
Abstract
Owing to the prevalence of the JAK2V617F mutation in myeloproliferative neoplasms (MPNs), its constitutive activity, and ability to recapitulate the MPN phenotype in mouse models, JAK2V617F kinase is an attractive therapeutic target. We report the discovery and initial characterization of the orally bioavailable imidazopyridazine, LY2784544, a potent, selective and ATP-competitive inhibitor of janus kinase 2 (JAK2) tyrosine kinase. LY2784544 was discovered and characterized using a JAK2-inhibition screening assay in tandem with biochemical and cell-based assays. LY2784544 in vitro selectivity for JAK2 was found to be equal or superior to known JAK2 inhibitors. Further studies showed that LY2784544 effectively inhibited JAK2V617F-driven signaling and cell proliferation in Ba/F3 cells (IC50=20 and 55 nM, respectively). In comparison, LY2784544 was much less potent at inhibiting interleukin-3-stimulated wild-type JAK2-mediated signaling and cell proliferation (IC50=1183 and 1309 nM, respectively). In vivo, LY2784544 effectively inhibited STAT5 phosphorylation in Ba/F3-JAK2V617F-GFP (green fluorescent protein) ascitic tumor cells (TED50=12.7 mg/kg) and significantly reduced (P<0.05) Ba/F3-JAK2V617F-GFP tumor burden in the JAK2V617F-induced MPN model (TED50=13.7 mg/kg, twice daily). In contrast, LY2784544 showed no effect on erythroid progenitors, reticulocytes or platelets. These data suggest that LY2784544 has potential for development as a targeted agent against JAK2V617F and may have properties that allow suppression of JAK2V617F-induced MPN pathogenesis while minimizing effects on hematopoietic progenitor cells.
Similar content being viewed by others

Introduction
The myeloproliferative neoplasms (MPNs) are a group of hematologic malignancies characterized by clonal proliferation of one or more myeloid lineages that arise from a polyclonal stem cell pool. The three most common Philadelphia chromosome-negative MPNs are polycythemia vera, essential thrombocythemia and primary myelofibrosis.1 Bone marrow cells from patients with an MPN can form erythroid colonies in the absence of exogenous cytokines.2 Although MPN colony formation is an erythropoietin-independent phenomenon, it retains sensitivity to erythropoietin-signaling pathway inhibitors, including the janus kinase 2 (JAK2) inhibitor, AG490.3
An acquired JAK2 mutation occurs in most polycythemia vera patients (>95%) and a majority of patients with essential thrombocythemia or primary myelofibrosis (50–60%).4, 5, 6, 7, 8 This acquired point mutation (JAK2 1849G→T) results in a missense substitution of phenylalanine for valine at amino acid position 617 (V617F) within the autoinhibitory pseudokinase domain of JAK2. This substitution results in a gain-of-function activation of JAK2 with subsequent phosphorylation of STAT5 (signal transducer and activator of transcription 5), one of a family of proteins regulating the transcription of genes integral to cell growth, death and differentiation.4, 5, 6, 7, 8, 9, 10 The JAK2V617F mutant isoform is oncogenic, resulting in growth factor-independent growth in cells, and sufficient for generating MPN phenotypes in murine models.5, 11, 12, 13, 14 Collectively, these findings define a fundamental role for the JAK2V617F mutation in the pathogenesis of many MPN cases and suggest that this mutant JAK2 kinase isoform is a promising therapeutic target for the treatment of MPNs. Given the pivotal role that wild-type JAK2 has in multiple stages of hematopoiesis, it is postulated that the optimal treatment approach would seek to selectively inhibit JAK2V617F while minimizing inhibition of wild-type JAK2.
In this manuscript, we report the discovery and initial evaluation of LY2784544, an orally bioavailable imidazopyridazine aminopyrazole, that potently inhibits JAK2V617F-induced pathogenesis in vitro and in vivo.
Materials and methods
Screening for active molecules
To identify JAK2 inhibitor leads, a 52,000-compound library from the Lilly compound collection was screened using the Ź-LYTE kinase assay with the Tyrosine 6 peptide (2 μM) (Life Technologies, Grand Island, NY, USA).
Second-tier screening of 1000 of the most active molecules was performed using JAK2 or JAK3 biochemical assays with various formats (radioactive filter-binding, Transcreener ADP assay (Cisbio Bioassays, Bedford, MA, USA) with fluorescence polarization detection, or LanthaScreen Kinase assay (Life Technologies, Grand Island, NY, USA) and a multipoint evaluation. After synthesis and characterization of additional lead compounds, efforts focused on an active molecule referred to as ‘Compound 1’ and later to another active molecule referred to as ‘Compound 2’ or LY2784544. LY2784544 was then compared with ruxolitinib, SAR302503, AZD1480 and CYT387, all synthesized as reported in the literature.15, 16, 17, 18, 19, 20
In vitro evaluations
JAK2-STAT5 pathway cell-based assays
Test compounds and known JAK clinical inhibitors were evaluated for their ability to inhibit JAK-mediated phosphorylation of STAT5 as measured with a Cellomics-detection system and an AlphaScreen SureFire assay (TGR Biosciences, Thebarton, Adelaide, South Australia, Australia) in cell types expressing wild-type or constitutively active JAK1, JAK2 and/or JAK3.
Cellomics imaging assays
TF-1 cells expressing JAK2 were incubated overnight in RPMI with 0.6% fetal bovine serum to eliminate stimulation by endogenous cytokines, then plated (2 × 105 cells/96-well) in RPMI containing either vehicle (DMSO) or serial diluted test compounds (10—point, 1:3 serial dilutions from 20 μM to 0.001 μM). After a 10-min incubation, erythropoietin (1.6 units/ml) (R&D Systems, Minneapolis, MN, USA) was added for 20 min, then the cells were fixed, incubated with Mouse-antiphosphorylated-STAT5 (pY694) Alexa Fluor 647 (1:10 dilution) (BD Biosciences, San Jose, CA, USA), stained with Hoechst solution (2 μg/ml), and analyzed using Cellomics Arrayscan VTi (Thermo Scientific Cellomics Products, Pittsburgh, PA, USA). The relative IC50 was calculated using a four-parameter logistic curve-fitting analysis with ActivityBase 4.0 (IDBS, Guildford, Surrey, UK).
NK-92 cells expressing the JAK3/JAK1 heterodimer were incubated overnight in MEM Alpha medium with 0.3% fetal bovine serum and 0.3% horse serum. The assay proceeded as described for the TF-1 cells, except after the incubation with the test compound inhibitors, interleukin-2 (IL-2) (0.5 μg/ml) was substituted for erythropoietin. Measurement and data analysis were performed as described for TF-1 cells above.
AlphaScreen SureFire assays
Murine pro-B-cells (Ba/F3) cells expressing the constitutively active mutant JAK2 (Ba/F3-JAK2V617F-GFP (green fluorescent protein) cells) were plated in DMEM/high medium (2 × 104 cells/96-well) with either vehicle (DMSO) or test compounds (0.001–20 μM). After 5 h, cells were lysed, transferred to a 384-well Proxiplate, incubated (2 h) with AlphaScreen SureFire reaction mixture, then evaluated for phosphorylated STAT5 (EnVision 06 plate reader, PerkinElmer, Waltham, MA, USA) (Ex680 nm, Em520–620 nm). Ba/F3 wild-type JAK2 cells were assayed in a similar fashion, except the assay was preceeded by overnight culture in RPMI 1640 medium containing 0.6% fetal bovine serum to maximize IL-3 reactivity, then stimulated with mouse IL-3 (2 ng/ml) for 20 min after the 5-h incubation with test compounds. ActivityBase 4.0 was used to fit the percentage inhibition and 10-point compound concentration data to a four-parameter logistic equation.
Ba/F3-expressing constitutively active TEL-JAK1, TEL-JAK2 and TEL-JAK3 were assayed for phosphorylated STAT5 similarly to Ba/F3-JAK2V617F cells.21
In vivo target inhibition
Dose- and time-dependent in vivo inhibition of JAK2V617F signaling was assessed by measuring inhibition of STAT5 phosphorylation in a mouse ascitic tumor model. Ba/F3-JAK2V617F-GFP cells (1 × 107) were implanted in the intraperitoneal cavity of severe combined immunodeficiency mice (SCID, strain-CB17, Taconic, Hudson, NY, USA) and allowed to develop into ascitic tumors for 7 days. For dose-response studies (six animals/group), LY2784544 was administered once by oral gavage (2.5, 5.0, 10.0, 20.0, 40.0, or 80.0 mg/kg), then 30 min later, ascitic tumor cells were collected, fixed, incubated for 2 h with Mouse-anti-pSTAT5 (pY694) Alexa Fluor 647 (1:10 dilution), and analyzed by flow cytometry (Cytomic FC 500, BD Bioscience). Time course studies were performed similarly, except the animals were treated with LY2784544 at 20, 40 or 80 mg/kg and ascitic tumor cells collected at prespecified intervals of 0.25–6.0 h after dosing. Data were analyzed by the one-way analysis of variance, and Dunnett’s test (α=0.05). Dose response data were analyzed with a four-parameter logistic curve-fitting program (JMP Statistical Analysis software; SAS Institute Inc., Cary, NC, USA).
In vivo efficacy studies
Intravenous injection of Ba/F3-JAK2V617F-GFP cells into SCID mice results in intrasplenic accumulation and proliferation of the JAK2V617F-GFP cells.5 In this hematologic disease model, LY2784544 was administered twice daily (BID) by oral gavage (2.5, 5.0, 10.0, 20.0, 40.0, or 80.0 mg/kg), beginning 7 days after tumor cell infusion (1 × 107 cells/mouse, six mice/group). After 14 days of treatment, complete blood counts were obtained from cardiac puncture samples (Advia 120 Hematology Analyzer, Siemens, Washington, DC, USA). Spleens were weighed and splenocytes were assayed by fluorescence-activated cell sorting (FACS, FACSCanto, BD Biosciences) to determine the total number and percentage of GFP-positive cells (α=0.05; Dunnett’s test).
The effect of LY2784544 on hematopoiesis in vivo was examined in an 8-day version of the same mouse hematologic disease model, with 8 days specified as it corresponds to the timing necessary for IL-3- and granulocyte-macrophage colony-stimulating factor-mediated development of erythroid lineages from early burst-forming unit-erythrocytes to red cells.22 LY2784544 was administered at 5, 20, 40 or 80 mg/kg one day before tumor cell infusion and continued BID for 8 days, after which the cells were processed as in the 14-day study. FACS assays were then used to measure the reduction of Ba/F3-JAK2V617F-GFP-positive tumor cell burden and the effect on erythroid progenitor cells (stained with allophycocyaninconjugated Ter119 and phycoerythrin-conjugated CD71 monoclonal antibodies/BD Bioscience) in the splenocytes. A 7-amino-actinomycin D (BD Biosciences) was used to detect nonviable cells. Peripheral blood cell counts were also performed.
Pharmacokinetic (PK)/efficacy modeling
Population PK/efficacy modeling used the nonlinear mixed-effects modeling program (NONMEM, version 6.2) (ICON Development Solutions, Ellicott City, MD, USA). A murine PK model was developed using data from SCID mice with and without implanted ascitic tumors as the PK profile did not differ between the two groups of animals. A murine PK/efficacy model was also developed using data from the 14-day in vivo efficacy study in order to predict steady-state plasma concentration and corresponding exposures (area under the curve during the dosing interval, AUC0–12) at the end of the experiment (day 14). The decrease of the tumor burden (Ba/F3-JAK2V617F-GFP cells) in the spleen was described using an indirect response model,23 which assumed the drug produced an inhibitory effect on Ksyn, the zero-order rate constant, representing the rate of expansion of the tumor burden.
Additional methods
A detailed description of other methods used in this study, but not summarized here, may be found in the Supplementary Data document found online.
Results
Discovery of LY2784544
Screening of the Lilly compound library, resulted in the identification of potent JAK2 inhibitors from the indanyl imidazopyridine aminopyrazole series such as Compound 1: 3-[(1R)-6-fluoro-2,3-dihydro-1H-inden-1-yl]-N-(3-methyl-1H-pyrazol-5-yl)-3H-imidazo[4,5-b]pyridin-5-amine dihydrochloride (Figure 1a). Compound 1 had a fourfold selectivity in the second-tier screening assay for JAK2 (IC50=2 nM) over JAK3 (IC50=8 nM) (Table 1). Specificity for JAK2 over JAK3 was confirmed using cell-based assays monitoring STAT5 phosphorylation after stimulation with erythropoietin in TF-1 cells (JAK2 homodimer-mediated signal, IC50=35 nM) or IL-2 in NK-92 cells (JAK3/JAK1 heterodimer-mediated signal, IC50=228 nM) (Table 1).

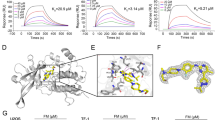
Chemical structures of JAK1/2 dual inhibitor Compound 1 and JAK2 inhibitor Compound 2 (LY2784544). (a) Chemical structure of imidazopyridine Compound 1: 3-[(1R)-6-fluoro-2,3-dihydro-1H-inden-1-yl]-N-(3-methyl-1H-pyrazol-5-yl)-3H-imidazo[4,5-b]pyridin-5-amine dihydrochloride. (b) Chemical structure of imidazopyridazine aminopyrazole Compound 2 (LY2784544): 3-(4-chloro-2-fluorobenzyl)-2-methyl-N-(5-methyl-1H-pyrazol-3-yl)-8-(morpholinomethyl)imidazo[1,2-b]pyridazin-6-amine. (c) High-resolution (1.8 Å) X-ray crystal structure of LY2784544 in JAK2 kinase. JAK, janus kinase.
Evaluation of Compound 1 in the 14-day rat immunotoxicology screen revealed a statistically significant decrease in the percentage of peripheral blood and splenic T-cytotoxic cells at pharmacologically active exposures (Table 1). These results suggested the need for a molecule with greater selectivity for JAK2 over JAK3 to minimize potential of JAK3-mediated effects on T-cell populations. Our strategy focused on identifying compounds that were potent in the TF-1 JAK2 cell-based assay (IC50<50 nM) and had an IC50>900 nM in the NK-92 JAK3/JAK1 heterodimer cell-based assay.
A high-resolution X-ray crystal structure of JAK2 with an indanyl imidazopyridine aminopyrazole revealed that the indanyl substituent was in a pocket close to the Gly993 residue. In JAK3, Ala966 is in the same relative position as Gly993 in JAK2. On the basis of the more sterically-demanding ATP-binding pocket in JAK3, it was hypothesized that incorporation of larger substituents on the phenyl ring at the position para to the connection with the bicyclic core would result in compounds with improved selectivity for JAK2 over JAK3. A series of compounds with various para-substituted aryl analogs and core modifications were designed, synthesized, and tested, resulting in the identification of a promising candidate, Compound 2 (LY2784544), the 4-chloro-2-fluorobenzyl imidazopyridine (Figure 1b). The high-resolution (1.8 Å) X-ray crystal structure of LY2784544, confirmed that the bulky chloro-substituent at the para position of the phenyl ring binds close to Gly993. In addition, the structure exhibited a favorable Bürgi-Dunitz interaction24 between the 4-chlorophenyl moiety of LY2784544 and two carbonyls of JAK2 (Gly993 and Asn981) (Figure 1c).
In the second-tier biochemical screening assay for JAK2, LY2784544 demonstrated 8- and 16-fold selectivity for JAK2 over JAK1 and JAK3, respectively (Table 1 and Supplementary Table 1). We assessed LY2784544 activity against a CEREP kinase panel (total of 99 kinases). Of these, only 20 kinases were inhibited by LY2784544 with IC50 (three concentration points) <0.3 μM (for details, see Supplementary Table 2). Furthermore, LY2784544 was potent in the cell-based TF-1 JAK2 assay (IC50=45 nM) and had the desired threshold selectivity in the NK-92 JAK3/JAK1 heterodimer assay (942 nM) (Table 1). The observed selectivity from the cellular assays was supported by the 14-day rat immunotoxicology studies, in which LY2784544 did not decrease the percentage of T-cytotoxic cells at pharmacologically active exposures (Table 1). No gastric- and intestine-related toxicity was observed in a 14-day rat pilot toxicology study after treatment with LY2784544 at 30, 60, or 100 mg/kg.
Evaluation of LY2784544 and four other JAK2 inhibitors in TEL-JAK1-, JAK2- and JAK3-expressing Ba/F3 cells
Initial understanding of the selectivity of LY2784544 for JAK2 and JAK3 was determined in cell-based assays mediated either by homodimerization of erythropoietin receptors and resulting in recruitment and activation of JAK2 proteins, or heterodimerization of IL-2 receptors (subunits α, ß, and γ) and resulting activation of JAK1 and JAK3 proteins. The complexity of JAK2 and JAK3 pathway activation in these assays may affect our understanding of JAK2 and JAK3 selectivity. To further examine selectivity, the inhibition of JAK-STAT5 signaling by LY2784544 and ruxolitinib were evaluated in Ba/F3 cells expressing ligand-independent, constitutively active TEL-JAK1, TEL-JAK2 and TEL-JAK3 fusion kinases by western blotting. As shown in Figure 2, both LY2784544 and ruxolitinib more effectively inhibited the phosphorylation of STAT5 in Ba/F3-TEL-JAK2 cells than in Ba/F3-TEL-JAK1 and JAK3 cells at the indicated concentrations. In comparison with ruxolitinib, LY2784544 was less effective at inhibiting STAT5 phosphorylation in TEL-JAK1 cells at indicated concentrations. To quantify the potency of LY2784544 and four other clinical JAK2 inhibitors in TEL-JAK cellular systems, more quantitative Alpha-Screen SureFire pSTAT5 assays were utilized to determine the IC50. As indicated in Table 2, LY2784544 was more selective for JAK2 than for JAK1 and JAK3 with an IC50 of 0.191 (JAK2), 2.904 (JAK1) and 4.744 μM (JAK3), suggesting 15- and 25-fold greater selectivity for JAK2 than for JAK1 and JAK3, respectively. In comparison, AZD1480 and ruxolitinib were not as selective as LY2784544 for JAK2 over JAK1 (Table 2).

LY2784544 selectively inhibits STAT5 phosphorylation in Ba/F3 cells expressing TEL-JAK2 than Ba/F3 cells expressing TEL-JAK1 and JAK3. Ba/F3 cells expressing TEL-JAK1, TEL-JAK2 and TEL-JAK3 were treated with the indicated concentrations of LY2784544, ruxolitinib or DMSO (0, vehicle control) for 2 h and cell lysates were probed for pSTAT5y694 and total STAT5 by western blotting. Ba/F3, murine pro-B-cells; DMSO, dimethylsulfoxide; JAK, janus kinase; STAT5, signal transducer and activator of transcription 5; TEL, translocated ets leukemia.
Assessment of LY2784544 inhibition of JAK2V617F-induced signaling and cell proliferation
As the JAK2V617F mutation has a fundamental role in the pathogenesis of several MPNs, we investigated inhibition of JAK2 signaling, as measured by STAT5 phosphorylation in JAK2V617F-induced hematological malignancy models. JAK2V617F and wild-type JAK2 complementary DNA were stably expressed in Ba/F3 cells. We demonstrated that V617F mutation activates the JAK2-STAT5 pathway, and induces cytokine-independent growth in Ba/F3 cells without expression of the erythropoietin receptor.25
As demonstrated by western blot (Figure 3a), both LY2784544 and ruxolitinib potently inhibited STAT5 phosphorylation in Ba/F3 cells expressing JAK2V617F across a concentration range from 50 nM–20 μM. LY2784544 showed marginal inhibition of STAT5 phosphorylation in IL-3-stimulated Ba/F3 cells expressing wild-type JAK2 at 50 and 200 nM concentrations. In contrast, ruxolitinib showed substantial inhibition of STAT5 phosphorylation (Figure 3b) at all three indicated concentrations (50 nM, 200 nM and 20 μM). Furthermore, LY2784544 effectively inhibited STAT5 phosphorylation in HEK293 cells expressing JAK2V617F and two other JAK2 mutants (H538Q and R564Q) with much less potency against the JAK2Y931C mutant (Figure 3c).

Inhibition of mutant JAK2 and IL-3-wild-type-JAK2-activated STAT5 phosphorylation by JAK2 inhibitors in Ba/F3 and HEK293T cells. (a) STAT5 phosphorylation in Ba/F3 cells expressing JAK2V617F was inhibited by LY2784544 and ruxolitinib for 2 h and then whole-cell lysates were probed for pSTAT5y694 and total STAT5 by western blotting. (b) Inhibition of IL-3-stimulated STAT5 phosphorylation in Ba/F3 cells expressing wild-type JAK2. Western blot of whole-cell lysates prepared from IL-3-stimulated Ba/F3 cells treated with the indicated concentrations of LY2784544 and ruxolitinib. Cells were pretreated with JAK2 inhibitors for 2 h, then stimulated with IL-3 (2 ng/ml) for 20 min. (c) Inhibition of STAT5 phosphorylation in HEK293T cells expressing JAK2 mutants. Western blot of whole-cell lysates prepared from HEK293T cells expressing JAK2V617F, H538Q, R564Q and Y931C mutants treated by LY2784544 for 1 h. Ba/F3, murine pro-B-cells; HEK, human embryonic kidney; IL, interleukin; JAK, janus kinase; STAT5, signal transducer and activator of transcription 5.
Inhibition of JAK2-STAT5 signaling by LY2784544 and other clinical JAK2 inhibitors was further evaluated in the robust and quantitative AlphaScreen SureFire pSTAT5 assay with Ba/F3 cells expressing either JAK2V617F or IL-3-activated wild-type JAK2. As seen in Table 3, LY2784544 potently inhibited the JAK2V617F signaling (IC50=20 nM) but, remarkably, showed very minimal activity against the IL-3-activated wild-type JAK2 signaling with an IC50 of 1183 nM. In comparison, ruxolitinib and AZD1480 were not only potent inhibitors of JAK2V617F signaling with an IC50 of 24 and 19 nM, respectively, but also were very active inhibitors of IL-3-wild-type JAK2-STAT5 signaling with an IC50 of 121 and 181 nM, respectively. SAR302503 and CYT387 inhibited IL-3-stimulated wild-type JAK2-dependent signaling at concentrations comparable to LY2784544, but both were less potent at inhibiting JAK2V617F compared with LY2784544 (102 and 200 nM, respectively).
As JAK2-STAT5 signaling has a role in MPN cell proliferation, LY2784544 was next evaluated in the MTS cell proliferation assay utilizing the same Ba/F3 cells expressing JAK2V617F or IL-3-activated wild-type JAK2. Consistent with findings from the signaling studies, LY2784544 inhibited the proliferation of JAK2V617F-expressing cells (IC50=55 nM) and was markedly less potent as an inhibitor of the proliferation of IL-3-stimulated wild-type JAK2 expressing Ba/F3 cells (IC50=1309 nM) (Table 3). Although JAK2 signaling is known to contribute to cell proliferation, the observed anti-proliferative effect of LY2784544 may also be related, at least in part, to an induction in apoptosis of JAK2V617F-expressing cells. LY2784544 effectively induced apoptosis in JAK2V617F-expressing cells with a comparable EC50 ±s.d. of 113±0.023 nM as measured by Caspase3/7-glo assays (for details, see Supplementary Figure 1 and Supplementary Table 3). In contrast to LY2784544, ruxolitinib and AZD1480 were more potent inhibitors of IL-3-dependent growth of wild-type JAK2 Ba/F3 cells (IC50=331 and 406 nM, respectively). The mechanisms of action of LY2784544 binding to wild-type and V617F JAK2 were analyzed by a kinetic ATP-Based MoA assay with using purified wild-type and V617F JAK2 proteins. The results did not display difference in the binding affinity of LY2784544 to V617F and wild-type JAK2 in these biochemical assays (for details, see Supplementary Table 4 and Figure 2).
Dose-, exposure- and time-dependent inhibition of JAK2V617F signaling in a mouse ascitic tumor model
An in vivo murine model utilizing Ba/F3-JAK2V617F ascitic tumors was developed as a FACS-based surrogate system with ready access to tumor cells for use in understanding the correlation between pharmacodynamics and PK of JAK2 inhibition. Testing of LY2784544 in SCID mice bearing Ba/F3-JAK2V617F-GFP, ascitic tumors demonstrated potent, dose- and exposure-dependent inhibition of STAT5 phosphorylation as demonstrated in Figure 4a (FACS histograms and bar graphs) and Figure 4b, respectively. The dose and plasma concentration necessary to inhibit 50% of STAT5 phosphorylation (threshold effective dose (TED50) and threshold effective concentration (TEC50)) were determined to be 12.7 mg/kg and 699.0 ng/ml (1.49 μM), respectively, at 30 min after dosing.

LY2784544 effectively inhibits JAK2V617F-STAT5 signaling in ascitic tumor cells from Ba/F3-JAK2V617F-GFP tumor-bearing mice. (a) Dose-dependent inhibition of JAK2V617F STAT5 signaling. Oral LY2784544 was administered at the indicated doses, with ascitic cells harvested 30 min later and STAT5 phosphorylation measured by FACS. Left: representative histograms showing decreased pSTAT5Y694-positive ascitic tumor cells collected from animals treated with LY2784544 in dose-dependent manner. Right: quantification of decreased pSTAT5Y694-positive ascitic tumor cells shown in the histograms. The horizontal line indicates the threshold effective dose (TED50). Standard error is indicated by the error bars with statistically significant inhibition, defined as P<0.05 using Dunnett’s test, indicated by an asterisk (*). (b) Plasma concentration–dependent inhibition of JAK2V617F activation. Blood was collected 30 min after oral LY2784544 exposure, and the concentration of LY2784544 was measured in the plasma and plotted versus percentage inhibition of STAT5 phosphorylation. Ba/F3, murine pro-B-cells; FACS, fluorescence-activated cell sorting; GFP, green fluorescent protein; JAK, janus kinase; STAT5, signal transducer and activator of transcription 5. TED50, threshold effective dose required to cause 50% inhibition.
The duration for the statistically significant inhibition of STAT5 phosphorylation was dose-dependent and ranged from at least 2 h after the 40 mg/kg dose (Figure 5a) to at least 6 h after the 80 mg/kg dose (Figure 5b, histograms and bar graph).

LY2784544 inhibits JAK2V617F signaling in a time-dependent manner in Ba/F3-JAK2V617F-GFP ascitic tumor cells after treatment with a single dose in SCID mice. LY2784544 was administered orally at (a) 40 mg/kg or (b) 80 mg/kg. The inhibition of STAT5 phosphorylation was measured by FACS at the indicated intervals from 0–6 h. Left: representative histograms showing decreased pSTAT5Y694-positive ascitic tumor cells collected from animals treated with LY2784544 in a time-dependent manner. Right: quantifications of pSTAT5Y694 FACS data shown in the histograms. Standard error is indicated by the error bars with statistically significant inhibition, defined as P<0.05 using Dunnett’s test, indicated by an asterisk (*). In some cases, there was a higher variability among replicas; this included the vehicle control group at 4 and 6 h at the 40-mg/kg dose. Ba/F3, murine pro-B-cells; FACS, fluorescence-activated cell sorting; GFP, green fluorescent protein; JAK, janus kinase; SCID, severe combined immunodeficiency; STAT5, signal transducer and activator of transcription 5.
In vivo efficacy of LY2784544 in JAK2V617F-induced mouse MPN model
The efficacy of LY2784544 was also evaluated in a previously described Ba/F3-JAK2V617F-induced SCID mouse hematologic disease model.26, 27 Following 14 days of BID treatment with LY2784544, tumor bearing mice exhibited a dose-dependent reduction in both splenomegaly and Ba/F3-JAK2V617F-GFP tumor burden (Figures 6a and b). Immunohistochemistry examination (Figure 6d) demonstrated a dramatic decrease in the GFP-immunolabeled Ba/F3-JAK2V617F cells in the 20mg/kg treatment group (∼50%; Figure 6b). Spleen mass was also reduced by about 20% in this treatment group (Figure 6a) suggesting the splenic weight reduction is largely due to the selective elimination of Ba/F3-JAK2V617-GFP-positive tumor cells. The TED50 for tumor burden reduction was determined to be 13.7 mg/kg, a result roughly similar to the TED50 for JAK2V617F signaling (TED50=12.7 mg/kg).

Reduction of splenomegaly and Ba/F3-JAK2V617F-GFP-positive tumor cell burden in a JAK2V617-induced mouse hematologic disease model after treatment with LY2784544, twice daily, for 14 days. LY2784544 was administered orally twice daily for 14 days at the specified doses, 7 days after intravenous infusion of tumor cells, with six mice per dosage or time point group, and a six-mouse vehicle control group. Standard error is indicated by the error bars with statistically significant inhibition, defined as P<0.05 using Dunnett’s test, indicated by an asterisk (*). (a) Dose-dependent inhibition of splenomegaly in LY2784544-treated groups. Percentage inhibition of splenomegaly was determined by comparing the mean spleen mass per dose group with the mean of the untreated control group. (b) Dose-dependent reduction of Ba/F3 JAK2V617F-GFP-positive tumor cell burden (total GFP-positive cells) in spleen as measured by flow cytometry. Splenocytes were isolated from the spleens of mice treated with LY2784544 at the indicated doses. Total GFP-positive Ba/F3-JAK2V617F cells were measured and determined by flow cytometry analysis. The horizontal line indicates the threshold effective dose (TED50). (c) Actual versus predicted efficacy as a function of the area under the plasma concentration-time curve for the dosing interval (AUCtau). A mouse PK/efficacy model was developed using the JAK2V617-induced MPN mouse model, in which LY2784544 was administered every 12 h (AUC0-12). The model-predicted data are represented with the actual data superimposed. (d) Immunostaining of spleen tissue from vehicle and LY2784544-treated Ba/F3-JAK2V617F-GFP mice with antibody against GFP. In the left column of the immunohistochemistry images, the brown staining indicates antibody binding. The right column of the fluorescent images reveals antibody staining as yellow, orange, and red. Brightfield microscopy images were acquired at room temperature using a whole-slide imaging system, Aperio's Scanscope XT (Aperio Technologies, Inc., Vista, CA, USA), at a × 20 magnification setting. The image analysis was performed using Aperio’s Positive Pixel Count Algorithm (version 9.1) (Aperio Technologies, Inc.) using the default parameter settings. AUC, area under the curve; JAK, janus kinase; Ba/F3, murine pro-B-cell; GFP, green fluorescent protein; MPN, myeloproliferative neoplasm.
The PK/efficacy model demonstrated the relationship between LY2784544 exposure and declining tumor burden in the spleen (Figure 6c) and can be used to predict the median exposure needed to achieve the 50% reduction in tumor cells and the dose range for human efficacy (up to 1660 mg daily).
To explore the effects of LY2784544 on the development of normal hematopoiesis, a similar experiment with this model was carried out. On the basis of previous study results that showed IL-3 and granulocyte-macrophage colony-stimulating factor cytokines mediated the development of erythroid lineages from early burst-forming unit-erythrocytes to red cells in 7–9 days,22 an 8 day study period was used and JAK2V617F-GFP positive and CD71+/Ter119+ erythroid progenitor cells were analyzed by FACS. This in vivo efficacy study again demonstrated a dose-dependent reduction in the percentage of Ba/F3-JAK2V617F-GFP-positive cells after treatment at the two highest doses tested (40 and 80 mg/kg, BID × 14 days; P=8 × 10−4 and 8 × 10−7, respectively) (Figure 7a). However, as shown in Figure 7b (representative histograms and bar graph), LY2784544 did not affect the normal erythroid progenitor (CD71+/Ter119+) cells at the dosing range of 5–80 mg/kg (P>0.99). In addition, a complete blood count was performed and showed that LY2784544 treatment did not reduce reticulocytes during this study period (Figure 7c).

LY2784544 selectively reduces Ba/F3-JAK2V617F-GFP-positive tumor cell burden in mice after oral treatment. LY2784544 was administered orally, twice daily for 8 days at the specified doses, starting 1 day before intravenous infusion of tumor cells, with six mice per dosage group, and a six-mouse vehicle control group. Standard error is indicated by the error bars with statistically significant inhibition, as defined as P<0.05 using Dunnett’s test, indicated by an asterisk (*). (a) LY2784544 significantly reduces Ba/F3-JAK2V617F-GFP tumor burden after treatment for 8 days. Ba/F3-JAK2V617F-GFP-positive cells in spleens were isolated and analyzed by flow cytometry. LY2784544 significantly reduced the percentage of Ba/F3-JAK2V617F-GFP-positive cells at 40 mg/kg (P=8 × 10−4) and 80 mg/kg (P=8 × 10−7). (b) LY2784544 shows no effect on erythroid progenitors. Left: representative FACS histograms showing no effect of LY2784544 (5–80 mg/kg) on CD71/Ter119 double positive cell populations (gated), however, tumor-bearing animals displayed a significantly reduced number of erythroid progenitors, as demonstrated in animals with no tumors versus vehicle controls. Right: quantification of CD71+/Ter119 FACS data. (c) LY2784544 shows no effect on peripheral blood reticulocytes in all treated groups. (d) LY2784544 restores the platelet count to levels observed in non-tumor-bearing mice. Peripheral blood platelet count in the tumor-bearing vehicle control animals was significantly reduced compared with non-tumor-bearing animals (P=6 × 10−5). Treatment with LY2784544 at 80 mg/kg significantly reversed the tumor-dependent suppression of platelet counts (P=1 × 10−6), returning the platelet count to levels observed in non-tumor-bearing mice. Ba/F3, murine pro-B-cells; CD, cluster of differentiation; FACS, fluorescence-activated cell sorting; GFP, green fluorescent protein; JAK, janus kinase.
The growth of Ba/F3-JAK2V617F-GFP cells disrupted murine erythropoiesis and thrombopoiesis. As shown in Figure 7b (histogram and bar graph), splenic erythroid progenitor cell numbers (CD71+/Ter119+) were significantly reduced in the tumor-bearing vehicle control animals (11.2±3.8% compared with 26.5±5.0% in the non-tumor-bearing animals; P=8 × 10−5) (Figure 7b). In addition, peripheral blood platelet counts are also significantly reduced, by 50%, in the tumor-bearing vehicle control animals as compared with non-tumor-bearing animals (from 1004 × 103–495 × 103/μl; P=6 × 10−5, Figure 7c). Treatment with LY2784544 at 80 mg/kg, BID, for 8 days significantly reversed the tumor-dependent suppression of platelet counts and allowed the platelet count to recover to 1043 × 103/μl (P=10−6) (Figure 7d). Taken together, these results showed that LY2784544 effectively reduced Ba/F3-JAK2V617F-GFP cells with minimal effects on the development of wild-type JAK2-mediated erythroid progenitors and reticulocytes in this period of observation.
Discussion
This report summarizes the discovery and initial characterization of LY2784544, an orally bioavailable, small-molecule inhibitor from the imidazopyridazine aminopyrazole series, capable of potent ATP-competitive inhibition of JAK2 tyrosine kinase. An evaluation of the inhibition of STAT5 phosphorylation in JAK2-dependent cell-based assays demonstrated that LY2784544 was equipotent to ruxolitinib and AZD1480, and more potent than SAR302503 and CYT387. Like the other JAK2 inhibitors tested, LY2784544 was found to lack significant activity in a TEL-JAK3 cell-based assay, but LY2784544 was found to have a greater degree of selection for JAK2 over JAK1 than either ruxolitinib or AZD1480. These results suggest that LY2784544 is a small molecule inhibitor that is more selective for JAK2 than for JAK1.
Further characterization showed that Ba/F3 cells expressing JAK2V617F were more sensitive to LY2784544 than were IL-3-stimulated Ba/F3 cells expressing wild-type JAK2. This sensitivity was evident for both the inhibition of JAK2V617F-STAT5 signaling and JAK2V617F-induced cell growth. LY2784544 was shown to be less potent in IL-3-treated wild-type JAK2-dependent cellular assays than ruxolitinib and AZD1480, suggesting it may have less effect on IL-3-dependent erythroid lineage development.
The JH1 inhibitor complex for LY2784544 has a different 3-D shape from the complex with ruxolitinib and AZD1480 (unpublished results and PDB accession code 2XA4). One explanation for the difference in the potency observed in the cell-based assay systems is the presumed different interaction between JH1 inhibitor complex and the JH2 domain, where the mutation occurs. Alternatively, other protein-protein interactions may be involved. For example, it has been previously reported that there is a difference in inhibitory regulation of wild-type and V617F-mutant JAK2. Suppressors of cytokine-signaling proteins, SOCS1 and SOCS3, are normally capable of binding to wild-type JAK2 and inhibiting its kinase activity;28, 29 however, expression of SOCS3 appears to paradoxically increase JAK2V617F protein stability, resulting in increased phosphorylation of both SOCS3 and JAK2V617F.30
LY2784544 was also found to effectively induce apoptosis and inhibit cell proliferation in Ba/F3-JAK2V617F-GFP cells at concentrations (EC50=113 nM and IC50=55 nM, respectively) that correlate well with the inhibition of STAT5 phosphorylation (20 nM) in the Ba/F3 cell model. These results suggest that the growth inhibition observed with LY2784544 treatment could be explained by induction of the apoptotic pathway through the inhibition of the JAK2V617F-STAT5 pathway in the mutant JAK2-expressing cells.30 Together, these in vitro data indicate that this molecule may have the potential to effectively reduce the JAK2V167F-induced MPN pathogenesis.
Good correlations were observed between in vivo and in vitro activity assessments of LY2784544. In both the JAK2V617F murine ascitic tumor model and a mouse JAK2V617F hematologic disease model, LY2784544 inhibited the JAK2V617F target in a dose- and time-dependent manner. The TED50 of 12.7 mg/kg identified in the murine ascitic tumor model that measures inhibition of STAT5 phosphorylation was similar to the TED50 of 13.7 mg/kg identified in the 14-day MPN disease model that measured reduction in splenic Ba/F3 cells expressing JAKV617F. The close agreement of these observations suggest that tumor cell reduction is linked with pharmacological inhibition of JAK2V617F signaling. LY2784544 was also found to significantly reduce the number of Ba/F3 cells expressing JAK2V617F by 50% or more at 20, 40 or 80 mg/kg, BID.
FACS analysis of the hematopoietic progenitor lineage of cells, isolated from the spleens of mice carrying JAK2V617F-expressing Ba/F3 cells, confirmed that LY2784544 effectively inhibits JAK2V617F-induced MPN phenotype. LY2784544 significantly reduced the Ba/F3-JAK2V617F-GFP tumor burden in the high-dose groups (40 and 80 mg/kg, BID) as compared with the vehicle-control mice. In contrast, this molecule had no effect on erythroid progenitor cells (CD71+/Ter119+) in all treated groups. The decrease in CD71+/Ter119+ cells (from 30 to 10%) and platelet count (from 1004 × 103 to 495 × 103/μl) (Figures 7b and d) in untreated tumor-bearing animals is evidence of the aggressive growth of Ba/F3-JAK2 V617F-GFP tumors and the associated disruption of normal hematopoiesis in SCID mice. It is therefore an intriguing observation that BID treatment with LY2784544 at 80 mg/kg restored platelet counts to the same levels (1043 × 103/μl, P=10−6) as that in the non-tumor-bearing animals. These results suggest that pharmacological exposures at the highest dose level tested in these studies may not suppress thrombopoiesis or erythropoiesis, consistent with the cell-based demonstrations of sensitivity of JAK2V617F-expressing cells to LY2784544. Collectively, these results indicate that LY2784544 blocks JAK2V617F signaling, suppressing the growth of malignant cells expressing mutant JAK2 without a significant effect on normal hematopoietic progenitors that express wild-type JAK2 at the doses and treatment duration studied.
In vitro and in vivo studies indicate that LY2784544, a JAK2 selective inhibitor, is very potent against JAK2V617-induced mouse hematological diseases. Although capable of inhibiting JAK2V716F-dependent signaling, cell proliferation and splenomegaly, and capable of inducing apoptosis of JAK2V617F-expressing cells, treatment with LY2784544 was observed to have little impact on wild-type JAK2-dependent cell numbers in the murine hematologic disease models. If these observations hold true in translation to human MPN clinical testing, LY2784544 may provide a potential clinical advantage in targeting JAK2V617F-induced MPN pathogenesis while minimizing effect on normal progenitor cells in patients with a JAK2V617F MPN. Currently, LY2784544 is being evaluated in MPN patients in two Phase I trials to investigate dose and schedule (I3X-MC-JHTA, NCT01134120; and I3X-MC-JHTC, NCT01520220), and a Phase II study to investigate efficacy (I3X-MC-JHTB, NCT01594723).
References
The International Agency for Research on Cancer. In Swerdlow S, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H et al (eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues 4th edn Lyon, France: International Agency for Research on Cancer (IARC), 2008.
Prchal JF, Axelrad AA . Letter: bone-marrow responses in polycythemia vera. N Engl J Med 1974; 290: 1382.
Ugo V, Marzac C, Teyssandier I, Larbret F, Lécluse Y, Debili N et al. Multiple signaling pathways are involved in erythropoietin-independent differentiation of erythroid progenitors in polycythemia vera. Exp Hematol 2004; 32: 179–187.
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365: 1054–1061.
James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434: 1144–1148.
Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352: 1779–1790.
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005; 7: 387–397.
Zhao R, Xing S, Li Z, Fu X, Li Q, Krantz SB et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem 2005; 280: 22788–22792.
Vannucchi AM, Antonioli E, Guglielmelli P, Rambaldi A, Barosi G, Marchioli R et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007; 110: 840–846.
Barosi G, Bergamaschi G, Marchetti M, Vannucchi AM, Guglielmelli P, Antonioli E et al. JAK2 V617F mutational status predicts progression to large splenomegaly and leukemic transformation in primary myelofibrosis. Blood 2007; 110: 4030–4036.
Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL . JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood 2006; 108: 1652–1660.
Bumm TG, Elsea C, Corbin AS, Loriaux M, Sherbenou D, Wood L et al. Characterization of murine JAK2V617F-positive myeloproliferative disease. Cancer Res 2006; 66: 11156–11165.
Zaleskas VM, Krause DS, Lazarides K, Patel N, Hu Y, Li S et al. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PLoS One 2006; 1: e18.
Xing S, Wanting TH, Zhao W, Ma J, Wang S, Xu X et al. Transgenic expression of JAK2V617F causes myeloproliferative disorders in mice. Blood 2008; 111: 5109–5117.
Burkholder TM, Clayton JR, Ma L Amino pyrazole compound. Eli Lilly and Company, USA. US patent 7,897,600 B2. 1 March 2011.
Rodgers JD, Shepard S Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors. Incyte Corporation, USA. US patent 7,598,257 B2. 6 October 2009.
Cao JJ, Hood J, Lohse D, Mak CC, McPherson A, Noronha G et alBi-aryl meta-pyrimidine inhibitors of kinases. TargeGen, Inc., USA. PCT Int. Appl. (2007), 336 pp. CODEN: PIXXD2 WO 2007053452 A1. 2007 May 10.
Feng X, Guan H, Kan Y, Ioannidis S, Peng B, Su M et alAstraZeneca AB, Sweden; AstraZeneca UK Limited. 4-(3-aminopyrazol) pyrimidine derivatives for use as tyrosine kinase inhibitors in the treatment of cancer. PCT Int. Appl. (2007), 92 pp. CODEN: PIXXD2 WO 2007049041 A1. 2007 May 3.
Burns CJ, Donohue AC, Feutrill JT Cytopia Research Pty Ltd, Australia. Phenyl amino pyrimidine compounds and uses thereof. PCT Int. Appl. (2008), 104 pp. CODEN: PIXXD2 WO 2008109943 A1. 2008 September 18..
Mitchell D, Cole KP, Pollock PM, Coppert DM, Burkholder TP, Clayton JR et al. Development and a practical synthesis of the JAK2 inhibitor LY2784544. Org Process Res Dev 2012; 16: 70–81.
Lacronique V, Boureux A, Monni R, Dumon S, Mauchauffé M, Mayeux P et al. Transforming properties of chimeric TEL-JAK proteins in Ba/F3 cells. Blood 2000; 95: 2076–2083.
Constantinescu SN, Ghaffari S, Lodish HF . The erythropoietin receptor: structure, activation and intracellular signal transduction. Trends Endocrinol Metab 1999; 10: 18–23.
Sharma A, Jusko WJ . Characteristics of indirect pharmacodynamics models and application to clinical drug responses. Br J Clin Pharmacol 1998; 45: 229–239.
Biirgi HB, Dunitz JD, Shefter E . Geometrical reaction coordinates. II. Nucleophilic addition to a carbonyl group. J Am Chem Soc 1973; 95: 5065–5067.
Funakoshi-Tago M, Pelletier S, Moritake H, Parganas E, Ihle JN . Jak2 FERM domain interaction with the erythropoietin receptor regulates Jak2 kinase activity. Mol Cell Biol 2008; 28: 1792–1801.
Pardanani A, Hood J, Lasho T, Levine RL, Martin MB, Noronha G et al. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia 2007; 21: 1658–1668.
Quintás-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA et al. Preclinical characterization of the selective JAK1/2 inhibitor RUXOLITINIB: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 2010; 115: 3109–3117.
Nicholson SE, Willson TA, Farley A, Starr R, Zhang JG, Baca M et al. Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. EMBO J 1999; 18: 375–385.
Sasaki A, Yasukawa H, Shouda T, Kitamura T, Dikic I, Yoshimura A . CIS3/SOCS-3 suppresses erythropoietin (EPO) signaling by binding the EPO receptor and JAK2. J Biol Chem 2000; 275: 29338–29347.
Hookham MB, Elliott J, Suessmuth Y, Staerk J, Ward AC, Vainchenker W et al. The myeloproliferative disorder-associated JAK2 V617F mutant escapes negative regulation by suppressor of cytokine signaling 3. Blood 2007; 109: 4924–4929.
Acknowledgements
The authors thank Lilly employees James A Cook, Bryan D Anderson and Sherri Hoover for performing the JAK LanthaScreen TR-FRET and additional biochemical assays; Xiu-Juan Yaun, Wayne D Blosser and Jeffery K Smallwood for the cell-based assay development; Lisa M Kays and Robert T Foreman for carrying out in vivo studies; Denis J McCann for PK study support; Yue-wei Qian and Mariam E Ehsani for expression and purification of JAK2 proteins; Gregory Donoho and Douglas J Zeckner for construction of JAK2V617F-expressing cell lines; James Alston for IHC staining; Jeffrey Hanson for imaging analysis support; Christopher A Slapak, Jonathan M Yingling, James J Starling, Richard B Gaynor, and Kerry L Blanchard for their guidance and support; and Marcio Chedid, Christopher A Slapak, Kerry Blanchard, James J Starling, Robert C Wild, Jonathan M Yingling, and Gregory D Plowman for critical review of the manuscript. In addition, the authors thank Mary Dugan Wood, who provided medical writing services on behalf of Eli Lilly and Company, and Svetlana Dominguez, ELS, employed by Eli Lilly and Company, for editorial assistance. This study was funded by Eli Lilly and Company.
Authors Contribution
Authors contribution are as follows: designed research: LM, JRC, ELK, MER, SP, JRG, LJH, HP, LFS, PI, TPB; performed research: LM, JRC, BZ, KMH-T, CP, RJE, WS, JMS, CH, MER, SP, JRG, LJH, HP, MSD; contributed vital new reagents or analytical tools: LM, JRC, BZ, MCS, KMH-T, LB, CP, RJE, WS, JMS, MER, SP, JRG, LJH, HP; collected data: LM, JRC, BZ, KMH-T, ELK, RJE, WS, JMS, MER, SP, JRG, LJH, HP, TPB; analyzed and interpreted data: LM, JRC, RAW, BZ, MCS, KMH-T, ELK, LB, CP, RJE, WS, JMS, CH, MER, SP, JRG, LJH, HP, LFS, MSD, TPB; wrote the manuscript with the assistance of a medical writer and editor: LM, JRC, RAW, TPB. All authors reviewed the manuscript and approved the final version.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
All authors are employed by Eli Lilly and Company.
Additional information
Some of the results detailed in this manuscript were presented at these conferences: (1) European School of Hematology International Conference on Myeloproliferative Neoplasms, September 30-October 02, 2010, Albufeira, Portugal (poster). (2) American Society of Hematology Annual Meeting, December 4-7, 2010, Orlando, Florida (poster). (3) American Association of Cancer Research Annual Meeting, April 2–6, 2011, Orlando, Florida (oral presentation)
Supplementary Information accompanies this paper on Blood Cancer Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Ma, L., Clayton, J., Walgren, R. et al. Discovery and characterization of LY2784544, a small-molecule tyrosine kinase inhibitor of JAK2V617F. Blood Cancer Journal 3, e109 (2013). https://doi.org/10.1038/bcj.2013.6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2013.6
Keywords
This article is cited by
-
JAK2 inhibitors for the treatment of Philadelphia-negative myeloproliferative neoplasms: current status and future directions
Molecular Diversity (2023)
-
Small molecules in targeted cancer therapy: advances, challenges, and future perspectives
Signal Transduction and Targeted Therapy (2021)
-
α7-Acetylcholine Receptor Signaling Reduces Neuroinflammation After Subarachnoid Hemorrhage in Mice
Neurotherapeutics (2021)
-
An improved synthesis of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine
Chemistry of Heterocyclic Compounds (2018)
-
Rethinking JAK2 inhibition: towards novel strategies of more specific and versatile janus kinase inhibition
Leukemia (2017)
