
Abstract
The advances in the treatment of chronic myeloid leukemia (CML) during the last years were also accompanied by the development of evading strategies by tumor cells, resulting in chemotherapy resistance in some patients. Patented organopalladium compounds derived from the reaction of N,N-dimethyl-1-phenethylamine (dmpa) with [1,2-ethanebis(diphenylphosphine)] (dppe) exhibited a potent antitumor activity in vivo and in vitro in melanoma cells. We showed here that the cyclopalladated derivative [Pd2(R(+))C2, N-dmpa)2(μ-dppe)Cl2], named compound 7b, was highly effective to promote cell death in the K562 human leukemia cells and its mechanisms of action were investigated. It was shown that compound 7b was able to promote exclusively apoptotic cell death in K562 cells associated to cytochrome c release and caspase 3 activation. This cytotoxic effect was not observed in normal peripheral mononuclear blood cells. The compound 7b-induced intrinsic apoptotic pathway was triggered by the protein thiol oxidation that resulted in the dissipation of the mitochondrial transmembrane potential. The preventive effect of the dithiothreitol on the compound 7b-induced cell death and all downstream events associated to apoptosis confirmed that death signal was elicited by the thiol oxidation. These findings contribute to the elucidation of the palladacycle 7b-induced cell death mechanism and present this compound as a promising drug in the CML antitumor chemotherapy.
Similar content being viewed by others

Main
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disorder of the hematopoietic system. Its incidence is estimated in 5000 new cases per year in the United States.1 This type of malignancy is characterized by a genetic abnormality, which is found in up to 95% of patients and considered the hallmark of this disease. In healthy individuals, the ABL gene is located on chromosome 9, whereas the BCR gene is located on chromosome 22. In the CML chromosome 9 and 22 exchange part of their DNA (translocation), and BCR and ABL join together, resulting in the abnormal gene BCR-ABL. This new mixed chromosome containing BCR-ABL is called Philadelphia chromosome.2 It is noteworthy that the consequence of this translocation is the fusion gene BCR-ABL, which has an enhanced tyrosine kinase activity and is responsible for the malignance of this type of cancer, as BCR-ABL alters the cell cycle.3
The treatment of CML has improved considerably in the last years. Advances in the field of drug development and the comprehension of the role of BCR-ABL tyrosine kinase activity in this cancer allowed the development of tyrosine kinase inhibitors (TKI). The first TKI approved for the treatment of CML was mesylate imatinib, a selective and potent inhibitor of BCR-ABL.4 Despite the successful results achieved with this drug, resistance is still a recurrent issue encountered in the treatment of this cancer in some patients. The mechanisms of imatinib resistance are classified according to its dependence on BCR-ABL. Drug resistance related to BCR-ABL are the most common and it is found in the majority of patients. It involves the amplification or overexpression of BCR-ABL and point mutations in the ABL kinase domain. Other mechanisms independent of BCR-ABL include multidrug-resistance expression and activation of downstream signaling molecules, such as Src kinases.5, 6, 7 Multiple strategies have been developed to overcome imatinib resistance, which includes the use of a second generation TKI-like dasatinib and nilotinib, other novel agents still in clinical trial, non-TKI-based therapy, dose escalation of imatinib and allogeneic stem cell transplant.8, 9 This highlights the need for the development of new agents in the treatment of CML able to selectively kill tumor cells and be an effective option in the treatment of this cancer.
Recently, the importance of the organopalladium chemistry was recognized with the Nobel Prize in Chemistry to the ‘Suzuki coupling’, a palladium-catalyzed cross coupling reaction by Professor Richard F. Heck of the University of Delaware in Newark, Delaware, USA; Professor Ei-ichi Negishi of Purdue University in West Lafayette, Indiana, USA and Professor Akira Suzuki of Hokkaido University in Sapporo, Japan.10 In fact, the versatility of such class of compounds has allowed its application in several fields, including Medicine. The synthesis of palladium compounds began in 1980 due to its similarity in structure to cisplatin. However, the first synthesized compounds exhibited little stability in biological systems, which was later solved by the use of ligands.11, 12
Cyclopalladated complexes derived from the reaction of N,N-dimethyl-1-phenethylamine (dmpa) with [1,2-ethanebis (diphenylphosphine)] (dppe) more stable and less toxic were synthesized and patented worldwide. Currently such compounds are under preclinical phase evaluation and they have exhibited a potent antitumor activity in vivo and in vitro at relatively low concentrations.13 It was demonstrated that this compound was able to induce mitochondrial permeabilization due to the oxidation of protein thiol groups in isolated rat liver mitochondria related to its cytotoxicity.14 Analog compounds with the coordinating ligand ferrocene have also exhibited its effects on lysosomes, resulting in lysosomal membrane permeabilization and cathepsin B release.15, 16
In this work, we investigated the induction of cell death by the palladacycle [Pd2(R(+)C2, N-dmpa)2(μ-dppe)Cl2], named compound 7b, in a well-known resistant leukemia cell line. Our results showed that the compound 7b induced a caspase-dependent apoptosis in K562 leukemia cells, with involvement of the intrinsic pathway triggered by mitochondrial thiol oxidation.
Results
Screening of the cytotoxicity of the palladacycle 7b in K562 cells
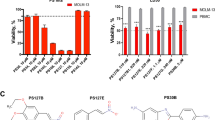
Cell viability was determined in order to screen the cytotoxic potencial of the organopalladated compound 7b against K562 leukemia cells. By using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) reduction test, it was shown that increasing concentrations of compound 7b decreased the cell viability in a concentration-dependent manner (Figure 1, closed circles). To avoid misinterpretation, cell viability was also measured by the trypan blue exclusion test and similar results were obtained (closed triangles). Both methods exhibited similar results excluding a possible redox interference of compound 7b with the MTT assay. The EC50 values obtained were about 1.2 μM for both methods, demonstrating the high cytotoxicity of the drug in this myeloid leukemia model. As our group had previously observed (Santana et al.14) that this compound was able to promote mitochondrial dysfunction due to the oxidation of protein thiol groups in isolated rat liver mitochondria, K562 cells were pre-incubated with dithiothreitol (DTT). As observed in Figure 1a, DTT was able to prevent compound 7b-induced cell death completely (open circles), indicating the involvement of thiol oxidation in the cytotoxicity of compound 7b. Also, the pre-incubation with N-acetylcysteine (NAC) and reduced glutathione (GSH) resulted in the inhibition of palladacycle-induced cell death (Figure 1b). Surprisingly, as observed in the Figure 1c, peripheral mononuclear blood cells were not affected by compound 7b (open circles) at the same concentration range in relation to K562 tumor cells (closed circles).

Viability of K562 and peripheral mononuclear blood cells after treatment with compound 7b for 24 h. (a) K562 cell viability was evaluated by the MTT reduction endpoint test (closed circles) and trypan blue exclusion assay (triangles). Cells (1 × 105/ml) were incubated with different concentrations of the drug. The effect of the cells-pre-incubation with 1 mM DTT was also assessed by MTT (open circles). The percentage of viable cells was calculated in relation to control (untreated), considered as 100%. (b) The effect of the thiol antioxidants 1 mM NAC and 1 mM GSH on K562 cells viability was assessed by MTT reduction assay. Cells (1 × 105/ml) were pre-incubated with NAC (white bars) or GSH (gray bars) for 30 min and then incubated with 0.5, 1 and 1.5 μM compound 7b for 24 h. The percentage of viable cells was calculated in relation to control (untreated), considered as 100%. The data are presented as mean±S.E.M. of three independent experiments. *Statistically different from control (P<0.05). (c) Mononuclear blood cells (1 × 105/ml) were stimulated with 5 μg/ml phytohemagglutinin and incubated with different concentrations of the drug (open circles) in comparison with K562 cells (closed circles)
Compound 7b induces DTT-sensitive apoptosis in K562 cells
K562 cells were doubled stained with annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) and analyzed by flow cytometry to investigate the type of cell death induced by compound 7b. As observed in Figure 2a, the cells incubated with compound 7b exhibited predominantly positive labeling for annexin V (black bars) and also annexin V-FITC/PI double staining (white bars), indicating the phophatidylserine externalization that occurs during apoptotic process. Quantification of the cytometry data of replicates revealed an expected concentration-dependent behavior and the decrease in the number of viable cells (An−/PI−) matched with MTT and Trypan blue results (Figure 2b). Compound 7b-induced apoptosis was completely prevented by DTT (Figures 2a and b). In addition, to confirm the occurrence of apoptotic cell death induced by compound 7b in these leukemia cells, the morphological nuclear alterations were observed in 4´,6-diamidino-2-phenylindole (DAPI)-stained K562 cells. Compound 7b induced nuclear fragmentation with chromatin condensation and apoptotic body formation, evidenced by the representative image obtained by confocal fluorescence microscopy (Figure 2c). Figure 2d shows the percentage of apoptotic nuclei in compound 7b-treated cells versus 7b non-treated cells by counting a hundred cells in widefield fluorescence microscopy. Compound 7b-treated cells exhibited more than 60% of apoptotic nuclei and the pre-incubation with DTT decreased this number below 10%.

Compound 7b-induced apoptosis in K562 cells. (a) Representative flow cytometry density plots of the double label with annexin V-FITC and PI. K562 cells (1 × 105/ml) were incubated for 24 h in the presence of different concentrations of the compound 7b. Cells were also pre-incubated with 1 mM DTT for 30 min. At least 10 000 cells were analyzed per sample and the figure is representative of three experiments made in triplicate. (b) The quantification was presented as mean±S.E.M.; An (Annexina V-FITC) and PI. (c) Nuclear alterations were accessed by DAPI staining and a representative image was acquired in a confocal microscopy. Each sample was done in duplicate and the number of apoptotic cells was expressed as a percentage of the total number of cells. White bars represent 5.0 μM. (d) The quantification of the apoptotic nuclei was performed by counting of 100 cells per field of view per sample in three different fields. * and # pairs are statistically different (P<0.05)
Compound 7b promotes protein thiol oxidation associated to mitochondrial depolarization in K562 cells
In order to directly access the participation of the thiol redox state in the compound 7b-induced cell death, the reduced thiol groups of cellular proteins and the GSH were measured after exposure to 1.5 and 3.0 μM compound 7b in the same experimental conditions of cell viability assays. Compound 7b oxidized protein thiol residues that is responsive to the concentration (Figure 3a) and, as expected, the pre-incubation with DTT prevented this effect. Thiol oxidation in Ca2+-loaded mitochondria is thought to trigger the opening of the mitochondrial permeability transition pore with the release of pro-apoptotic proteins.17, 18 The mitochondrial transmembrane potential (ΔΨ) was evaluated in order to investigate the occurrence of mitochondrial permeabilization related to apoptosis induced by the compound 7b. The addition of compound 7b in digitonin-permeabilized K562 cells resulted in an immediate dissipation of the mitochondrial ΔΨ (Figure 3b) at the same concentration range that induced cytotoxicity. Such dissipation was inhibited by DTT, showing that it is preceeded by the thiol oxidation. Ca2+ chelators EGTA or BAPTA-AM had no effect on compound 7b-induced ΔΨ dissipation (not shown). The same results were found in intact cells loaded with JC-1 analyzed by flow cytometry (Figure 3c). In this experiment, control cells (absence of drug) emitted red fluorescence due to high ΔΨ. However, compound 7b-treated cells showed progressive loss of red J-aggregate fluorescence with the increase of green fluorescence after 24 h incubation due to mitochondrial transmembrane dissipation and DTT prevented it.

Oxidation of the protein thiol groups and dissipation of the mitochondrial ΔΨ by compound 7b in K562 cells. (a) Cells (5 × 105/ml) were incubated for 24 h in the presence of the compound 7b. Reduced thiol groups were measured spectrophotometrically at 412 nm by using DTNB. Absorbance values were converted in percentage in relation to control and presented as mean±S.E.M. Numbers are drug concentrations in μM. *Statistically different from control (P<0.05) and #statistically different from compound 7b (P<0.05). The amount of protein thiol groups in control was 2.33 μM. (b) Cells (5 × 105/ml) were incubated in a buffer containing 250 mM sucrose, 10 mM HEPES, 2 mM KH2PO4, 0.5 mM EGTA, 0.5% (w/v) BSA, 5 mM MgCl2 (pH 7.2) plus 5 mM succinate and 1.0 μM rhodamine 123, in the presence of the compound 7b. Traces are representative of three independent experiments and the numbers are concentrations in μM. ΔΨ was generated by uptake of succinate by cells after selective permeabilization with 0.004% (w/v) digitonin. Fluorescence arbitrary units (a.u.). (c) Cells (1 × 105/ml) were incubated with compound 7b (1.25 and 1.5 μM) for 24 h. Later, JC-1 was added and the samples were analyzed by flow cytometry. The uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (10 μM) was used as positive control. Figure is representative of three experiments made in triplicate. DTT (1.0 mM) was pre-incubated with cells for 30 min
Compound 7b induced caspase-dependent apoptosis throughout the mitochondrial intrinsic pathway
The triggering of mitochondrial permeability transition may result in apoptotic or necrotic cell death.19 The evidences presented here pointed to compound 7b-induced apoptosis. In order to investigate whether thiol oxidation and mitochondrial depolarization were involved in the cell death, it was investigated the release of cytochrome c from mitochondrial to cytosol in K562 cells incubated with compound 7b. To estimate the cytochrome c release, we performed a double-staining confocal fluorescence imaging using MitoTracker red in combination with an anti-cytochrome c antibody immunostaining. As expected, in the absence of drug (control) cytochrome c (green) was co-localized with MitoTracker red as evidenced by yellow/orange color, indicating that this protein is confined in mitochondria (Figure 4a). After a 12-h exposure to compound 7b, cytochrome c was redistributed all over the cytosol and did not co-localize with MitoTracker, indicating that cytochrome c was no longer confined in mitochondria. Again, DTT reversed this effect as shown by the co-localization of cytochrome c with MitoTracker red. Cytochrome c is a mobile electron transporter in the respiratory chain and also participates in the apoptosis, triggering through the apoptosome formation and caspase activation. Thus, we also investigated the compound 7b-induced caspase 3 activation by flow cytometry (Figure 4b). The dislocation of the histogram to higher fluorescence values indicated caspase activation (black solid line) and the pre-incubation with DTT prevented it (dotted line), indicating that the oxidation of thiol residues results in mitochondrial permeabilization with release of pro-apoptotic proteins and caspase activation. The quantification was presented in the Figure 4c.

Cytochrome c release and caspase 3 activation promoted by compound 7b. (a) Confocal laser scanning microscope images showing MitoTracker red fluorescence (red), cytochrome c (green), DAPI staining (blue) in merged images (yellow indicates pixels of co-localization) in K562 cells. The cells were either untreated (control) or exposed to 1.25 μM compound 7b with or without DTT (1 mM) for 12 h. White scale bars represent 5.0 μM. (b) Cells (1 × 105/ml) were incubated for 24 h in the presence of 1.25 μM compound 7b. Cells were also pre-incubated with 1 mM DTT for 30 min. Caspase 3 activation was accessed by flow cytometry using an anti-active caspase 3 antibody conjugated with FITC and the data quantification was made by using geometrical mean of three independent experiments (c). * and # pairs are statistically different (P<0.05)
Discussion
Cancer cells exhibit genetic and epigenetic mutations that may result in resistance to cell death.20 Mutations in the p53 tumor suppressor gene are relatively common in a variety of tumors, including human myeloid leukemia, and result in the absence of DNA damage protection, leading to an extensive resistance to apoptosis.21, 22 Another genetic mutation responsible for the pathogenicity of CML is the Philadelphia chromosome, originated by a translocation between chromosomes 9 and 22. Such translocation originates the BCR-ABL gene that has an enhanced tyrosine kinase activity, leading to tumor progression and resistance to cell death.23 Therefore, drug resistance is the major challenge in cancer chemotherapy. It evidences the need for the development of new drugs or new targets to improve efficiency and selectivity to kill cancer cells. K562 cells exhibit both alterations mentioned above.
The application of organometallic compounds in the antitumor chemotherapy began in 1964 with cisplatin. Since then, several structural derivatives have been synthesized in order to increase the efficacy and to overcome the toxicity/side effects of the original drug.24, 25 Because of the chemical similarity between platinum and palladium, organopalladate complexes started to stand out as a promising alternative to cancer therapy due to their high potency and the reaching of molecular targets. However, the first synthesized compounds presented low stability in biological system that was solved with the use of chelating ligands.12 The organometallic palladacycle used in this study was synthesized and recently patented worldwide. It has been shown that the compounds derived from the reaction of dmpa with dppe exhibit a potent antitumor activity in vivo and in vitro at relatively low concentrations.13, 16 Nevertheless, few advances have been made concerning the molecular basis of the antitumor effect of these compounds. In this work, we screened the cytotoxic effects of the organopalladated compound 7b in a BCR/ABL-positive K562 leukemia cell line and the molecular mechanisms that account for cell death were also investigated.
Our results evidenced the in vitro cytotoxic activity of the palladacycle 7b in CML K562 cells at relatively low concentrations (below 1.5 μM). Cytotoxicity induced by the compound 7b exhibited selectivity, as normal cells were not affected at the same concentration. Such selectivity was also observed from ferrocen-derived palladacycles.16 The cytotoxicity of compound 7b was first reported in a melanoma model with apparent inhibition of the respiratory activity.13 It has been recently demonstrated that its enantiomer compound 7a exhibited in vitro and in vivo trypanocidal effects associated to the mitochondrion structure disruption and also cytotoxicity against B16F10-Nex melanoma cells.26, 27
Besides the role of mitochondria in energy production, theses organelles concentrate lethal weapons to orchestrate cell death, and such feature makes them a strategic target in the antitumor chemotherapy.28 Recently, we demonstrated the induction of mitochondrial permeabilization in isolated rat liver mitochondria by the compound 7b. Such permeabilization was promoted by the oxidation of protein thiol residues.14 The oxidation of protein thiol groups in mitochondrial membranes promoted by free radicals or thiol oxidizing agents results in the opening of the mitochondrial permeability transition pore.17, 29 The opening of this large conductance channel causes the dissipation of the mitochondrial ΔΨ, loss of the matrix components, increased reactive oxygen species production, swelling and outer membrane rupture.30 All these morphological and functional mitochondrial alterations may be accompanied by the release of apoptogenic factors to cytosol, including cytochrome c, that activate intermediate and effector caspases, resulting in the execution of cell death.31
The organopalladated compound 7b was able to trigger exclusively a caspase-dependent apoptotic cell death program in K562 leukemia cells through the intrinsic pathway, characterized by mitochondrial permeabilization, cytochrome c release and caspase 3 activation. This is a noteworthy feature as it was shown that leukemia cells were insensitive to cell death induction via the extrinsic pathway due to the downregulation of CD95 expression.32 Despite the potency and specificity of compound 7b to induce apoptosis in K562 leukemia cells, our results also elucidated the molecular mechanisms involved in the cell death pathway and the proposed sequence of events elicited by compound 7b that culminated in cell death is presented in the Scheme I.
The reactivity of compound 7b with protein thiol groups, including those located in mitochondrial membranes, was responsible for triggering cell death in this leukemia model. The cell death induced by compound 7b in K562 cells was completely inhibited by the thiol reducing agent DTT. Interestingly, DTT also inhibited the mitochondrial permeabilization (evaluated indirectly by the dissipation of the ΔΨ) and all downstream events, suggesting that mitochondria are the main target of the palladacycle. The specificity of this oxidation was proposed, as GSH was not oxidized by the compound 7b and probably was promoted by the partition of the drug between the hydrophilic compartments and membranes. Other thiol reactants GSH and NAC, but not the antioxidant butylhydroxytoluene, were able to inhibit the cell death promoted by compound 7b. Furthermore, after 24 h of incubation compound 7b did not promote the increase in the 2´,7´-dichlorofluorescein fluorescence (not shown), indicating that the thiol oxidation was not mediated by free radicals.
In summary, our results suggest that compound 7b is a potent and specific apoptosis inducer in K562 cells. Considering that apoptotic cell death is an important regulatory program to prevent cancer development and that leukemia cells have been able to escape from extrinsic apoptotic pathways, resulting in chemotherapy resistance, our study contributes to the preclinical screening of the cytotoxicity of the palladacycle 7b in a resistant CML model and provides a better comprehension of the molecular basis of cell death induced by this drug.
Materials and Methods
Cell culture
K562 cells, a CML cell line that expresses the BCR-ABL gene, were grown in suspensions in RPMI-1640 medium, pH 7.2, supplemented with 10% fetal bovine serum (Gibco SBF, Invitrogen, Grand Island, NY, USA), 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were maintained in culture flasks at 37 °C in 5% CO2 atmosphere (Sanyo MCO-20AIC, Osaka, Japan).
Biphosphinic palladacycle complex [Pd2(R(+)C2, N-dmpa)2(μ-dppe)Cl2]
The cyclopalladated complex 7b used in this study was derived from dmpa complexed to dppe and synthesized as described in Rodrigues et al.13 Its chemical structure is shown in Chart 1. The compound was diluted to 10 mM in DMSO (stock solution), and for in vitro assays it was diluted to the final concentration in water.

Chemical structure of the cyclopalladated compound 7b [Pd2(R(+))C2, N-dmpa)2(μ-dppe)Cl2]
Cytotoxicity assays
K562 cells or peripheral mononuclear blood cells (1 × 105/ml) were incubated for 24 h in 96-well microplates in the presence of different concentrations of compound 7b. The cytotoxicity was accessed by MTT reduction test and by trypan blue dye exclusion assay.33 The peripheral mononuclear blood cells was pre-incubated for 4 h with 5 μg/ml phytohemagglutinin before the addition of the drug. For the MTT reduction test, at the end of the incubation period, 0.25 mg/ml MTT was added followed by 4 h incubation. Then 100 μl of 10% SDS prepared in 0.01 M HCl was added and mixed thoroughly to dissolve the formazan crystals at 37 °C. Plates were read at 630 nm (Microplate Reader Biotek ELX 800, BioTek Instruments, Inc., Winooski, VT, USA) and the cell viability was determined in relation to control performed in the absence of compound 7b, and considered as 100%. For the Trypan blue exclusion assay, after the addition of 0.016% trypan blue in the cell suspension, the cells were counted using a hemacytometer under optical microscopy. Each compound 7b concentration was tested in three different experiments, ran in triplicate. It was also been evaluated that the effect of some modulators in compound 7b induced cell death: 1 mM DTT, 1 mM NAC, 1 mM GSH, 10 μM BAPTA-AM and 1 mM EGTA, by pre-incubation of K562 cells with these compounds for 30 min.
Annexin V-FITC/PI double-staining and flow cytometry analysis
After treatment with different concentrations of compound 7b for 24 h, K562 cells were centrifuged and resuspended in binding buffer (0.01 M HEPES (pH 7.4) 0.14 M NaCl and 2.5 mM CaCl2) at a ratio of 1 × 105 cells/ml. The suspensions were transferred to 5 ml tubes and 5 μl Annexin V-FITC (BD Biosciences, San Jose, CA, USA) plus 5 μg/ml PI (BD Biosciences) were added. The cells were incubated at room temperature for 20 min and after the addition of 0.3 ml of binding buffer, the analysis was performed in a FACSCalibur flow cytometer using the CellQuest software (BD Biosciences) (10 000 events were collected per sample). Experimental controls were performed with cells treated only with the medium. Data were presented as media±S.E.M. of the triplicates.
Measurement of active caspase 3
Caspase 3 activity was measured by flow cytometry. K562 cells (1 × 105 cells/ml) were treated with 1.25 μM compound 7b for 24 h. Then cells were fixed with 2% paraformaldehyde in PBS for 30 min and permeabilized with 0.01% saponin in PBS for 15 min at room temperature. Cells were then collected and incubated with Anti-Active-Caspase-3 monoclonal antibody conjugated with FITC (BD-Pharmingen, San Diego, CA, USA). After incubation for 40 min at 37 °C, the fluorescence was analyzed in a FACScalibur Flow Cytometer (BD Biosciences), using CellQuest software (10. 000 events were collected per sample). Alternations in the fluorescence intensity were determined by comparing the levels of the treated cells to those of the controls.
Mitochondrial ΔΨ
Δψ was determined in digitonin-permeabilized K562 cells by changes in the fluorescence of rhodamine 123 at 37 °C in a Hitachi F-2500 fluorescence spectrophotometer (Tokyo, Japan) at 505/535 nm excitation/emission wavelength pair with a slit width of 5/5 nm. Δψ was also assessed by flow cytometry using the lipophilic cationic dye JC-1 (5,5´,6,6´-tetrachloro-1,1´,3,3´-tetraethylbenzimidazolcarbocyanine iodide; Molecular Probes, Eugene, OR, USA). JC-1 is cationic dye that exhibits potential-dependent accumulation in mitochondria by fluorescence emission shift from green (∼520 nm) to red (∼590 nm). Consequently, mitochondrial depolarization is indicated by a decrease in the red-green fluorescence intensity ratio. Cells were stimulated (1 × 105 cells/ml) and after a 24-h incubation with the compound 7b, cells were centrifuged and 1.0 μg/ml JC-1 was added. To achieve total depolariztion, cells were treated with 10 μM carbonyl cyanide p-trifluoromethoxyphenylhydrazone. After the incubation period the fluorescence was analyzed in a FACScalibur Flow Cytometer (BD Biosciences), using CellQuest software (10 000 events were collected per sample).
Cytochrome c release and nuclear alterations analyses
The release of cytochrome c from mitochondria was estimated by using double-labeled confocal cell imaging with MitoTracker Red CMXRos (Invitrogen, Carlsbad) and a cytochrome c antibody. K562 cells (1 × 105/ml) were incubated for 12 h with 1.25 μM compound 7b in the absence and presence of pre-incubation with 1.0 mM DTT. Then, cells were primarily labeled with 100 nM MitoTracker Red (Molecular Probes) for 30 min at 37 °C. Then cells were washed with PBS and fixed with 2% paraformaldehyde in PBS for 30 min. Fixed cells were permeabilized with 0.01% saponin in PBS for 15 min and washed with PBS followed by a 2 h incubation at room temperature with a monoclonal mouse antibody against human cytochrome c 1 : 100 (0.5 μg/ml, RD Systems, Minneapolis, MN, USA). After washing, cells were incubated for 40 min with Alexa Fluor 488-conjugated goat anti-mouse IgG (1 : 500; Molecular Probes) and 5.0 μg/ml DAPI (Sigma-Aldrich, St. Louis, MO, USA). Labeled cells were placed onto glass coverslips covered and mounted in Fluoromount (Electron Microscopy Sciences, Hatfield, PA, USA). Microscopy analyses were performed with a confocal laser scanning microscope (Leica TCS-SP8 Microscope, Benshein, Germany) equipment with a × 63 objective (Plan-Neofluar, 1.4 numerical aperture) under oil immersion using LAS-AF software (Leica). The pinhole device was adjusted to capture fluorescence of one airy unit in one focal section. Alexa Fluor 488 was excited using an argon laser (λex=488 nm) and its emissions was detected from 500–550 nm. MitoTracker Red was excited using HeNe laser (λex=543 nm) and its emissions was detected from 560–610 nm. Alexa Fluor 488 was excited using an argon laser (λex=488 nm) and its emissions was detected from 500–550 nm. DAPI was excited with a UV laser (λex=353 nm) and the emission was detected from 370–460 nm. In addition, DAPI staining was used to observe the apoptotic nuclear morphology. The frequency of cells presenting nuclear alterations was visually determined by observing and counting at least 100 cells per field of view in three different fields per sample. Each sample was done in duplicate and the number of cells with nuclear alterations was expressed as a percentage of the total number of cells.
Reduced protein thiol content and GSH levels
Protein SH groups were quantified by using 5,5′-dithiobis(2-nitrobenzoic) acid (DTNB, Ellman’s reagent). After incubation time with compound 7b, cells were centrifuged during 10 min at 700 × g. The pellet was treated with 0.2 ml of 6% trichloroacetic acid and centrifuged at 6000 × g during 15 min to precipitate the cellular proteins. The final pellet was suspended with 1 ml of 0.5 M potassium phosphate buffer, pH 7.6. After addition of 0.1 mM DTNB, absorbance was determined at 412 nm and the amount of thiol groups was calculated from ɛ=13 600/M.34 GSH levels were determined spectrofluorometrically using o-phthalaldehyde.35 The supernatant obtained after acid precipitation (0.1 ml) was added to 1.9 ml of buffer, and after the addition of 0.05 mg/ml o-phthalaldehyde, the fluorescence was acquired at 350 and 420 nm, excitation and emission wavelengths, respectively.
Statistical analyses
Values are the mean of at least three independent experiments run in triplicate. Data for each assay are expressed as mean±S.E.M. and were statistically analyzed by ANOVA. Multiple comparisons among group mean differences were tested by using the Tukey post test. Differences were considered significant when P<0.05. All graphical and statistical analyses were performed with Origin Pro 8 software (Micronal Software, Inc., Northampton, MA, USA).

Proposed mechanism for the compound 7b-induced apoptosis in K562 leukemia cells. Compound 7b promotes the oxidation of the protein thiol groups inside the cells. The oxidation of the protein thiol residues in the mitochondrial membranes results in mitochondrial permeabilization associated to the mitochondrial ΔΨ and cytochrome c release to the cytosol. In the presence of dATP, cytochrome c induces the assembly of apoptosome and activation of pro-caspase 9, which, in turn, activates the effector caspase 3. Thiol reducers (DTT, NAC and GSH) inhibit the thiol oxidation and all downstream events that culminate in apoptosis, showing the cell death is triggered by compound 7b-induced thiol oxidation in K562 leukemia cells
Abbreviations
- CML:
-
chronic myeloid leukemia
- DAPI:
-
4´,6-diamidino-2-phenylindole
- dmpa:
-
N,N-dimethyl-1-phenethylamine
- dppe:
-
1,2-ethanebis(diphenylphosphine)
- DTNB:
-
5,5´-dithiobis(2-nitrobenzoic acid)
- DTT:
-
dithiothreitol
- FITC:
-
fluorescein isothiocyanate
- GSH:
-
reduced glutathione
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide
- NAC:
-
N-acetylcysteine
- OPT:
-
o-phthalaldehyde
- PI:
-
propidium iodide
- TKI:
-
tyrosine kinase inhibitor
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D . Cancer statistics, 2009. CA Cancer J Clin 2009; 59: 225–249.
Faderl S, Talpaz M, Estrov Z, O´Brien S, Kurzrock R, Kantarjian HM . The biology of chronic myeloid leukemia. N Engl J Med 1999; 341: 164–172.
Ren R . Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer 2005; 5: 172–183.
Deininger M, Buchdunger E, Druker BJ . The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 2005; 105: 2640–2653.
Roychowdhury S, Talpaz M . Managing resistance in chronic myeloid leukemia. Blood Rev 2011; 25: 279–290.
Ramirez P, Dipersio JF . Therapy options in imatinib failures. Oncologist 2008; 13: 424–434.
Kumari A, Brendel C, Hochhaus A, Neubauer A, Burchert A . Low BCR-ABL expression levels in hematopoietic precursor cells enable persistence of chronic myeloid leukemia under imatinib. Blood 2012; 119: 530–539.
Radich JP . Measuring response to BCR-ABL inhibitors in chronic myeloid leukemia. Cancer 2012; 118: 300–311.
Eiring AM, Khorashad JS, Morley K, Deininger MW . Advances in the treatment of chronic myeloid leukemia. BMC Med 2011; 9: 1–6.
Seechurn CCCJ Kitching MO, Colacot TJ, Snieckus V . Palladium-catalysed cross-coupling: a historical contextual perspective to the 2010 Nobel Prize. Angew Chem Int Ed Engl 2012; 51: 5062–5085.
Gao E, Liu C, Zhu M, Lin H, Wu Q, Liu L . Current development of Pd(II) complexes as potential antitumor agents. Anticancer Agents Med Chem 2009; 9: 356–368.
Caires AC . Recent advances involving palladium (II) complexes for the cancer therapy. Anticancer Agents Med Chem 2007; 7: 484–491.
Rodrigues EG, Silva LS, Fausto DM, Hayashi MS, Dreher S, Santos EL et al. Cyclopalladated compounds as chemotherapeutic agents: antitumor activity against a murine melanoma cell line. Int J Cancer 2003; 107: 498–504.
Santana DP, Faria PA, Paredes-Gamero EJ, Caires ACF, Nantes IL, Rodrigues T . Palladacycles catalyze the oxidation of critical thiols of the mitochondrial membrane proteins and lead to mitochondrial permeabilization and cytochrome c release associated with apoptosis. Biochem J 2009; 417: 247–256.
Barbosa CMV, Oliveira CR, Nascimento FD, Smith MCM, Fausto DM, Soufen MA et al. Biphosphinic palladacycle complex mediates lysosomal-membrane permeabilization and cell death in K562 leukaemia cells. Eur J Pharmacol 2006; 542: 37–47.
Oliveira CR, Barbosa CM, Nascimento FD, Lanetzki CS, Meneghin MB, Pereira FEG et al. Pre-clinical antitumour evaluation of Biphosphinic Palladacycle Complex in human leukaemia cells. Chem Biol Interact 2009; 177: 181–189.
Castilho RF, Kowaltowski AJ, Meinicke AR, Bechara EJH, Vercesi AE . Permeabilization of the inner mitochondrial membrane by Ca2+ ions is stimulated by t-butyl hydroperoxide and mediated by reactive oxygen species generated by mitochondria. Free Radic Biol Med 1995; 18: 479–486.
Tait SW, Green DR . Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 2010; 11: 621–632.
Rasola A, Bernardi P . The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 2007; 12: 815–833.
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674.
Harris CC . p53: at the crossroads of molecular carcinogenesis and risk assessment. Science 1993; 262: 1980–1981.
Brosh R, Rotter V . When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 2009; 9: 701–713.
Mello JV, Gordon DE, Cross NC, Goldman JM . The ABL-BCR fusion gene is expressed in chronic myeloid leukemia. Blood 1993; 81: 158–165.
Rosenberg B, Van Camp L . The successful regression of large solid sarcoma 180 tumors by platinum compounds. Cancer Res 1970; 30: 1799–1802.
Wang D, Lippard SJ . Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 2005; 4: 307–320.
Matsuo AL, Silva LS, Torrecilhas AC, Pascoalino BS, Ramos TC, Rodrigues EG et al. In vitro and in vivo trypanocidal effects of the cyclopalladated compound 7a, a drug candidate for treatment of Chagas’ disease. Antimicrob Agents Chemother 2010; 54: 3318–3325.
Serrano FA, Matsuo AL, Monteforte PT, Bechara A, Smaili SS, Santana DP et al. A cyclopalladated complex interacts with mitochondrial membrane thiol-groups and induces the apoptotic intrinsic pathway in murine and cisplatin-resistant human tumor cells. BMC Cancer 2011; 11: 296–312.
Fulda S, Galluzzi L, Kroemer G . Targeting mitochondria for cancer therapy. Nat Rev Drug Discov 2010; 9: 447–464.
Kowaltowski AJ, Castilho RF, Vercesi AE . Mitochondrial permeability transition and oxidative stress. FEBS Lett 2001; 495: 12–15.
Zoratti M, Szabò I . The mitochondrial permeability transition. Biochim Biophys Acta 1995; 1241: 139–176.
Taylor RC, Cullen SP, Martin SJ . Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 2008; 9: 231–241.
Friesen C, Fulda S, Debatin KM . Deficient activation of the CD95 (APO-1/Fas) system in drug-resistant cells. Leukemia 1997; 11: 1833–1841.
Mosmann T . Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assay. J Immunol Methods 1983; 65: 55–63.
Jocelyn PC . Spectrophotometric assay of thiols. Methods Enzymol 1987; 143: 44–67.
Hissin PJ, Hilf R . A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal Biochem 1976; 74: 214–226.
Acknowledgements
This work was supported by grants from the Brazilian agencies FAPESP (2012/12247-8) and CNPq (476824/2010-9). We thank Prof. Rodrigo LOR Cunha for the drawing of the chemical structure.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
ACFC is the inventor on patents and patent applications related to use of compound 7b. The other authors declare no conflict of interest.
Additional information
Edited by A Stephanou
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Moraes, V., Caires, A., Paredes-Gamero, E. et al. Organopalladium compound 7b targets mitochondrial thiols and induces caspase-dependent apoptosis in human myeloid leukemia cells. Cell Death Dis 4, e658 (2013). https://doi.org/10.1038/cddis.2013.190
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.190
