
Abstract
Cryogenic electron microscopy (cryo-EM) data represent density maps of macromolecular systems at atomic or near-atomic resolution. However, building and refining 3D atomic models by using data from cryo-EM maps is not straightforward and requires significant hands-on experience and manual intervention. We recently developed StarMap, an easy-to-use interface between the popular structural display program ChimeraX and Rosetta, a powerful molecular modeling engine. StarMap offers a general approach for refining structural models of biological macromolecules into cryo-EM density maps by combining Monte Carlo sampling with local density-guided optimization, Rosetta-based all-atom refinement and real-space B-factor calculations in a straightforward workflow. StarMap includes options for structural symmetry, local refinements and independent model validation. The overall quality of the refinement and the structure resolution is then assessed via analytical outputs, such as magnification calibration (pixel size calibration) and Fourier shell correlations. Z-scores reported by StarMap provide an easily interpretable indicator of the goodness of fit for each residue and can be plotted to evaluate structural models and improve local residue refinements, as well as to identify flexible regions and potentially functional sites in large macromolecular complexes. The protocol requires general computer skills, without the need for coding expertise, because most parts of the workflow can be operated by clicking tabs within the ChimeraX graphical user interface. Time requirements for the model refinement depend on the size and quality of the input data; however, this step can typically be completed within 1 d. The analytical parts of the workflow are completed within minutes.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others
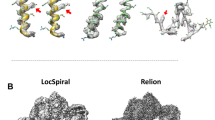
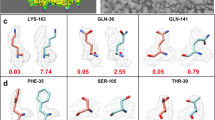
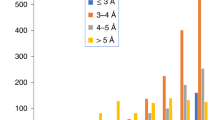
Code availability
StarMap is available under libre/open source license (BSD 2-Clause ‘Simplified’ License) from GitHub (https://github.com/wlugmayr/chimerax-starmap). StarMap version 1.1.66 used to generate data from Box 3 is available from the Supplement.
References
Kühlbrandt, W. Biochemistry. The resolution revolution. Science 343, 1443–1444 (2014).
Milne, J. L. S. et al. Cryo-electron microscopy: a primer for the non-microscopist. FEBS J. 280, 28–45 (2013).
Henderson, R. & McMullan, G. Problems in obtaining perfect images by single-particle electron cryomicroscopy of biological structures in amorphous ice. Microscopy 62, 43–50 (2013).
Baker, T. S. & Henderson, R. Electron cryomicroscopy of biological macromolecules. In International Tables for Crystallography Vol. F (eds. Arnold, E., Himmel, D. M. & Rossmann, M. G.) 593–614 (John Wiley & Sons, Ltd, 2012).
Scheres, S. H. W. Processing of structurally heterogeneous cryo-EM data in RELION. In Methods in Enzymology Vol. 579 (ed. Crowther, R. A.) 125–157 (Academic Press, 2016).
Ludtke, S. J. Single-particle refinement and variability analysis in EMAN2.1. In Methods in Enzymology Vol. 579 (ed. Crowther, R. A.) 159–189 (Academic Press, 2016).
Rosenthal, P. B. & Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003).
Cowtan, K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D. Biol. Crystallogr. 62, 1002–1011 (2006).
Chen, M., Baldwin, P. R., Ludtke, S. J. & Baker, M. L. De novo modeling in cryo-EM density maps with Pathwalking. J. Struct. Biol. 196, 289–298 (2016).
Chojnowski, G., Sobolev, E., Heuser, P. & Lamzin, V. S. The accuracy of protein models automatically built into cryo-EM maps with ARP/wARP. Acta Crystallogr. D. Struct. Biol. 77, 142–150 (2021).
Terwilliger, T. C., Adams, P. D., Afonine, P. V. & Sobolev, O. V. A fully automatic method yielding initial models from high-resolution cryo-electron microscopy maps. Nat. Methods 15, 905–908 (2018).
Frenz, B., Walls, A. C., Egelman, E. H., Veesler, D. & DiMaio, F. RosettaES: a sampling strategy enabling automated interpretation of difficult cryo-EM maps. Nat. Methods 14, 797–800 (2017).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D. Biol. Crystallogr. 74, 531–544 (2018).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D. Biol. Crystallogr. 74, 519–530 (2018).
Kim, D. N. et al. Cryo_fit: democratization of flexible fitting for cryo-EM. J. Struct. Biol. 208, 1–6 (2019).
Kidmose, R. T. et al. Namdinator – automatic molecular dynamics flexible fitting of structural models into cryo-EM and crystallography experimental maps. IUCrJ 6, 526–531 (2019).
Simons, K. T., Kooperberg, C., Huang, E. & Baker, D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 268, 209–225 (1997).
Simons, K. T. et al. Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins 34, 82–95 (1999).
Leman, J. K. et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods 17, 665–680 (2020).
Alford, R. F. et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 13, 3031–3048 (2017).
DiMaio, F. et al. Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement. Nat. Methods 12, 361–365 (2015).
DiMaio, F., Tyka, M. D., Baker, M. L., Chiu, W. & Baker, D. Refinement of protein structures into low-resolution density maps using rosetta. J. Mol. Biol. 392, 181–190 (2009).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Frenz, B. et al. Automatically fixing errors in glycoprotein structures with Rosetta. Structure 27, 134–139.e3 (2019).
Pavlovicz, R. E., Park, H. & DiMaio, F. Efficient consideration of coordinated water molecules improves computational protein-protein and protein-ligand docking discrimination. PLoS Comput. Biol. 16, e1008103 (2020).
Renfrew, P. D., Choi, E. J., Bonneau, R. & Kuhlman, B. Incorporation of noncanonical amino acids into Rosetta and use in computational protein-peptide interface design. PLoS One 7, e32637 (2012).
Wang, R. Y.-R. et al. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. eLife 5, e17219 (2016).
Xu, J. et al. Structural basis for the initiation of eukaryotic transcription-coupled DNA repair. Nature 551, 653–657 (2017).
Usluer, G. D. et al. Cryo-EM structure of the bacterial actin AlfA reveals unique assembly and ATP-binding interactions and the absence of a conserved subdomain. Proc. Natl Acad. Sci. USA 115, 3356–3361 (2018).
James, Z. M. et al. CryoEM structure of a prokaryotic cyclic nucleotide-gated ion channel. Proc. Natl Acad. Sci. USA 114, 4430–4435 (2017).
Kotov, V., Lunelli, M., Wald, J., Kolbe, M. & Marlovits, T. C. Helical reconstruction of Salmonella and Shigella needle filaments attached to type 3 basal bodies. Biochem. Biophys. Rep. 27, 101039 (2021).
Wald, J. et al. Cryo-EM structure of pleconaril-resistant rhinovirus-B5 complexed to the antiviral OBR-5-340 reveals unexpected binding site. Proc. Natl Acad. Sci. USA 116, 19109–19115 (2019).
Bunduc, C. M. et al. Structure and dynamics of a mycobacterial type VII secretion system. Nature 593, 445–448 (2021).
Miletic, S. et al. Substrate-engaged type III secretion system structures reveal gating mechanism for unfolded protein translocation. Nat. Commun. 12, 1546 (2021).
Goessweiner-Mohr, N. et al. Structural control for the coordinated assembly into functional pathogenic type-3 secretion systems. Preprint at https://www.biorxiv.org/content/10.1101/714097v1.full (2019).
Wald, J. et al. Mechanism of AAA+ ATPase-mediated RuvAB–Holliday junction branch migration. Nature 609, 630–639 (2022).
Loquet, A. et al. Atomic model of the type III secretion system needle. Nature 486, 276–279 (2012).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Terwilliger, T. C., Sobolev, O. V., Afonine, P. V., Adams, P. D. & Read, R. J. Density modification of cryo-EM maps. Acta Crystallogr. D. Biol. Crystallogr. 76, 912–925 (2020).
Sanchez-Garcia, R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 4, 1–8 (2021).
Lawson, C. L. et al. EMDataBank.org: unified data resource for CryoEM. Nucleic Acids Res. 39, D456–D464 (2011).
De Rosier, D. J. & Klug, A. Reconstruction of three dimensional structures from electron micrographs. Nature 217, 130–134 (1968).
Zhang, X., Jin, L., Fang, Q., Hui, W. H. & Zhou, Z. H. 3.3 Å cryo-EM structure of a nonenveloped virus reveals a priming mechanism for cell entry. Cell 141, 472–482 (2010).
Dube, P., Tavares, P., Lurz, R. & van Heel, M. The portal protein of bacteriophage SPP1: a DNA pump with 13-fold symmetry. EMBO J. 12, 1303–1309 (1993).
Costa, A. & Patwardhan, A. A novel mirror-symmetry analysis approach for the study of macromolecular assemblies imaged by electron microscopy. J. Mol. Biol. 378, 273–283 (2008).
Marco, S. et al. The molecular chaperone TF55. FEBS Lett. 341, 152–155 (1994).
He, S. & Scheres, S. H. W. Helical reconstruction in RELION. J. Struct. Biol. 198, 163–176 (2017).
Zhang, X. Python-based Helix Indexer: a GUI program for finding symmetry of helical assembly through Fourier-Bessel indexing of electron microscopic data. Protein Sci. 31, 107–117 (2022).
Tykač, M., Černý, J. & Murshudov, G. N. De novo detection of symmetry in cryo-EM density maps. Acta Crystallogr. A 77, C692 (2021).
Reboul, C. F., Kiesewetter, S., Elmlund, D. & Elmlund, H. Point-group symmetry detection in three-dimensional charge density of biomolecules. Bioinformatics 36, 2237–2243 (2020).
DiMaio, F. et al. Improved low-resolution crystallographic refinement with Phenix and Rosetta. Nat. Methods 10, 1102–1104 (2013).
Terwilliger, T. C. et al. phenix.mr_rosetta: molecular replacement and model rebuilding with Phenix and Rosetta. J. Struct. Funct. Genomics 13, 81–90 (2012).
Pražnikar, J., Afonine, P. V., Gunčar, G., Adams, P. D. & Turk, D. Averaged kick maps: less noise, more signal…and probably less bias. Acta Crystallogr. D. Biol. Crystallogr. 65, 921–931 (2009).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D. Biol. Crystallogr. 68, 352–367 (2012).
Skubák, P., Murshudov, G. N. & Pannu, N. S. Direct incorporation of experimental phase information in model refinement. Acta Crystallogr. D. Biol. Crystallogr. 60, 2196–2201 (2004).
Bourne, P. E. et al. Macromolecular crystallographic information file. In Methods in Enzymology Vol. 277 (eds. Abelson, J. N., Simon, M. I., Carter, Jr., C. W. & Sweet, R. M.) 571–590 (Academic Press, 1997).
Westbrook, J. D. & Fitzgerald, P. M. D. The PDB format, mmCIF formats, and other data formats. In Structural Bioinformatics (eds. Bourne, P. E. & Weissig, H.) 159–179 (John Wiley & Sons, Ltd, 2003).
Park, H., Zhou, G., Baek, M., Baker, D. & DiMaio, F. Force field optimization guided by small molecule crystal lattice data enables consistent sub-angstrom protein–ligand docking. J. Chem. Theory Comput. 17, 2000–2010 (2021).
Wlodawer, A., Li, M. & Dauter, Z. High-resolution cryo-EM maps and models: a crystallographer’s perspective. Structure 25, 1589–1597.e1 (2017).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Wang, S., Sun, S., Li, Z., Zhang, R. & Xu, J. Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Comput. Biol. 13, e1005324 (2017).
Yang, J. et al. The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 (2015).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Keating, K. S. & Pyle, A. M. RCrane: semi-automated RNA model building. Acta Crystallogr. D. Biol. Crystallogr. 68, 985–995 (2012).
Kappel, K. et al. De novo computational RNA modeling into cryo-EM maps of large ribonucleoprotein complexes. Nat. Methods 15, 947–954 (2018).
Jossinet, F., Ludwig, T. E. & Westhof, E. Assemble: an interactive graphical tool to analyze and build RNA architectures at the 2D and 3D levels. Bioinformatics 26, 2057–2059 (2010).
Chou, F.-C., Echols, N., Terwilliger, T. C. & Das, R. RNA structure refinement using the ERRASER-Phenix pipeline. Methods Mol. Biol. 1320, 269–282 (2016).
Fleishman, S. J. et al. RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS ONE 6, e20161 (2011).
van Zundert, G. C. P., Bonvin, A. M. J. J., van Zundert, G. C. P. & Bonvin, A. M. J. J. Fast and sensitive rigid-body fitting into cryo-EM density maps with PowerFit. AIMS Biophys. 2, 73–87 (2015).
Feng, Z. et al. Ligand Depot: a data warehouse for ligands bound to macromolecules. Bioinformatics 20, 2153–2155 (2004).
Dimitropoulos, D., Ionides, J. & Henrick, K. Using MSDchem to search the PDB ligand dictionary. Curr. Protoc. Bioinforma. 14, 14.3.1–14.3.21 (2006).
Long, F. et al. AceDRG: a stereochemical description generator for ligands. Acta Crystallogr. D. Struct. Biol. 73, 112–122 (2017).
Landrum, G. et al. RDKit: Open-source cheminformatics. Available at https://zenodo.org/record/6961488#.YyplJOzML_U (Zenodo, 2021).
Yoshikawa, N. & Hutchison, G. R. Fast, efficient fragment-based coordinate generation for Open Babel. J. Cheminform. 11, 49 (2019).
Terwilliger, T. C., Klei, H., Adams, P. D., Moriarty, N. W. & Cohn, J. D. Automated ligand fitting by core-fragment fitting and extension into density. Acta Crystallogr. D. Biol. Crystallogr. 62, 915–922 (2006).
Evrard, G. X., Langer, G. G., Perrakis, A. & Lamzin, V. S. Assessment of automatic ligand building in ARP/wARP. Acta Crystallogr. D. Biol. l Crystallogr. 63, 108–117 (2007).
Nicholls, R. A. Ligand fitting with CCP4. Acta Crystallogr. D. Biol. Crystallogr. 73, 158–170 (2017).
Kahraman, A. et al. Cross-link guided molecular modeling with ROSETTA. PLoS ONE 8, e73411 (2013).
Ovchinnikov, S., Kamisetty, H. & Baker, D. Robust and accurate prediction of residue–residue interactions across protein interfaces using evolutionary information. eLife 3, e02030 (2014).
Marks, D. S. et al. Protein 3D structure computed from evolutionary sequence variation. PLoS ONE 6, e28766 (2011).
Veusz - A Scientific Plotting Package. Available at https://veusz.github.io/
Liu, K. et al. Structural basis of CXC chemokine receptor 2 activation and signalling. Nature 585, 135–140 (2020).
The UniProt Consortium. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 42, D191–D198 (2014).
Olieric, V. et al. Data-collection strategy for challenging native SAD phasing. Acta Crystallogr. D. Biol. Crystallogr. 72, 421–429 (2016).
Santos, R. et al. Structure of human immunoproteasome with a reversible and noncompetitive inhibitor that selectively inhibits activated lymphocytes. Nat. Commun. 8, 1692 (2017).
Grieben, M. et al. Structure of the polycystic kidney disease TRP channel Polycystin-2 (PC2). Nat. Struct. Mol. Biol. 24, 114–122 (2017).
Zhou, X. et al. Cryo-EM structures of the human endolysosomal TRPML3 channel in three distinct states. Nat. Struct. Mol. Biol. 24, 1146–1154 (2017).
Ruiz Carrillo, D. et al. Crystallization and preliminary crystallographic analysis of human aquaporin 1 at a resolution of 3.28 Å. Acta Crystallogr. F. Struct. Biol. Commun. 70, 1657–1663 (2014).
Yuan, B. et al. Structural dynamics of the functional nonameric type III translocase export gate. J. Mol. Biol. 433, 167188 (2021).
Roh, S.-H. et al. Subunit conformational variation within individual GroEL oligomers resolved by Cryo-EM. Proc. Natl Acad. Sci. USA 114, 8259–8264 (2017).
Acknowledgements
We thank all current and former members of the Marlovits laboratory for their support in this project. High-performance computing was possible through access to the computing clusters at Deutsches Elektronen Synchrotron (DESY)/Hamburg (Germany) and the Vienna Scientific Cluster (Austria). This project was supported by funds available to T.C.M. through the Behörde für Wissenschaft, Forschung und Gleichstellung of the city of Hamburg at the Institute of Structural and Systems Biology at the University Medical Center Hamburg–Eppendorf (UKE), DESY, the Institute for Molecular Biotechnology (IMBA) of the Austrian Academy of Sciences and the Research Institute of Molecular Pathology (IMP).
Author information
Authors and Affiliations
Contributions
Conceptualization: F.D.M. and T.C.M. Methodology: W.L., F.D.M. and T.C.M. Software: W.L. and F.D.M. Investigation: N.G.M., J.W., V.K. Formal analysis: all authors. Resources: T.C.M. Writing—original draft: V.K. and W.L. Writing—review and editing: all authors. Visualization: V.K. and W.L. Supervision: T.C.M. Funding acquisition: T.C.M.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Protocols thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key papers related to this protocol
DiMaio, F. et al. Nat. Methods 12, 361–365 (2015): https://doi.org/10.1038/nmeth.3286
Wang, R. Y.-R. et al. eLife 5, e17219 (2016): https://doi.org/10.7554/eLife.17219
Kotov, V. et al. Biochem. Biophys. Rep. 27, 101039 (2021): https://doi.org/10.1016/j.bbrep.2021.101039
Wald, J. et al. Nature 609, 630–639 (2022): https://doi.org/10.1038/s41586-022-05121-1
Supplementary information
Supplementary Video 1
This short video shows how to set up a StarMap refinement and how to inspect outputs
Supplementary Software 1
A distribution of StarMap (any platform)
Supplementary Data 1
Input and output files for an exemplary StarMap run (Box 3)
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lugmayr, W., Kotov, V., Goessweiner-Mohr, N. et al. StarMap: a user-friendly workflow for Rosetta-driven molecular structure refinement. Nat Protoc 18, 239–264 (2023). https://doi.org/10.1038/s41596-022-00757-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-022-00757-9
This article is cited by
-
Multi-scale structures of the mammalian radial spoke and divergence of axonemal complexes in ependymal cilia
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
