in prediction error compared to the median k-fold CV estimate. Despite this improvement, we found no relationship between sample density and prediction error (Appendix 4).
Discussion
The neotropical tree species Cedrela odorata is a target of illegal logging, and has been heavily used for its timber at least since the description of the genus Cedrela in 1756 (Browne 1756; Pennington and Muellner 2010). Illegal logging of C. odorata typically involves fabrication of source on export documentation (Urrunaga et al. 2012), and since 2001, C. odorata has been protected in at least one country under CITES Appendix III (Ferriss 2014), increasing the importance of technologies that predict the origin of C. odorata wood. Country-of-origin declarations on import documents for wood can be difficult for wood importers to verify, and it is even more challenging for customs and border patrol agents to corroborate, making independent assessment methods a high-priority tool to aid the legal evaluation of wood products. Although, C. odorata and all Cedrela species have been elevated to CITES Appendix II, geographic localization of C. odorata wood remains relevant for seizures predating CITES Appendix II listing. Geographic localization is also essential to discovering the networks responsible for illegal logging, as has been shown for animal poaching (Wasser et al. 2018). We demonstrated that SNPs have the power to at least partially resolve the geographic origin of C. odorata across much of its range in Central America and western South America, and we present results from discrete classification of geographic origin with random forests (Breiman 2001) and continuous spatial prediction with SPASIBA (Guillot et al. 2016).
Using SNPs to predict the geographic region origin for C. odorata via discrete classification
A number of methods have been used for discrete assignment of genotypes to geographic groups (Manel et al. 2005; Ogden and Linacre 2015). Some of these methods use criteria or assumptions that are explicitly ‘genetic’ (e.g., Hardy–Weinberg and linkage equilibrium) (Rannala and Mountain 1997; Pritchard et al. 2000; Piry et al. 2004), but others are agnostic with regard to the input data type or the process(es) underlying the input data (Chen et al. 2017; Schrider and Kern 2018). ‘Model-free’ methods like random forests can use high-dimensional genetic data and non-genetic data to produce predictive functions that are robust to any type of data and distribution (e.g., non-normal distributions, zero-truncated, continuous, or categorical data), allowing genetic data to be combined with other information that provides independent evidence of geographic origin such as specifically stable isotope profiles (Kagawa and Leavitt 2010; Gori et al. 2015, 2018). This is especially important in cases involving plantation grown timber, where genotypic data may correctly identify the ancestral geographic origin, but not the growing location for a specific tree (e.g., plantation-grown C. odorata from Africa). This flexibility has led to the adoption of random forest methods for multiple applications in ecology and evolution (Boulesteix et al. 2012; Brieuc et al. 2018), genomics and genetic association analysis (Goldstein et al. 2011; Stephan et al. 2015), and population assignment based on genetic variation (Bertolini et al. 2015; Chen et al. 2017; Sylvester et al. 2018) and parasite community (Perdiguero-Alonso et al. 2008; Pérez-Del-Olmo et al. 2010). Random forests have been less frequently used for spatial classification, with current published cases based on reflection and chemical spectra rather than genotypes (Li et al. 2012; Finch et al. 2017).
In our specific analysis, we determined that random forest classification based on SNP genotypes can predict whether C. odorata s. l. specimens originated in Central or South America with 5.8% classification error (Table 1). This method offers high discrimination accuracy for broad-scale geographic source validation, and could serve as a ‘first-pass’ test for questions related to provenance on trade documentation. We found that random forest classification was less precise for identifying finer-scale questions, such ‘country-of-origin’ or ‘department-sized regions-of-origin’ within a country (34.3% error and 34.7% error, respectively). We suspect that within-class sample size was at least partly responsible for the relatively high error estimations at this scale, since both of these analyses (target country and narrow regional) indicated that some geographic signal was available for classification with our SNP assay (Table 1). It is important to note that while our method did not show high precision for identifying the country-of-origin, the four South American countries that listed C. odorata as CITES Appendix III (Bolivia, Brazil, Columbia, Peru) account for > 63% of the land area of the continent. With denser sampling across northern and eastern South America, it should be possible to test this classification method using CITES III protection status (protected versus non-protected) as the classifier, as exports from all of these countries are highly restricted.
Using SNPs to predict the geographic origin of C. odorata by continuous assignment
Methods have also been developed for estimating the origin of genotypes using continuous assignment (Wasser et al. 2004; Yang et al. 2012; Rañola et al. 2014; Guillot et al. 2016). These methods assume Hardy–Weinberg and linkage equilibrium and only use genetic data, but these limitations are balanced by the potential power of providing a precise geospatial source for a sample, rather than a categorical assignment (Degen et al. 2017; Chen et al. 2017). Continuous assignment with methods like SPASIBA are particularly relevant for questions involving wood legality because harvest locations are frequently not included in genetic reference populations, especially for geographically widespread species.
Our median prediction error for continuous assignment of C. odorata in South America was ~ 189 km via the LOOCV method, and ~ 260 km via the more conservative k-fold CV method (Table 2). These error estimates show promise for country-of-origin predictions, but may be less helpful for smaller countries and areas near international borders. Assignment errors ranged from 170 to 598 km for target countries (with Peru showing the lowest mean error; Fig. 3b), and from 133 to 598 km for department-sized geographic regions in our study area (with NE and SW regions in Peru showing the lowest errors; Fig. 3c). These errors – while large – are comparable to the continuous geographic assignment errors from other organisms based on similarly-sized datasets. For example, the mean error for the placement of humans based on 100 SNPs was ~ 430 km (Rañola et al. 2014), the 75% placement error of Arabidopsis based on 100 SNPs was 375 km (Guillot et al. 2016), and the median error for the geographic assignment of elephants based on 16 microsatellites ranged from 267 km (savannah) to 301 km (forest populations) (Wasser et al. 2015).
Despite the practical advantages of SPASIBA for providing continuous origin prediction, a potential disadvantage of the method lies in the assumption that spatial auto-covariance of allele frequencies diminishes with geographic distance. The assumptions of this simple gradient function may introduce error if allele frequency surfaces are irregular or lack a dominant cline. In our study, we also stratified samples by precipitation, temperature, and latitude (Fig. 1) to identify genes that might be responsive to different climate factors over small geographic distances, as might be the case due to heterogeneous elevation gradients imposed by the Andes Mountains. In doing so, SNPs selected for this assay appear biased towards genes showing stronger differentiation by climatic gradients than to spatial gradients of geographic distance; we observed that pairwise genetic distance was more strongly associated with pairwise MAT and AP distances than pairwise geographic distance (genetic distance ~ MAT distance Mantel r = 0.42; genetic distance ~ AP distance Mantel r = 0.26; genetic distance ~ geographic distance Mantel r = 0.08; Appendix 5) (Goslee and Urban 2007). This bias may have reduced the accuracy of our SPASIBA predictions, either by violating assumptions of simple gradients, or by selecting genes that show weaker correlations with geographic distances than they do to climatic distances. We will explore solutions to this in the future, by examining additional loci that show higher correlations to geography than climate, and testing continuous assignment methods that relax the assumption of isolation-by-distance allele frequency gradients (Rañola et al. 2014; Battey et al. 2019).
Recommendations for improving SNP-based geographic predictions for Cedrela
The accuracy of assignment can be dramatically influenced by the pattern of geographic sampling and density of genome coverage, especially for continuous assignment methods. Although we did not observe a relationship between sample density and prediction error (Appendix 4), continuous methods have been shown to provide highest accuracies when training datasets include individuals from the same genetic background as test individuals (Guillot et al. 2016). Additionally, the impact of the size of the genetic database on assignment accuracy can also be substantial. For example, two independent analyses have shown that increasing genomic density from 100 to 1,000 SNPs leads to significant reductions in prediction error (Rañola et al. 2014; Guillot et al. 2016). Our foundation dataset of 144,083 SNPs for C. odorata offers a rich resource that can be used to further refine SNP assays for geographic assignment. In this context, we note that additional SNPs (~ 350) are currently being evaluated for spatial assignment of C. odorata and C. fissilis, and this includes different nuclear and organelle markers from different source populations (Blanc-Jolivet et al., unpublished; Paredes-Villanueva et al. 2019). Joint analysis of these two marker sets using common samples should show whether simply doubling the number of genotypes and SNPs offers significant improvements in geographic assignment accuracy. Finally, different Cedrela species can show different allele frequencies across loci, and this may distort allele frequency surfaces used in spatial assignment and lead to less accurate geographic predictions for C. odorata. In this regard, we recommend exploration of genetic structure across reference specimens with DAPC (Appendix 3) or a similar method. In our reference database for C. odorata, we identified examples of specimens that were taxonomically misidentified, and this is common in natural history collections of tropical plants (Goodwin et al. 2015). Additional “C. odorata” reference specimens should be assessed for taxonomic cohesiveness with C. odorata before usage with evidence.
Conclusions
We identified greater than 100,000 SNPs that can be used to develop and refine assays for geographic localization of C. odorata wood specimens. From this database, we designed and tested a 140 SNP assay to predict the geographic origin of C. odorata, and we evaluated discrete (random forest) and continuous (SPASIBA) prediction methods. These methods make different assumptions with regard to the Hardy–Weinberg and linkage equilibrium; as such, they may show different performance depending on the degree to which SNPs track selective (environmental) gradients or deviate from genetic assumptions. Although the observed error estimates from our geographic predictions are too large for fine-scale geographic assignment, the assay shows high accuracy for determining the continent of origin and promise for country-level verification of specimens. This assay provides a tangible first step for determining the origin and legality of C. odorata wood, and these SNP resources and methods should provide the wood products industry with new (and developing) tools to improve the legality of C. odorata and closely allied species in wood trade.
Data Availability
Raw sequence data associated with this study is available from the NCBI GenBank BioProject Archive under accession number PRJNA369105. Also see the data set available via the Oregon State University Scholars Archive https://doi.org/10.7267/TQ57NX45Z. This supplementary directory contains a reduced representation nuclear genome reference for C. odorata, a VCF file containing 144,083 high quality SNPs for C. odorata which may be used to further refine out SNP genotyping assay for geographic assignment or other purposes, 140 SNP primers used for the Agena™ MassARRAY®, and data sets and R code to replicate our statistical analysis.
References
Abe H, Hayano A, Inoue-Murayama M (2012) Forensic species identification of large macaws using DNA barcodes and microsatellite profiles. Mol Biol Rep 39:693–699. https://doi.org/10.1007/s11033-011-0787-1
Asif MJ, Cannon CH (2005) DNA extraction from processed wood: a case study for the identification of an endangered timber species (Gonystylus bancanus). Plant Mol Biol Rep 23:185–192. https://doi.org/10.1007/BF02772709
Bankevich A, Nurk S, Antipov D et al (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. https://doi.org/10.1089/cmb.2012.0021
Bargent J (2017) Report exposes inner workings of timber trafficking in Peru. In: Insight crime. https://www.insightcrime.org/news/analysis/reportexposes-inner-workings-timber-trafficking-peru/. Accessed 10 Dec 2019
Battey CJ, Ralph PL, Kern AD (2019) Predicting geographic location from genetic variation with deep neural networks. GrbioRxiv. https://doi.org/10.1101/2019.12.11.872051
Bertolini F, Galimberti G, Calò DG et al (2015) Combined use of principal component analysis and random forests identify population-informative single nucleotide polymorphisms: application in cattle breeds. J Anim Breed Genet 132:346–356. https://doi.org/10.1111/jbg.12155
Blanc-Jolivet C, Yanbaev Y, Kersten B, Degen B (2018) A set of SNP markers for timber tracking of Larix spp. in Europe and Russia. Forestry 91:614–628. https://doi.org/10.1093/forestry/cpy020
Boulesteix A-L, Janitza S, Kruppa J, König IR (2012) Overview of random forest methodology and practical guidance with emphasis on computational biology and bioinformatics. Wires Data Min Knowl 2:493–507. https://doi.org/10.1002/widm.1072
Breiman L (2001) Random forests. Mach Learn 45:5–32
Brieuc MSO, Waters CD, Drinan DP, Naish KA (2018) A practical introduction to Random Forest for genetic association studies in ecology and evolution. Mol Ecol Resour 18:755–766. https://doi.org/10.1111/1755-0998.12773
Browne P (1756) The civil and natural history of Jamaica: in three parts. Printed for the author, and sold by T. Osborne and J. Shipton in Gray's-Inn, London, England, UK, pp 158
Chen K-Y, Marschall E, Sovic M et al (2017) assignPOP: An R package for population assignment using genetic, non-genetic, or integrated data in a machine-learning framework. Methods Ecol Evol 9:439–446. https://doi.org/10.1111/2041-210X.12897
Clemento AJ, Crandall ED, Garza JC, Anderson EC (2014) Evaluation of a single nucleotide polymorphism baseline for genetic stock identification of Chinook Salmon (Oncorhynchus tshawytscha) in the California Current large marine ecosystem. Fish-B NOAA 112:112–130. https://doi.org/10.7755/FB.112.2-3.2
Coghlan ML, White NE, Parkinson L et al (2012) Egg forensics: An appraisal of DNA sequencing to assist in species identification of illegally smuggled eggs. Forensic Sci Int Genet 6:268–273. https://doi.org/10.1016/j.fsigen.2011.06.006
Conniff R (2017) Invisible forest: chasing the illegal loggers looting the Amazon. Wired. https://www.wired.com/story/on-the-trail-of-the-amazonianlumber-thieves/. Accessed 9 Dec 2019
Cronn R, Knaus BJ, Liston A et al (2012) Targeted enrichment strategies for next-generation plant biology. Am J Bot 99:291–311. https://doi.org/10.3732/ajb.1100356
Danecek P, Auton A, Abecasis G et al (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Degen B, Blanc-Jolivet C, Stierand K, Gillet E (2017) A nearest neighbour approach by genetic distance to the assignment of individual trees to geographic origin. Forensic Sci Int Genet 27:132–141. https://doi.org/10.1016/j.fsigen.2016.12.011
Degen B, Ward SE, Lemes MR et al (2013) Verifying the geographic origin of mahogany (Swietenia macrophylla King) with DNA-fingerprints. Forensic Sci Int Genet 7:55–62. https://doi.org/10.1016/j.fsigen.2012.06.003
Dick CW, Pennington RT (2019) History and geography of neotropical tree diversity. Annu Rev Ecol Evol 50:279–301. https://doi.org/10.1146/annurevecolsys-110617-062314
Dormontt EE, Boner M, Braun B et al (2015) Forensic timber identification: It’s time to integrate disciplines to combat illegal logging. Biol Conserv 191:790–798. https://doi.org/10.1016/j.biocon.2015.06.038
Dormontt EE, Jardine DI, van Dijk K-J et al (2020) Forensic validation of a SNP and INDEL panel for individualisation of timber from bigleaf maple (Acer macrophyllum Pursch). Forensic Sci Int Genet 46:102252. https://doi.org/10.1016/j.fsigen.2020.102252
Doukakis P, Pikitch EK, Rothschild A et al (2012) Testing the effectiveness of an international conservation agreement: marketplace forensics and CITES caviar trade regulation. PLoS ONE 7:e40907. https://doi.org/10.1371/journal.pone.0040907
Dumolin S, Demesure B, Petit RJ (1995) Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor Appl Genet 91:1253–1256. https://doi.org/10.1007/BF00220937
Dumolin-Lapègue S, Pemonge M-H, Gielly L et al (1999) Amplification of oak DNA from ancient and modern wood. Mol Ecol 8:2137–2140. https://doi.org/10.1046/j.1365-294x.1999.00788.x
Elias P (2012) Logging and the law: How the U.S. Lacey Act helps reduce illegal logging in the Tropics. Union of Concerned Citizens, Cambridge, MA, USA. https://www.ucsusa.org/resources/logging-and-law. Accessed 17 May 2017
Ferriss S (2014) An analysis of trade in five CITES-listed taxa. The Royal Institute of International Affairs, Chatham House, London. https://www.chathamhouse.org/publication/analysis-trade-five-cites-listed-taxa. Accessed 21 Oct 2019
Fick SE, Hijmans RJ (2017) WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas. Int J Climat 37:4302–4315. https://doi.org/10.1002/joc.5086
Finch KN (2018) Dataset for genomic resources for the neotropical tree genus Cedrela (Meliaceae) and its relatives. Oregon State University. https://doi.org/10.7267/NV935820Q
Finch KN (2019) Dataset for predicting the geographic origin of Spanish Cedar (Cedrela odorata L.) based on DNA variation (Version 1). Oregon State University. https://doi.org/10.7267/TQ57NX45Z
Finch KN (2019b) Genomic resources for phylogenetics, species delimitation, and geographic localization of neotropical tree species Cedrela odorata L. (Meliaceae). Dissertation, Oregon State University. https://ir.library.oregonstate.edu/concern/graduate_thesis_or_dissertations/3197xt184
Finch KN, Espinoza E, Jones FA, Cronn R (2017) Source identification of western Oregon Douglas-fir wood cores using mass spectrometry and random forest classification. Appl Plant Sci 5:1600158. https://doi.org/10.3732/apps.1600158
Finch KN, Jones FA, Cronn RC (2019) Genomic resources for the Neotropical tree genus Cedrela (Meliaceae) and its relatives. BMC Genom 20:58. https://doi.org/10.1186/s12864-018-5382-6
Gasson P (2011) How precise can wood identification be? Wood anatomy’s role in support of the legal timber trade, especially CITES. IAWA J 32:137–154. https://doi.org/10.1163/22941932-90000049
Goldstein BA, Polley EC, Briggs FB (2011) Random forests for genetic association studies. Stat Appl Genet Mol Biol 10:32. https://doi.org/10.2202/1544-6115.1691
Goodwin ZA, Harris DJ, Filer D et al (2015) Widespread mistaken identity in tropical plant collections. Curr Biol 25:R1066–R1067. https://doi.org/10.1016/j.cub.2015.10.002
Gori Y, Stradiotti A, Camin F (2018) Timber isoscapes: a case study in a mountain area in the Italian Alps. PLoS ONE 13:e0192970. https://doi.org/10.1371/journal.pone.0192970
Gori Y, Wehrens R, La Porta N, Camin F (2015) Oxygen and hydrogen stable isotope ratios of bulk needles reveal the geographic origin of Norway spruce in the European Alps. PLoS ONE 10:e0118941. https://doi.org/10.1371/journal.pone.0118941
Goslee SC, Urban DL (2007) The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw 22:1–19. https://doi.org/10.18637/jss.v022.i07
Goudet J (2005) Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol Ecol Notes 5:184–186. https://doi.org/10.1111/j.1471-8286.2004.00828.x
Guillot G, Jónsson H, Hinge A et al (2016) Accurate continuous geographic assignment from low- to high-density SNP data. Bioinformatics 32:1106–1108. https://doi.org/10.1093/bioinformatics/btv703
Gupta SK, Bhagavatula J, Thangaraj K, Singh L (2011) Establishing the identity of the massacred tigress in a case of wildlife crime. Forensic Sci Int Genet 5:74–75. https://doi.org/10.1016/j.fsigen.2010.05.004
Healey A, Furtado A, Cooper T, Henry RJ (2014) Protocol: a simple method for extracting next-generation sequencing quality genomic DNA from recalcitrant plant species. Plant Methods 10:21. https://doi.org/10.1186/1746-4811-10-21
Heyduk K, Stephens JD, Faircloth BC, Glenn TC (2016) Targeted DNA region re-sequencing. In: Aransay AM, Trueba JLL (eds) Field guidelines for genetic experimental designs in high-throughput sequencing. Springer International Publishing, Switzerland, pp 43–68
Hijmans RJ (2016) geosphere: spherical trigonometry. R package version 1.5–5. See http://www.cranr-projectorg/package=geosphere org/package=geosphere
Hoare A (2015) Tackling illegal logging and the related trade: What progress and where next? The Royal Institute of International Affairs, London, England, UK. https://www.chathamhouse.org/publication/tackling-illegal-logging-and-related-trade-what-progress-and-where-next. Accessed 10 May 2019
Jiao L, Yin Y, Xiao F et al (2012) Comparative analysis of two DNA extraction protocols from fresh and dried wood of Cunninghamia lanceolata (Taxodiaceae). IAWA J 33:441–456. https://doi.org/10.1163/22941932-90000106
Jiao L, Yu M, Wiedenhoeft AC et al (2018) DNA barcode authentication and library development for the wood of six commercial Pterocarpus species: the critical role of xylarium specimens. Sci Rep 8:1–10. https://doi.org/10.1038/s41598-018-20381-6
Jombart T, Ahmed I (2011) adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071. https://doi.org/10.1093/bioinformatics/btr521
Kagawa A, Leavitt SW (2010) Stable carbon isotopes of tree rings as a tool to pinpoint the geographic origin of timber. J Wood Sci 56:175–183. https://doi.org/10.1007/s10086-009-1085-6
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. https://doi.org/10.7717/peerj.281
Kitpipit T, Tobe SS, Kitchener AC et al (2012) The development and validation of a single SNaPshot multiplex for tiger species and subspecies identification—implications for forensic purposes. Forensic Sci Int 6:250–257. https://doi.org/10.1016/j.fsigen.2011.06.001
Kuravadi NA, Yenagi V, Rangiah K et al (2015) Comprehensive analyses of genomes, transcriptomes and metabolites of neem tree. PeerJ 3:e1066. https://doi.org/10.7717/peerj.1066
Lever J, Krzywinski M, Altman N (2016) Points of significance: Model selection and overfitting. Nat Meth 13:703–704. https://doi.org/10.1038/nmeth.3968
Li B, Wei Y, Duan H et al (2012) Discrimination of the geographical origin of Codonopsis pilosula using near infrared diffuse reflection spectroscopy coupled with random forests and k-nearest neighbor methods. Vib Spectrosc 62:17–22. https://doi.org/10.1016/j.vibspec.2012.05.001
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:13033997
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26:589–595. https://doi.org/10.1093/bioinformatics/btp698
Li H, Handsaker B, Wysoker A et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Linacre A, Tobe SS (2008) On the trial of tigers–tracking tiger in Traditional East Asian Medicine. Forensic Sci Int 1:603–604. https://doi.org/10.1016/j.fsigss.2007.10.112
Manel S, Gaggiotti OE, Waples RS (2005) Assignment methods: matching biological questions with appropriate techniques. Trends Ecol Evol 20:136–142. https://doi.org/10.1016/j.tree.2004.12.004
McKenna A, Hanna M, Banks E et al (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. https://doi.org/10.1101/gr.107524.110
Nellemann C (2012) Green carbon, black trade: illegal logging, tax fraud and laundering in the world’s tropical forests. United Nations Environment Programme, GRID-Arendal, Norway. https://www.grida.no/publications/rr/green-carbon-black-trade/. Accessed 9 Nov 2017
Ogden R, Dawnay N, McEwing R (2009) Wildlife DNA forensics—bridging the gap between conservation genetics and law enforcement. Endang Species Res 9:179–195. https://doi.org/10.3354/esr00144
Ogden R, Gharbi K, Mugue N et al (2013) Sturgeon conservation genomics: SNP discovery and validation using RAD sequencing. Mol Ecol 22:3112–3123. https://doi.org/10.1111/mec.12234
Ogden R, Linacre A (2015) Wildlife forensic science: a review of genetic geographic origin assignment. Forensic Sci Int 18:152–159. https://doi.org/10.1016/j.fsigen.2015.02.008
Ogden R, McGough HN, Cowan RS et al (2008) SNP-based method for the genetic identification of ramin Gonystylus spp. timber and products: applied research meeting CITES enforcement needs. Endang Species Res 9:255–261. https://doi.org/10.3354/esr00141
Paredes-Villanueva K, Blanc-Jolivet C, Mader M et al (2019) Nuclear and plastid SNP markers for tracing Cedrela timber in the tropics. Conservation Genet Resour. https://doi.org/10.1007/s12686-019-01110-1
Paredes-Villanueva K, Espinoza E, Ottenburghs J et al (2018) Chemical differentiation of Bolivian Cedrela species as a tool to trace illegal timber trade. Forestry 91:603–613. https://doi.org/10.1093/forestry/cpy019
Pennington RT, Lavin M (2016) The contrasting nature of woody plant species in different neotropical forest biomes reflects differences in ecological stability. New Phytol 210:25–37. https://doi.org/10.1111/nph.13724
Pennington TD, Muellner AN (2010) A monograph of Cedrela (Meliaceae). dh books, Milborne Port, England, p 7
Pennington RT, Hughes M, Moonlight PW (2015) The origins of tropical rainforest hyperdiversity. Trends Plant Sci 20:693–695. https://doi.org/10.1016/j.tplants.2015.10.005
Perdiguero-Alonso D, Montero FE, Kostadinova A et al (2008) Random forests, a novel approach for discrimination of fish populations using parasites as biological tags. Int J Parasitol 38:1425–1434. https://doi.org/10.1016/j.ijpara.2008.04.007
Pérez-Del-Olmo A, Montero FE, Fernández M et al (2010) Discrimination of fish populations using parasites: random forests on a ‘predictable’ host-parasite system. Parasitology 137:1833–1847. https://doi.org/10.1017/S0031182010000739
Piry S, Alapetite A, Cornuet J-M et al (2004) GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539. https://doi.org/10.1093/jhered/esh074
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. https://doi.org/10.1093/bioinformatics/btq033
R. Core Team et al (2013) R: a language and environment for statistical computing
Rachmayanti Y, Leinemann L, Gailing O, Finkeldey R (2006) Extraction, amplification and characterization of wood DNA from Dipterocarpaceae. Plant Mol Biol Rep 24:45–55. https://doi.org/10.1007/BF02914045
Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci USA 94:9197–9201. https://doi.org/10.1073/pnas.94.17.9197
Rañola JM, Novembre J, Lange K (2014) Fast spatial ancestry via flexible allele frequency surfaces. Bioinformatics 30:2915–2922. https://doi.org/10.1093/bioinformatics/btu418
Saunders J, Reeve R (2014) The EU timber regulation and CITES. Chatham House, London. https://www.cifor.org/library/4503/. Accessed 28 Nov 2016
Schrider DR, Kern AD (2018) Supervised machine learning for population genetics: a new paradigm. Trends Genet 34:301–312. https://doi.org/10.1016/j.tig.2017.12.005
Sheikh PA, Bermejo LF, Procita K (2019) International illegal logging: background and issues. Congressional Research Service, Washington DC, USA
Stephan J, Stegle O, Beyer A (2015) A random forest approach to capture genetic effects in the presence of population structure. Nat Commun 6:7432. https://doi.org/10.1038/ncomms8432
Sun Y, Wong AKC, Kamel MS (2009) Classification of imbalanced data: a review. Int J Pattern Recognit Artif Intell 23:687–719. https://doi.org/10.1142/S0218001409007326
Sylvester EVA, Bentzen P, Bradbury IR et al (2018) Applications of random forest feature selection for fine-scale genetic population assignment. Evol Appl 11:153–165. https://doi.org/10.1111/eva.12524
Tnah LH, Lee SL, Ng KKS et al (2012) DNA extraction from dry wood of Neobalanocarpus heimii (Dipterocarpaceae) for forensic DNA profiling and timber tracking. Wood Sci Technol 46:813–825. https://doi.org/10.1007/s00226-011-0447-6
Urrunaga JM, Johnson A, Orbegozo ID, Mulligan F (2012) The laundering machine. Environmental Investigation Agency, Washington. https://eia-global.org/reports/the-laundering-machine. Accessed 9 Dec 2019
van Zonneveld M, Thomas E, Castañeda-Álvarez NP et al (2018) Tree genetic resources at risk in South America: a spatial threat assessment to prioritize populations for conservation. Divers Distrib 00:1–12. https://doi.org/10.1111/ddi.12724
Wasser SK, Brown L, Mailand C et al (2015) Genetic assignment of large seizures of elephant ivory reveals Africa’s major poaching hotspots. Science 349:84–87. https://doi.org/10.1126/science.aaa2457
Wasser SK, Shedlock AM, Comstock K et al (2004) Assigning African elephant DNA to geographic region of origin: applications to the ivory trade. Proc Natl Acad Sci USA 101:14847–14852. https://doi.org/10.1073/pnas.0403170101
Wasser SK, Torkelson A, Winters M et al (2018) Combating transnational organized crime by linking multiple large ivory seizures to the same dealer. Sci Adv 4:eaat0625. https://doi.org/10.1126/sciadv.aat0625
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370. https://doi.org/10.2307/2408641
White NE, Dawson R, Coghlan ML et al (2012) Application of STR markers in wildlife forensic casework involving Australian black-cockatoos (Calyptorhynchus spp.). Forensic Sci Int Genet 6:664–670. https://doi.org/10.1016/j.fsigen.2011.10.003
Wiedenhoeft AC, Simeone J, Smith A et al (2019) Fraud and misrepresentation in retail forest products exceeds U.S. forensic wood science capacity. PLoS ONE 14:e0219917. https://doi.org/10.1371/journal.pone.0219917
Wimmer V, Albrecht T, Auinger H-J, Schön C-C (2012) synbreed: a framework for the analysis of genomic prediction data using R. Bioinformatics 28:2086–2087. https://doi.org/10.1093/bioinformatics/bts335
Yang W-Y, Novembre J, Eskin E, Halperin E (2012) A model-based approach for analysis of spatial structure in genetic data. Nat Genet 44:725–731. https://doi.org/10.1038/ng.2285
Yu M, Jiao L, Guo J et al (2017) DNA barcoding of vouchered xylarium wood specimens of nine endangered Dalbergia species. Planta 246:1165–1176. https://doi.org/10.1007/s00425-017-2758-9
Zarraonaindia I, Iriondo M, Albaina A et al (2012) Multiple SNP markers reveal fine-scale population and deep phylogeographic structure in European anchovy (Engraulis encrasicolus L.). PLoS ONE 7:e42201. https://doi.org/10.1371/journal.pone.0042201
Acknowledgements
The authors thank the Missouri Botanical Garden Herbarium (MO) and the New York Botanical Garden (NYBG) for aiding in specimen collection, especially James Solomon of MO, Dennis Stevenson, Samantha Frangos, and Lisa DeGironimo of NYBG. Field collections and identification of samples from Peru would not have been possible without the help of Carlos Reynel and Aniceto Daza Yomona of the Herbario Weberbauer at Universidad Nacional Agraria La Molina. Sampling in Bolivia was carried out under the MMAYA/VMABCCGDF/DGBAP/MEG N° 0280/2016 authorization and samples were identified by Noel Kempff Mercado (Museo de Historia Natural, Santa Cruz, Bolivia). Research permits R.D. No. 001-2016- SERNANP-DGANP, R.D. No. 230-2016-SERFOR-DGGSPFFS, and Contrato No. 001-2016-SERFORDGGSPFFS-DGSPF were granted for field work and genetic analyses to the von Thüenen Institute of Forest Genetics and Instituto de Investigaciones de la Amazonía Peruana in Peru. We are grateful to Gabriel Hidalgo, Gerardo Flores, David Aldana, Luisa Huaratapairo, and Eduardo Mejía of for their support in collecting samples and extracting DNA. Ecuadorian samples provided by Thüenen were collected with the coordination of Stephen Cavers as part of the ‘SEEDSOURCE’ project, F96-2002-INCO-DEV-1 contract number 003708. We appreciate project coordination from Ashley Warriner and A. J. Doty (U.S. Forest Service International Programs), and informatics and sequencing assistance from the Center for Genome Research and Biocomputing at Oregon State University. Funding for this study was provided by U. S. Agency for International Development (Award 19318814Y0010-140001) to the U.S. Forest Service International Programs, the U.S.D.A. Forest Service Pacific Northwest Research Station, and the Moldenke Endowment (Botany and Plant Pathology Department, Oregon State University).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Finch, K.N., Cronn, R.C., Ayala Richter, M.C. et al. Predicting the geographic origin of Spanish Cedar (Cedrela odorata L.) based on DNA variation. Conserv Genet 21, 625–639 (2020). https://doi.org/10.1007/s10592-020-01282-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-020-01282-6