
Abstract
Otopalatodigital spectrum disorders (OPDSD) constitute a group of dominant X-linked osteochondrodysplasias including four syndromes: otopalatodigital syndromes type 1 and type 2 (OPD1 and OPD2), frontometaphyseal dysplasia, and Melnick–Needles syndrome. These syndromes variably associate specific facial and extremities features, hearing loss, cleft palate, skeletal dysplasia and several malformations, and show important clinical overlap over the different entities. FLNA gain-of-function mutations were identified in these conditions. FLNA encodes filamin A, a scaffolding actin-binding protein. Here, we report phenotypic descriptions and molecular results of FLNA analysis in a large series of 27 probands hypothesized to be affected by OPDSD. We identified 11 different missense mutations in 15 unrelated probands (n=15/27, 56%), of which seven were novel, including one of unknown significance. Segregation analyses within families made possible investigating 20 additional relatives carrying a mutation. This series allows refining the phenotypic and mutational spectrum of FLNA mutations causing OPDSD, and providing suggestions to avoid the overdiagnosis of OPD1.
Similar content being viewed by others


Introduction
Otopalatodigital (OPD) spectrum disorders (OPDSD) are dominant X-linked osteochondrodysplasias with great clinical and radiological overlap including five syndromes: otopalatodigital syndromes type 1 and type 2 (OPD1, MIM#311300; and OPD2, MIM#304120); Melnick–Needles syndrome (MNS, MIM#309350); frontometaphyseal dysplasia (FMD, MIM#305620); and terminal osseous dysplasia with pigmentary defects (MIM#300244).1, 2, 3 The most characteristic features are facial dysmorphism, extremities anomalies, hearing loss, cleft palate, skeletal dysplasia (leading to S-shaped bowing of long bones, limb deformations and short stature) and various malformations (including cardiac, respiratory airway, genitourinary, gastrointestinal, ocular and central nervous system anomalies). In males, OPD1 represents the mildest form of OPDSD, FMD is more severe, and most OPD2 and MNS patients die in utero or during the first weeks of life.4 Phenotypic severity is highly variable in females. Mutations in FLNA (MIM *300017) were identified in all five syndromes,3, 5 most of them being missense mutations. They can affect many of the multiple domains of filamin A (FLNA), but some regions are more likely to be mutated, that is calponin-homology domain 2 (CH2) encoded by exons 3–5 in OPD1, OPD2 and FMD and rod domain 10 (RD10) encoded by exon 22 in MNS and FMD.5 These mutations are thought to modify FLNA interactions with its partners,6 leading to the hypothesis of a gain-of-function mechanism.7 Of note, only one FLNA mutation was associated with terminal osseous dysplasia with pigmentary defects in three unrelated families and three sporadic cases; it was responsible for activation of a cryptic splice site, removing the last 48 nucleotides from exon 31 and resulting in a loss of 16 amino acids.3
Here, we report from the data collected in our diagnostic laboratory the clinical description and results of FLNA gene analysis in a large series of suspected OPDSD patients. This series allows refining the clinical spectrum of these disorders, describing new OPDSD-causing mutations and proposing diagnosis criteria for OPD1, the mildest syndrome. Discriminating between the different entities is of interest to provide accurate genetic counseling, particularly when only females are affected within a family.
Subjects and methods
Patients
Patients’ DNAs were referred to our diagnostic laboratory with a signed informed consent in agreement with the French law, a complete specific clinical and paraclinical form, face and extremities pictures and X-rays. For each patient, FLNA analysis was validated by three clinical geneticists (CG, DL and MAD). All patients were diagnosed as OPD1, OPD2, MNS or FMD based on clinical and radiological features. When only females were affected within an OPD family, we chose to refer them as to OPD syndrome without specification on type 1 or type 2 if there was no major congenital anomaly, since the classification is made according to phenotypic severity in males.
Genetic analyses
FLNA gene (NM_001110556.1) was analyzed using previously published conditions.7, 8 First, we looked for point mutations by direct Sanger sequencing in the hotspots (exons 2, 3, 4, 5 and 22) and then, if not contributory, in the remaining exons.8 Second, customized array-comparative genomic hybridization (CGH) was performed when sufficient amount of DNA was available, to look for gene-dosage anomalies, using previously published conditions.9 Briefly, the custom microarrays (8 × 60 K) were designed with e-array web software (Agilent Technologies, Santa Clara, CA, USA) using the Similarity Score Filter in order to select highly specific probes. A high resolution was achieved in the FLNA gene and 50 kb around the gene (one probe every 100 bp, 880 probes including 357 exonic probes), and the resolution was one probe every 350 bp in the 300 kb regions on either side of the FLNA gene (1,714 probes). X-chromosome inactivation was studied from female DNA testing the androgen receptor polymorphic locus as previously described,8 but also recently validated loci (CNKSR2 gene on chromosome Xp22.12, HMGB3 and TMEM185A genes on locus Xq28) when the androgen receptor locus was not informative.10
Mutations have been submitted to LOVD (Leiden Open Variation Database) in the FLNA locus-specific mutation databases (http://grenada.lumc.nl/LSDB_list/lsdbs/FLNA).3
Results
Patients
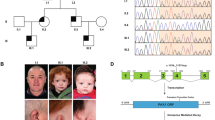
Twenty-seven probands (16 males and 11 females) were recruited in this study. Familial history was informative in 9 of them; 24 probands were referred by geneticists from the French Federation of Reference Centers of Developmental Anomalies and the 3 others were from Sweden, Lebanon and Germany. All patients were diagnosed as OPD1, OPD2, OPD (when only females with no major congenital anomaly were affected within a family), MNS or FMD based on clinical and radiological features. In some of the patients, diagnosis was difficult to assign in a specific category, such as P11 and P20, because of clinical overlap between the various OPDSD entities. Among the probands, there were 9 OPD1 males, 4 OPD females, 1 OPD2 male, 2 OPD2 females, 4 MNS females and 7 FMD patients (including 6 males). Genetic segregation analysis allowed subsequent assessment of 20 relatives carrying a FLNA mutation (overall, 35 patients carrying a mutation were included in the study). Comprehensive clinical data are described in Appendix S1 and summarized in Table 1 and Supplementary Tables S1, S2, S3 and S4. Pedigrees are shown in Figure 1 and photographs in Figure 2 (faces), Supplementary Figures S1, S2, S3 and S4 (extremities and X-rays).

Main pedigrees of this series: arrows indicate the proband in each family, stars indicate available DNA for segregation analysis. Gray icons represent affected or carrier females with a milder phenotype than affected boys of the family.

Main photographs of several patients with otopalatodigital spectrum disorders described in this series. Full black lines, full gray lines, dashed black lines and dashed gray lines delineate OD1, OPD/OPD2 female, Melnick–Needles syndrome and frontometaphyseal dysplasia patients, respectively. A full color version of this figure is available at the Journal of Human Genetics journal online.
Molecular results
We identified 11, including 7 novel, FLNA missense variations in 15 probands (n=15/27, 56%): 6 different mutations in 7 out of 16 patients with suspected OPD1 or OPD2, 2 different mutations in all 4 MNS patients, and 3 different mutations in 4 out of the 7 FMD patients, all of them carried by male probands (Table 2). Regarding the novel missenses, amino acid conservation is illustrated in Supplementary Figure S5. No gene-dosage anomalies using customized array-CGH were identified in patients P5, P10 and P25. A skewed X inactivation pattern was observed in all but 2 (P12.II.1 and her asymptomatic mother) informative females carrying a FLNA variation (91%, n=20/22) (Table 2). Finally, 12 probands had no identifiable sequence variant in FLNA that could explain their phenotype.
Clinical findings in patients carrying a FLNA mutation
All 5 males affected by OPD1 or OPD2 syndromes (P1.III.6, P1.IV.3, P2.III.1, P2.II.3 and P14) shared typical facial and extremity features, and hearing loss (Supplementary Tables S1 and S2). Facial dysmorphism included hypertelorism (n=5/5), downslanted palpebral fissures (n=4/5) and cleft palate (central and posterior including bifid uvula) (n=4/5). Anomalies of the extremities were represented by first ray hypoplasia (n=5/5), widely spaced digits (n=5/5), clubbed fingers (n=5/5) and distal phalangeal hypoplasia (n=4/5). Joint limitations, especially of elbows, were constant (n=5/5). Bone dysplasia and cryptorchidism were frequent (n=4/5). Among the 10 OPD/OPD2 females carrying a mutation from 4 families (P11, P12.II.1, P12.II.2, P13.II.1, P13.I.1, P14.I.2, P15.IV.6, P15.III.3, P15.II.7 and P15.III.25), facial dysmorphism (n=7/10), extremities anomalies (n=5/8), hearing loss (n=4/5), posterior cleft palate (or bifid uvula) (n=4/6) and bone dysplasia (n=2/4) were frequent. Otherwise, in OPD1 females, hearing loss was not described and facial features were hardly detectable.
Most frequent features in the 8 MNS females were narrow forehead (n=6/7), proptosis and micro/retrognathia (n=6/8), bone dysplasia (n=6/7), skull base sclerosis (n=3/4), teeth anomalies (n=4/5), cheekbone prominence (n=4/7) and hypertelorism (n=3/8). Two patients (P17 and P19) carrying the recurrent p.A1188T substitution3 had bleeding manifestations. Conventional hematological explorations were normal. The other MNS patients had no such manifestations.
The 4 FMD males carrying a FLNA mutation had typical facial and extremities features, hearing loss and bone dysplasia. None had cleft palate. Specific additional anomalies were found in FMD in comparison to other OPDSD including craniosynostosis (n=3/3), major prominent supraorbital ridges (n=4/4), antero-inferior mandibular spur (n=4/4), joint limitations especially restricted elbow extensions (n=3/3), long slender digits (n=4/4), muscular underdevelopment of upper limb girdle and/or intrinsic muscles of hands (n=4/4) and urethral obstruction (n=3/4). Other constant features were teeth anomalies (n=3/3) and scoliosis (n=6/6). Thoracic deformation (n=3/4) with restrictive pulmonary disease (n=2/2) was also reported. In familial cases (P21, P23, P24), the mothers had mild manifestations.
In addition, 5 OPDSD patients had ptosis, oculomotor paresis and/or strabismus in our series (P2.III.1, P13.I.1, P14.II.1, P17 and P19).
Discussion
In our series, we identified a FLNA mutation in 52% (n=14/27) of all OPDSD probands, 30% (n=3/10) of OPD1/OPD2 males, 50% (n=3/6) of OPD/OPD2 females, 100% (n=4/4) of MNS females and 57% (n=4/7) of FMD patients. A variant was also identified in P12.II.1 and its pathogenicity is discussed below.
The 11 different identified variations were missense. They were located with higher frequency in the already known OPDSD clusters of mutations, therefore confirming previously reported hotspots (Table 2). Four novel mutations were associated with OPD1 or OPD2: one located within CH2 (P14), one immediately close to CH2 (P13), and the 2 others in RD6 (RD: repeated domain) (P11) and RD22 (P12). No OPDSD mutation had been reported in RD6 to date, while RD22 has been implicated in a single FMD female patient.11 We found a novel MNS mutation in the exon 22 cluster. The 2 novel FMD mutations were located within RD9 (P22) and RD16 (P21). RD16 was involved in a sporadic FMD female11 and a neonatally deceased OPD2 male.4
X inactivation was skewed in all but one affected female (P12.II.1) and her asymptomatic carrier mother (91%, n=20/22). Skewing was mostly partial, ranging from 80/20% to 90/10%, in females carrying an OPD1 mutation (n=3/4). Although skewing is usually more pronounced in OPD2, MNS and FMD (between 90/10% and 100/0%),5 it was partial at several loci in several members of family 15. Bone dysplasia severity and the uncommon associated features, such as cortical dysplasia and glaucoma displayed by P12.II.1, may be due to random X inactivation but a differential diagnosis could be considered as the variant pathogenicity was not definitively established.
This study is the first to report results of FLNA analysis in a large series from a diagnostic laboratory. Previous studies estimated the rate of FLNA mutation detection to be 100% in OPD1 and MNS, 70% in OPD22 and 57% in FMD.11 Here, the mutational rate was markedly lower in OPD1, probably because most patients referred for OPD1 had atypical presentation that made this diagnosis unlikely, in particular P3, P5, P6, P7 and P8. Likewise, P10 and P16 were not really convincing. This tendency to overdiagnose OPD syndrome is probably explained because the minimal diagnostic criteria of the mildest form of the spectrum are established from only few case reports, no series are available and clinicians therefore referred atypical patients. It is noteworthy that in the next-generation sequencing era, phenotypic spectrum of many syndromes has been extended. On the basis of this possibility for FLNA-related disorders, especially the mildest OPD1 syndrome, we chose to accept atypical patients for FLNA testing. However, this hypothesis was not confirmed. Therefore, the diagnosis should be established and FLNA analysis requested when sufficient highly suggestive features are found in a patient, a fortiori if the pedigree is consistent with X-linked inheritance: facial dysmorphism (prominent supraorbital ridges, hypertelorism, downslanted palpebral fissures and microretrognathia), extremities anomalies (first ray hypoplasia, widely spaced digits, clubbed fingers, distal phalangeal hypoplasia and nail agenesis), posterior and central cleft palate (including bifid uvula, but not involving neither primary palate nor upper lip), bone dysplasia (limitations of joint mobility, especially elbows, deformations and/or undertubulation of long bones and cortical irregularities), conductive or mixed hearing loss. Other less specific features include teeth anomalies, cutaneous syndactyly, splayed digits and overlapping digits. Retrospectively, all OPD1 males carrying a mutation had typical presentation. Only P9 and P4 really had combination of clinical features suggestive of OPD1 but other differential diagnoses could be considered, especially Freeman–Sheldon syndrome (P9) and Larsen syndrome (P4). Other unconfirmed OPD1 males referred for FLNA testing had insufficient criteria. According to our series, the most distinctive features between confirmed (that is, carrying a FLNA mutation) and suspected (no detected mutation) OPD1 male patients were extremities anomalies, posterior cleft palate and hearing loss. In OPD/OPD2 females, facial dysmorphism, although inconstant, was of better value to predict a mutation. Instead, all FMD males had a homogeneous phenotype according to previously defined criteria,11 whatever their FLNA status. It is noteworthy that the patients were not secondarily reassigned according to their genotype even if overlaps between syndromes make it difficult for some patients to be assigned to a specific diagnosis, and molecular findings can be taken into account (see below). One could criticize the variant pathogenicity and OPD diagnosis in P12.II.1. Indeed, she displayed compatible features but other atypical findings such as cortical dysplasia, and above all, X inactivation pattern was random. The fact that her asymptomatic mother carried the variant and had no X inactivation skewing argue against its pathogenicity. Moreover, although the variant was not found in Exome Variant Server, it was found in ExAc with a minor allelic frequency of 0.00008 (7 out of 86,932 chromosomes, rs727503930). No larger segregation analysis could be performed in the family. Of note, the only OPD2 female patient we found to carry a FLNA mutation without a skewed X inactivation is fetus 2 mother from Naudion et al. (personal data from the new studied loci: CNKSR2 65/35, HMGB3 80/20, TMEM185A 72/28). All these data led us to consider this variant to be of unknown significance.
Our study also refines the clinical spectrum of OPDSD. First, previous reports, as ours, show that eye anterior segment anomalies also belong to the clinical spectrum of these syndromes. The manifestations include Peters anomaly, glaucoma, cataract, iris coloboma, sclerocornea, congenital or post-natal corneal clouding.4, 12, 13, 14, 15, 16 Second, craniosynostosis is not unfrequent in FMD (at least four affected males in our series) as previously noted.17 Third, ptosis, oculomotor paresis and strabismus noted in 5 OPDSD patients may be related to skull bone dysplasia and subsequent nerve compression. Finally, we noted that 2 MNS patients carrying the recurrent p.Ala1188Thr displayed bleeding manifestations. It is noteworthy that macrothrombocytopenia and platelet dysfunctions were described in patients carrying loss-of-function mutations, either in isolation or associated with periventricular nodular heterotopia.9, 18 The frequency of such symptoms may be underestimated as they are rarely spontaneously mentioned by patients. A link between bleeding and MNS is therefore possible and clinicians should be aware although more data are required to enable definitive confirmation.
This series emphasizes two main characteristics of OPDSD, namely clinical overlap between the different conditions and intrafamilial clinical heterogeneity. P11 and patient 1 described by Zenker et al.19 display prominent supraorbital ridges typical of FMD and extremities suggestive of OPD syndrome. In both of them, the pathogenic mutation, within RD6 and RD15, respectively, is located outside the classical OPD CH2 cluster. A common pathophysiological mechanism involving these 2 different RD can be hypothesized. Likewise, P22 suffers from FMD with a phenotypic severity partially overlapping MNS. In this latter patient, the missense mutation is located in RD9 (next to MNS and FMD RD10 clusters). In family 20, P20.II.1 was referred for MNS and the novel p.Val1163Leu substitution was identified. Segregation analysis led us to note that the severity of bone dysplasia and dysmorphic features ranged from campomelia and characteristic facial features in the proband to isolated slight bowing of tibias in her mother, while her sister had an intermediate phenotype. This familial heterogeneity is classically found in FMD. Likewise, in families 12 and 15, females were variably affected, P12.I.1 and P15.II.7 being asymptomatic. Although the diagnosis is not definitely established in family 12, we can observe the wide phenotypic variability in females suffering from OPDSD. X inactivation pattern seems to correlate in family 15 but not in families 12 and 20. Other factors are probably involved to account for this clinical variability among females.
The absence of FLNA mutation in some patients may also be explained by the lack of sensitivity of the molecular techniques used or by genetic heterogeneity, especially in FMD.
Correct discrimination of the different OPDSD entities is of major interest in order to provide accurate genetic counseling. When only females are affected within a family, clinical severity and level of X inactivation skewing may help to anticipate phenotypic severity in males.
Thus, this series describes new mutations, provide additional arguments to include ophthalmological and craniosynostosis as signs of OPDSD, and maybe bleeding manifestations as part of MNS. We emphasize some clues relevant for OPD1 diagnosis. Final diagnosis among the different conditions should consider and integrate data from the clinical presentation, the nature of the variant, X inactivation pattern and segregation analysis, although overlaps are sometimes important.
References
Verloes, A., Lesenfants, S., Barr, M., Grange, D. K., Journel, H., Lombet, J. et al. Fronto-otopalatodigital osteodysplasia: clinical evidence for a single entity encompassing Melnick-Needles syndrome, otopalatodigital syndrome types 1 and 2, and frontometaphyseal dysplasia. Am. J. Med. Genet. 90, 407–422 (2000).
Robertson, S. P. Otopalatodigital syndrome spectrum disorders: otopalatodigital syndrome types 1 and 2, frontometaphyseal dysplasia and Melnick-Needles syndrome. Eur. J. Hum. Genet. 15, 3–9 (2007).
Sun, Y., Almomani, R., Aten, E., Celli, J., van der Heijden, J., Venselaar, H. et al. Terminal osseous dysplasia is caused by a single recurrent mutation in the FLNA gene. Am. J. Hum. Genet. 87, 146–153 (2010).
Naudion, S., Moutton, S., Coupry, I., Sole, G., Deforges, J., Guerineau, E. et al. Fetal phenotypes in otopalatodigital spectrum disorders. Clin. Genet. 89, 371–377 (2015).
Robertson, S. P., Twigg, S. R., Sutherland-Smith, A. J., Biancalana, V., Gorlin, R. J., Horn, D. et al. Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat. Genet. 33, 487–491 (2003).
Nakamura, F., Stossel, T. P. & Hartwig, J. H. The filamins: organizers of cell structure and function. Cell Adh. Migr. 5, 160–169 (2011).
Clark, A. R., Sawyer, G. M., Robertson, S. P. & Sutherland-Smith, A. J. Skeletal dysplasias due to filamin A mutations result from a gain-of-function mechanism distinct from allelic neurological disorders. Hum. Mol. Genet. 18, 4791–4800 (2009).
Fergelot, P., Coupry, I., Rooryck, C., Deforges, J., Maurat, E., Sole, G. et al. Atypical male and female presentations of FLNA-related periventricular nodular heterotopia. Eur. J. Med. Genet. 55, 313–318 (2012).
Nurden, P., Debili, N., Coupry, I., Bryckaert, M., Youlyouz-Marfak, I., Sole, G. et al. Thrombocytopenia resulting from mutations in filamin A can be expressed as an isolated syndrome. Blood 118, 5928–5937 (2011).
Bertelsen, B., Tumer, Z. & Ravn, K. Three new loci for determining x chromosome inactivation patterns. J. Mol. Diagn. 13, 537–540 (2011).
Robertson, S. P., Jenkins, Z. A., Morgan, T., Ades, L., Aftimos, S., Boute, O. et al. Frontometaphyseal dysplasia: mutations in FLNA and phenotypic diversity. Am. J. Med. Genet. A 140, 1726–1736 (2006).
Weh, E., Reis, L. M., Happ, H. C., Levin, A. V., Wheeler, P. G., David, K. L. et al. Whole exome sequence analysis of Peters anomaly. Hum. Genet. 133, 1497–1511 (2014).
Kondoh, T., Okamoto, N., Norimatsu, N., Uetani, M., Nishimura, G. & Moriuchi, H. A Japanese case of oto-palato-digital syndrome type II: an apparent lack of phenotype-genotype correlation. J. Hum. Genet. 52, 370–373 (2007).
Stratton, R. F. & Bluestone, D. L. Oto-palatal-digital syndrome type II with X-linked cerebellar hypoplasia/hydrocephalus. Am. J. Med. Genet. 41, 169–172 (1991).
Murphy-Ryan, M., Babovic-Vuksanovic, D. & Lindor, N. Bifid tongue, corneal clouding, and Dandy-Walker malformation in a male infant with otopalatodigital syndrome type 2. Am. J. Med. Genet. A 155A, 855–859 (2011).
Santos, H. H., Garcia, P. P., Pereira, L., Leao, L. L., Aguiar, R. A., Lana, A. M. et al. Mutational analysis of two boys with the severe perinatally lethal Melnick-Needles syndrome. Am. J. Med. Genet. A 152A, 726–731 (2010).
Fennell, N., Foulds, N., Johnson, D. S., Wilson, L. C., Wyatt, M., Robertson, S. P. et al. Association of mutations in FLNA with craniosynostosis. Eur. J. Hum. Genet. 23, 1684–1688 (2015).
Berrou, E., Adam, F., Lebret, M., Fergelot, P., Kauskot, A., Coupry, I. et al. Heterogeneity of platelet functional alterations in patients with filamin A mutations. Arterioscler. Thromb. Vasc. Biol. 33, e11–e18 (2013).
Zenker, M., Nahrlich, L., Sticht, H., Reis, A. & Horn, D. Genotype-epigenotype-phenotype correlations in females with frontometaphyseal dysplasia. Am. J. Med. Genet. A 140, 1069–1073 (2006).
Acknowledgements
We sincerely thank all families who contributed to the study, all physicians who referred their patients to our laboratory (Jean-Luc Alessandri, Patricia Blanchet, Géraldine Viot, Delphine Heron, Chloé Quélin, Yves Alembik, Philippe Parent, Laurent Besson-Leaud and S Bernhard), physicians, genetic counselors and medical students who contributed to data collection (Laètitia Lambert, Charlène Rambaud and Linda Pons). We thank NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010). We thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Moutton, S., Fergelot, P., Naudion, S. et al. Otopalatodigital spectrum disorders: refinement of the phenotypic and mutational spectrum. J Hum Genet 61, 693–699 (2016). https://doi.org/10.1038/jhg.2016.37
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.37
This article is cited by
-
Periventricular nodular heterotopias is associated with mutation at the FLNA locus-a case history and a literature review
BMC Pediatrics (2023)
-
Homozygous loss-of-function variants in FILIP1 cause autosomal recessive arthrogryposis multiplex congenita with microcephaly
Human Genetics (2023)
-
Prune belly syndrome in surviving males can be caused by Hemizygous missense mutations in the X-linked Filamin A gene
BMC Medical Genetics (2020)
