
Abstract
The study objective was to determine the effects of two treatment regimens on quantities of ceftiofur and tetracycline resistance genes in feedlot cattle. The two regimens were ceftiofur crystalline-free acid (CCFA) administered to either one or all steers within a pen and subsequent feeding/not feeding of therapeutic doses of chlortetracycline. A 26-day randomized controlled field trial was conducted on 176 steers. Real-time PCR was used to quantify blaCMY-2, blaCTX-M, tet(A), tet(B) and 16S rRNA gene copies/gram of feces from community DNA. A significant increase in ceftiofur resistance and a decrease in tetracycline resistance elements were observed among the treatment groups in which all steers received CCFA treatment, expressed as gene copies/gram of feces. Subsequent chlortetracycline administration led to rapid expansion of both ceftiofur and tetracycline resistance gene copies/gram of feces. Our data suggest that chlortetracycline is contraindicated when attempting to avoid expansion of resistance to critically important third-generation cephalosporins.
Similar content being viewed by others

Introduction
Cephalosporins are used in both animals and humans to treat a wide variety of bacterial diseases. For the year 2011, cephalosporins sold or distributed in the United States for use in food-producing animals amounted to 26,611 kgs1. Ceftiofur, a third-generation cephalosporin, is used in beef cattle to treat bovine respiratory disease and acute bovine interdigital necrobacillosis. Ceftiofur is classified as a critically important antimicrobial by the World Health Organization2. It belongs to the same general class of antibiotics as ceftriaxone, which has been highly valued in human medicine for treating serious and life-threatening infections. Ceftriaxone is one of the few drugs available to treat invasive salmonellosis in children3,4. Emergence and spread of Salmonella enterica (non-Typhi), Escherichia coli and other gut bacteria resistant to third-generation cephalosporins have been linked by some authors to ceftiofur use in veterinary practice and animal husbandry5,6,7,8. Data from the 2010 U.S. National Antimicrobial Resistance Monitoring System (NARMS) indicate that 2.8% of the non-Typhi Salmonella isolates arising from human clinical cases were resistant both to ceftiofur and ceftriaxone9. At the same time, 21.5% of non-Typhi Salmonella isolates obtained from food-producing animals at slaughter were resistant to both ceftiofur and ceftiaxone10. This is a substantial increase when compared with 14.5% of non-Typhi Salmonella animal isolates that were resistant in 2009. The prevalence of ceftriaxone resistance among human isolates has been observed to be relatively stable since 20049; however, in 2010 ceftriaxone resistance among non-Typhi Salmonella cattle isolates (22%) was observed to be at its highest since NARMS testing began in 199710. In response to observed increases in cephalosporin resistance among food animal non-Typhi Salmonella isolates and to preserve antibiotic efficacy both in veterinary and human medicine, the U.S. Food and Drug Administration released an order in April 2012 restricting certain extra-label uses of cephalosporin drugs in food-producing animals11.
Restricting or banning the use of some less critical antimicrobial classes in animal agriculture may result in unintended consequences, such as an increase in therapeutic use of antimicrobials that are deemed to be critical for human medicine12,13. Strategies to control ceftiofur resistance need to be designed and tested at the farm level in order to protect both animal and human health.
The first objective of the study was to evaluate pen-level antimicrobial treatment effects as measured by quantifying levels of resistance genes present in cattle feces. Differential ceftiofur treatment was employed to mimic the practice of housing cattle in their home pen (post-antimicrobial treatment) versus keeping them in a hospital pen with all other animals also having recently been treated. The cumulative load of resistant enteric bacteria voided into the feedlot environment affects the ‘dose’ that other cattle are exposed to through the fecal-oral route. Models incorporating bacterial metapopulations are now being used to assess the relative and absolute exposure levels of cattle to resistant versus susceptible bacteria in agricultural environments14,15. It has been suggested that readily available and more susceptible bacteria, can aid in out-competing the resistant bacterial populations in the gut of treated animals, thus promoting their recolonization with susceptible bacteria16. Although not universally accepted, many scientists believe that emergent resistance genes are initially associated with a fitness cost to the host bacterium, especially soon after the introduction of new drugs to the market17,18,19,20. It was expected that the baseline equilibrium could be reestablished more quickly in pens where treated and non-treated animals were housed together than in pens where all animals were treated with a third-generation cephalosporin. In order to maximize any differential antimicrobial effects we choose the extreme ratios of treated versus non-treated animals at 1:10 vs 11:0, respectively.
The second objective of the study was to evaluate the effect of feeding chlortetracycline on levels of ceftiofur and tetracycline resistance elements detected during the post-therapeutic period of the two third-generation cephalosporins pen-level regimens described above. Published papers in the human medical literature have demonstrated a protective effect of fluoroquinolone as an alternative intervention against the emergence of cephalosporin resistance21,22. Our previous work suggested that chlortetracycline in the cattle feed significantly decreased the prevalence of ceftiofur-resistant E. coli23. The outcome measure in that study was strictly phenotypic, utilizing culture-based approaches and measuring the proportion of bacteria resistant to a panel of 15 antimicrobials (NARMS CMV1AGNF Gram negative panel, Trek Diagnostics, Cleveland, OH). The total bacterial load was not quantified, so the effect of chlortetracycline on the background bacterial population could not be determined. The current study was designed to evaluate the effects of chlortetracycline on ceftiofur and tetracycline resistance gene copies, as well as the 16S rRNA gene copies, using quantitative qPCR assays on community DNA extracted from the bovine fecal metagenome.
Relying strictly on phenotypic, or even genotypic, data from cultured isolates restricts the focus to a very small subset of the gut microbiome. Resistance elements are rarely restricted to one genus (e.g., Escherichia or Salmonella) or even to a family (e.g., Enterobacteriaceae). If results from our previous work23 proved to be robust across the entire metagenome, chlortetracycline would be expected to minimize the proliferation and accumulation of ceftiofur-resistant bacteria and their resistance elements in production-animal agriculture settings. Overall, in the present study, samples were evaluated both at phenotypic24 and genotypic levels, the latter involving both analysis of fecal bacterial isolates24,25,26 as well as the broader metagenome. This manuscript focuses on the results obtained from the analysis of resistance genes present in fecal community DNA and will compare and contrast those findings with the isolate-based work. This quantitative study further explores the differential selection and co-selection of the resistance genes found in the fecal metagenome, based upon pen-level antimicrobial regimens. Several studies, including this study on E. coli isolates, have demonstrated the phenomenon of differential selection of resistance genes based on chlortetracycline exposure24,26,27. Other studies have likewise reported associations among various tetracycline and ceftiofur-resistance genes28,29,30,31.
The objective of this study was to investigate the effects of two treatment strategies: 1) the pen-level effects of the ratio of CCFA-treated animals housed with untreated animals and 2) the subsequent pen-level feeding of therapeutic doses of chlortetracycline, on the levels of cephalosporin and tetracycline resistance gene copies present in the bovine fecal metgenome. Quantitative real-time PCR was performed on fecal community DNA to determine the changes in microbial populations as measured by changes in target gene copies [blaCMY-2, blaCTX-M, tet(A), tet(B) and 16S rRNA genes] under the influence of various treatments. The study results were essentially quantitative; that is, the results are expressed in terms of absolute resistance gene copies per gram of feces or else standardized to the 16S rRNA gene copies representing the total bacterial population in each gram of feces. Our hypothesis was that both treatment strategies would help decrease the levels of ceftiofur-resistance elements in the bovine fecal metagenome.
Results
To initially determine the dynamics of the primary resistance gene of interest for the full study period, blaCMY-2 and 16S rRNA gene copies per gram of wet feces were determined from community DNA extracted from all 2,424 fecal samples obtained from the trial. There were 176 steers studied in this trial over 26 days. Fecal samples were collected every other day. Forty samples from 7 animals could not be collected because the animal either became sick or else died during the trial. Later, blaCTX-M, tet(A) and tet(B) gene copies per gram of wet feces were determined only from those 694 fecal samples obtained from Days 0, 4, 12 and 26 during the trial. These 4 days represented maximum expected effects of pre-treatment, differential CCFA-treatment ratios, chlortetracycline administration and the post-treatment washout phases, respectively. All community DNA qPCR assays were run in triplicate.
In our study, depending on the gene, there were as few as zero and as many as 435 (approximately 21%) missing observations (observations below the limit of quantification (LOQ) of the qPCR assay). The blaCMY-2 and 16S rRNA gene quantity datasets derived from all 2,424 community DNA samples (7,272 total observations) had 179 (2.5%) and zero missing observations, respectively. The data obtained from a subset of community DNA samples from the representative four days had 44, 435, 2 and 431 missing observations for blaCMY-2, blaCTX-M, tet(A), tet(B) genes, respectively, from a total of 2,084 observations. A multiple imputation technique was efficient in imputing reasonable values to the missing observations in the dataset. This technique avoided an artificially left-truncated distribution that is commonly observed with low-copy qPCR since the assay cannot provide gene quantity values below the LOQ; the LOQ of the assay being the lowest value estimated among DNA samples as shown in Figures 1 and 2.

Histograms illustrating (A) overall raw distribution of log10blaCTX-M gene copies per gram feces and, (B) distribution of log10blaCTX-M gene copies per gram feces of the imputed estimates of missing observations obtained from a single imputation, subgraphed by the number of missing observations (a = 0, b = 1, c = 2, d = 3) among the PCR triplicate assays.
Note: *represents missing observations arbitrarily assigned a single low non-zero value.

Histograms illustrating: (A) overall raw distribution of log10tet(B) gene copies per gram feces and (B) distribution of log10tet(B) gene copies per gram feces of the imputed estimates of missing observations obtained from a single imputation, subgraphed by the number of missing observations (a = 0, b = 1, c = 2, d = 3) among the PCR triplicate assays.
Note: *represents missing observations arbitrarily assigned a single low non-zero value.
In Figure 1 (A, B), the histograms demonstrate the log10blaCTX-M gene quantity distribution from raw and imputed data, respectively, from the first imputation subdivided by the number of missing observations (n = 0, 1, 2, or 3) among the triplicates of any single community DNA extract (sub graphs: a, b, c and d). Of the 20 imputations performed and utilized in subsequent multivariate regression analyses, the histograms shown here are the results of the first imputation simply to illustrate the distribution of the gene copies before and after the procedure. Sub graphs a, b, c and d represent the distributions of the observations that had: no missing triplicates, one missing triplicate, two missing triplicates and all three missing triplicates, respectively. The arbitrarily assigned non-zero peak at −1.15 log10 gene copies in the non-imputed histograms represents those observations that were missing in each category. The calculated LOQ for the blaCTX-M gene real-time PCR quantification assay was 1.82 log10 gene copies per gram. The distribution is therefore left-truncated at this quantification limit (Figure 1 A, subgraphs a, b and c). Figure 1A, subgraph d shows the distribution of observations that had three missing triplicates. The imputation process distributed these observations in a way that yielded many more observations below the 1.82 log10 gene copies quantification limit compared with the single or double missing triplicates (Figure 1 and supplementary Table 1). Similarly, raw and imputed gene distributions alone for the tet(B) gene are shown in Figure 2 A, B. The limit of quantification for the tet(B) gene was −0.56 log10 gene copies. Overall, the distribution of missing observations was shifted much more to the left (i.e., fewer gene copies) as the number of missing observations among triplicates increased.
From regression analyses, the marginal predicted means of blaCMY-2 log10 gene copy numbers per gram wet feces alone, standardized to 16S rRNA gene copy numbers and stratified by the four treatment groups, are shown in Figure 3A and B. The baseline, Day 0, predicted mean log10blaCMY-2 gene copies per gram wet feces (±SE) for the four treatment groups were 4.29 ± 0.12, 4.52 ± 0.12, 4.55 ± 0.12 and 4.70 ± 0.12. The two treatment groups in which only one steer in a pen received CCFA treatment on Day 0 showed a non-significant decline (P > 0.05) from Day 0 to Day 4 in the log10blaCMY-2 gene copies per gram at the pen level. This was in contrast to the other two groups where all the steers in a pen received CCFA treatment (Figure 3, subgraphs A and B) and the gene copies increased significantly (P < 0.05). As seen on Day 12, chlortetracycline treatment starting after sampling on Day 4 was associated with further expansion of log10blaCMY-2 gene copies per gram, regardless of whether CCFA was administered to one or all eleven steers within a pen.

Line graphs illustrating (A) non-standardized log10blaCMY-2 (B) standardized (as ratio to log10 16S rRNA) log10blaCMY-2 and (C) log10 16S rRNA gene copies per gram wet feces, by treatment groups and over days.
Dark navy line (with solid square) is for the pens in which CCFA was administered to all the steers within pens followed by CTC administration at the pen level; green (with hollow square) is for the pens in which CCFA was administered to a single steer within the pens followed by CTC administration at the pen level; maroon line (with solid triangle) represents pens in which CCFA was administered to all steers within pens without subsequent CTC administration at the pen level; and orange lines (with hollow triangle) represents pens in which CCFA was administered to a single steer within the pens without subsequent CTC administration at the pen level. (Data are marginal means with 95% confidence intervals, represented here as lowess smoothed curves).
Figures 3C, 4A and 5A show the16S rRNA gene copies alone. The baseline, Day 0, predicted mean log10 16S rRNA gene copies per gram ± SE for the four treatment groups were 10.06 ± 0.05, 10.13 ± 0.05, 10.10 ± 0.05 and 10.01 ± 0.05. CCFA administration at different housing ratios did not lead to any significant changes in the total 16S rRNA gene copies per gram feces.

Line graphs illustrating (A) non-standardized log10blaCTX-M (short dashed lines), non-standardized log10blaCMY-2 (solid lines) and log10 16S rRNA (long dashed lines) gene copies per gram wet feces (B) standardized (as ratio to log10 16S rRNA) log10blaCTX-M (short dashed lines) and log10blaCMY-2 (solid lines) gene copies per gram wet feces, by treatment groups and over days. See Figure 3 for treatment groups key.
(Data are marginal means and error bars represent 95% confidence intervals).

Line graphs illustrating (A) non-standardized log10tet(B) (short dashed lines), non-standardized log10tet(A) (solid lines) and log10 16S rRNA (long dashed lines) gene copies per gram wet feces (B) standardized (as ratio to log10 16S rRNA) log10tet(B) (short dashed lines) and log10tet(A) (solid lines) gene copies per gram wet feces, by treatment groups and over days. See Figure 3 for treatment groups key.
(Data are marginal means and error bars represent 95% confidence intervals).
Effects of treatments on absolute and standardized ceftiofur-resistance (blaCMY-2 and blaCTX-M) gene copy numbers per gram wet feces for Days 0, 4, 12 and 26 are represented in Figures 4A and B, respectively. Day 4 illustrates only the effect of CCFA regimen on the ceftiofur-resistance gene copies. Fecal samples collected from animals in pens in which CCFA was administered to all animals within the pen (Figure 4A and B, Day 4, depicted by dark navy and maroon lines) had a greater number of ceftiofur-resistance determinants, blaCMY-2 and blaCTX-M gene copies per gram, compared with the other two treatment groups in which CCFA was administered to only one steer among 11. Day 12 in Figures 4A and B illustrates the effect of in-feed chlortetracycline on the absolute and standardized ceftiofur-resistance gene copies, respectively. Chlortetracycline treatment was found to increase both blaCMY-2 and blaCTX-M gene copies per gram compared with the pens that did not receive chlortetracycline treatment (Figure 4A and B, Day12, depicted by dark navy and green lines), regardless of the pen-level CCFA treatment ratio.
The effect of treatments on absolute and standardized tetracycline resistance [tet(A) and tet(B)] gene copy numbers per gram wet feces for the four days tested are represented in Figure 5A and B, respectively. Initially, CCFA administered to all steers in a pen significantly decreased both tet(A) and tet(B) gene copies per gram feces (Figure 5A and B, Day 4, depicted by dark navy and maroon lines) at the pen level compared with pens in which CCFA was administered to just one steer among 11. More expectedly, there also was a marked increase in each of the tetracycline resistance tet(A) and tet(B) gene copies per gram feces following in-feed chlortetracycline treatment (Figure 5A and B, Day12, depicted by dark navy and green lines). The dataset, as well as graphs and tables illustrating comparative results from imputed and non-imputed data, have been provided as part of the supplementary material. Model estimates using non-imputed data for all five genes also are included in the supplementary material.
Discussion
The hypothesis for this study was based on previous work that suggested that therapeutic doses of chlortetracycline in feed lowered the proportion of ceftiofur-resistant E. coli isolates found in bovine feces23. In that study, chlortetracycline appeared to differentially favor a population of E. coli that were singly, doubly, or even triply resistant (including to tetracycline) versus those multi-drug resistant strains in which ceftiofur and tetracycline resistance both were present and always with at least 6 other resistance types23. The results of that study suggested the intriguing possibility for using chlortetracycline as an intervention to control or reduce ceftiofur-resistance among pens of animals previously treated with ceftiofur, if only in the short term, by taking advantage of fitness cost differences among tetracycline-resistant strains.
The present study involved 176 steers. The full study was conducted in two replicates of 88 steers each. Fecal samples were evaluated both by non-type-specific (NTS) E. coli culture-based24,25,26 as well as metagenome-based assays. This manuscript focuses on results obtained from an additional analysis of resistance genes copies present in the entire fecal metagenome. The NTS E. coli culture-based study was performed only on the first replicate (n = 88 steers), while the metagenome-based assays described in this manuscript involved both replicates. In the metagenome-based study, 16S rRNA gene copies were quantified to take into account the effect of treatment strategies on the total bacterial population.
In the present study, chlortetracycline did not reduce ceftiofur-resistance in the fecal metagenome; in fact, gene copy numbers of both blaCMY-2 and blaCTX-M genes significantly increased following chlortetracycline treatment. There are aspects of this study design that could have caused discrepancy with the results of Platt et al. (2008)23. In the present study, either one or else all animals within each of 8/16 pens received ceftiofur treatment prior to chlortetracycline treatment. Initial ceftiofur exposure among individual animals might have changed the microbial gut population at the pen level enough to have modified the effect of CTC26. Importantly, any chlortetracycline effect that would have been expected, unlike the previous study, would have been observed on the microbial population at disequilibrium.
The blaCMY-2 gene has previously been quantified in two longitudinal trials, one in beef cattle32 and one in dairy cattle33, to evaluate the effect of ceftiofur treatment. The former trial was an experimental study in which three different dose regimens of ceftiofur were compared. One of the regimens consisted of a single dose of 6.6 mg/kg; this was similar to that used in the current study. Alali et al. (2009)32 showed that ceftiofur treatment significantly increased both the standardized as well as non-standardized log10blaCMY-2 gene copies per gram feces; in fact, this is similar to what was observed in our study. The quantity of blaCMY-2 gene in our study also was comparable to the earlier feedlot cattle study32; however, the blaCMY-2 gene quantities were lower in the dairy cattle study reported by Boyer et al.(2012)33. The authors attributed this difference to the likelihood that feedlot cattle harbor higher blaCMY-2 gene copies as compared to dairy cattle because of variation in management and environment and especially treatment pressures. All three studies recorded a temporary increase in blaCMY-2 gene copies immediately following ceftiofur treatment.
We did not observe any significant effects on the 16S rRNA gene copies following ceftiofur treatment. In the earlier feedlot cattle study32, the treatment group that received a similar ceftiofur dose to this study had overall higher predicted mean log1016S rRNA gene copies per gram when compared to the control group. Results from both studies indicate that a selective advantage favored expansion of ceftiofur-resistance elements (and likely bacteria harboring same) as opposed to a significant reduction in overall bacterial community DNA following ceftiofur treatment. The dairy cattle study33 did not report quantification of a bacterial reference gene.
The blaCTX-M gene was detected in E. coli25,26 as reported earlier and was also quantified from community DNA samples derived from feces in this study. For both of the targeted ceftiofur-resistance genes, blaCMY-2 and blaCTX-M gene copies per gram feces significantly increased in treatment groups where all animals were administered CCFA compared with the pens in which only one animal received CCFA treatment. The short-term selection of blaCMY-2 gene copies following CCFA administration has been previously documented, as mentioned before32,33. However, this is the first study to quantify the blaCTX-M gene in feedlot cattle in the United States. This gene has been noticeably absent from North American agriculture for some time now.
The quantities of tetracycline genes tet(A) and tet(B) significantly decreased on Day 4 in treatment pens where all animals were administered CCFA as compared to the pens where only one animal received CCFA treatment. Several phenotypic studies on E. coli have recorded that all ceftiofur-resistant isolates were found to be co-resistant to tetracycline5,23,34. Therefore, although the decrease in copies per gram wet feces of both tetracycline genes from community DNA was unexpected, it is readily explainable. First, E. coli bacteria constitute a small fraction of the entire microbial community; that is, the gut flora comprises both culturable and non-culturable organisms that may or may not carry ceftiofur and tetracycline determinants together on one plasmid. Second, even though those E. coli that remain following ceftiofur treatment are resistant both to ceftiofur and tetracycline, those bacteria that are resistant only to tetracycline do not survive, or else are suppressed. The complexity of the co-selection or co-resistance mechanisms has been discussed by Doyle et al. (2013)35. Microbial populations, along with their constituent organisms, may evolve differently under specific selection pressures in different niches, depending on both the complexity of the niches and the selection pressure, particularly if the starting bacterial population is not exactly the same. Competitive bacterial interactions play a critical role in niches such as the host gut that harbor large numbers of varied microbes35.
The blaCTX-M gene in the culture-based study was always found to be harbored on an IncN plasmid among the 1,050 E. coli isolates examined from the 88 first replicate animals25. This plasmid was shown to be self-transmissible without conferring any additional fitness cost to the recipient E. coli25. This indicates the presence of E. coli (and likely other Gram-negative bacteria) that harbor plasmids which possess resistance determinants directed towards beta-lactams (including third-generation cephalosporins) alone; in contrast, the blaCMY-2 gene in U.S. cattle has previously almost always been reported to be associated with multiple drug resistance and located on an IncA/C plasmid35,36. Furthermore, E. coli isolates derived from 29 out of 88 first replicate study animals tested positive for the blaCTX-M gene25. Our metagenome-based assay quantified blaCTX-M gene copies from 87 out of 88 animals from at least one community DNA sample. All DNA samples derived from 12 animals had measurable blaCTX-M gene copies (greater than the threshold limit of the qPCR assay). Of 1050 community DNA samples from the first replicate, 760 had measurable blaCTX-M gene copies (data not shown). These results clearly indicate a higher sensitivity of metagenome-based assays than culture-based approaches alone.
Distinct differences were observed between the E. coli (culture-based assay) results24,26 and the gene quantification results obtained in this study. The qPCR results from community DNA indicated an increase in both blaCMY-2 and blaCTX-M gene copies per gram wet feces following chlortetracycline treatment. This runs counter to the results obtained from E. coli isolates in which the blaCMY-2 gene was differentially favored over the blaCTX-M gene when chlortetracycline was fed26. Similarly, tet(A) and tet(B) gene copies per gram wet feces significantly decreased on Day 4; this was in direct contrast to an observed increase in the proportion of E. coli isolates harboring tet(A) and tet(B) following ceftiofur treatment24.
These results clearly indicate the bias that can be introduced if conclusions regarding microbial populations are based solely on particular culturable organisms. There is a huge unculturable microbial population that might well be expected to behave differently under similar selective pressures. Furthermore, it has been indicated before that the bacterial cells can also lose the entire plasmid or parts of the plasmid carrying resistance determinants, or the resistance genes may not be expressed under culturing stress32,37. Therefore, results from specific culturable organisms may incorrectly estimate the presence and quantity of target resistance genes among the gut flora. The results from metagenome-based assays also have their own limitations; for example, different bacterial species may have multiple and variable amounts of target gene copies per bacterial cell (relative abundance)32,38. The metagenome-based assay gives information about the overall target gene load in community DNA, but information about variations in specific bacterial species carrying target genes is not provided from this community DNA-based assay. At present, most antimicrobial resistance monitoring and surveillance systems across the world test isolates and/or samples for antimicrobial resistance by using culture-based methods. Extrapolating the results obtained from specific culturable organisms (e.g. E. coli, Salmonella, or Campylobacter) to the entire microbial community may not be valid. This can heavily influence the policies formulated by regulatory bodies to control antimicrobial resistance in humans and animals. Therefore, a metagenome-based component should be considered as one part of a more holistic monitoring system to identify when contradictory or paradoxical results are seen, especially to avoid any such biases when making important policy decisions.
A multiple imputation procedure, a simulation-based method, was used to deal with missing data in this study. Several plausible imputed datasets are created in this procedure and results from each are combined in subsequent regression analyses by taking the average over the distribution of these missing data. This technique permits accounting for the uncertainty linked with the missing data39. The imputed datasets for missing values were sampled from a predictive distribution which was based on observed data. Therefore, this procedure is essentially based on a Bayesian approach39. This procedure has been suggested to produce less-biased estimates and more accurate standard errors as compared to other approaches such as single imputation, mean imputation and complete case analysis40. According to one paper41 a relative efficiency of 99% could be achieved by utilizing 20 imputations when 20% of the observations are missing. Another study proposed that multiple imputation techniques did not improve a linear mixed model when missing data represented up to 40% of the sample set42. However, the authors acknowledged that a multiple imputation procedure may be useful if such models included relevant predictor variables, which were not included in their linear mixed models but were included in ours. Multiple imputation procedures should utilize known covariates that are believed to be associated with the missing values43.
The maximum missing observations in the study were approximately 21% for any given gene. Twenty imputations were utilized to get reasonable estimates of all missing observations. We based our imputation model on unique animal identifier (which comprised pen as well as CCFA and CTC treatment status), sampling day and the number of observations missing among the triplicate of each sample (i.e., 1, 2, or all 3 triplicates were missing). The number of missing observations among the triplicates was considered as an important predictor, similar to the previous dairy study that imputed missing quantitative real-time PCR data44. The histograms as well as the supplementary Table 1 resulting from the imputed datasets, demonstrating log10 distributions of gene copies per wet gram feces and based on the number of missing observations per triplicate, reveal that the distribution of missing observations shifts towards the left (including additional imputed observations at lower gene copies) as the number of missing values among triplicates increases. Observations in which all three data points among the triplicates were missing were more likely to be assigned a lower value and also a value below the assay limit of quantification when contrasted with observations with either double or single missing values among the triplicates. Multiple imputation is a useful tool to handle missing data and obtain a valid inference from parametric statistical models similar to the ones used in this study and whose assumptions depend on the integrity of underlying distributions. It is an effective procedure to analyze qPCR data, which often have moderate levels of missing values.
Overall, our study results indicate that chlortetracycline should not be used in an attempt to control ceftiofur-resistance following treatment with a cephalosporin; rather, such an approach would appear to be misguided at best. Chlortetracycline treatment led to significant expansion of both blaCMY-2 and blaCTX-M genes copies per gram wet feces, especially in cattle pens where all animals were previously exposed to ceftiofur. Pen-level ceftiofur-resistance elements were present in lower numbers in pens where just one CCFA treated animal was co-housed with many more non-treated pen-mates. Further studies with mixing at differing treatment ratios should be conducted in order to establish the threshold of the co-mingling effect on levels of antimicrobial resistance. In addition, distinct differences were observed when comparing culture-based results reported elsewhere24,26 and the metagenome-based results obtained herein; this warrants further investigation as current worldwide antimicrobial resistance monitoring and surveillance systems tend to be based solely on culture-based approaches. Such approaches may provide a biased picture of the changes occurring in the microbiome because they depend largely on the organisms being observed. The incorporation of gene quantification and other metagenome-based approaches in surveillance systems may dramatically increase a more holistic understanding in the sphere of antimicrobial resistance.
Methods
Study design
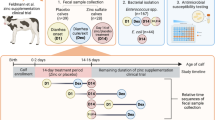
Based on the animal study location and relevant institutional oversight of the co-investigators' roles, the protocols and procedures were approved by the Clinical Research Review Committee at Texas A&M University (CRRC # 09-35) and the Amarillo-Area Cooperative Research, Education and Extension Triangle Animal Care and Use Committee (Protocol No. 2008-07). An Institutional Biosafety Committee approval was in place at Kansas State University (IBC No. 794) where the laboratory analyses took place. Bovine fecal samples were obtained from feeder cattle in a 26-day field trial. The randomized controlled longitudinal study was conducted in accordance with the approved protocols on 176 steers. The steers were housed in two replicates of 88 steers each. For each replicate, steers were assigned to eight pens of 11 steers such that the average pen weights were similar. A total of four treatment groups resulted from the random allocation of two treatment strategies in a complete two-way full factorial manner. The two strategies were: 1) differential pen-level CCFA treatment ratios (Mix) and, 2) subsequent pen-level chlortetracycline treatment (CTC). Pen-level CCFA treatment ratios (Mix) referred to whether long acting CCFA (Excede®, Zoetis Animal Health, Florham Park, NJ, U.S.A.) was administered to either one or else all 11 animals within pens. Pen-level CTC treatment (Aureomycin®, pre-mix complex equivalent to 220.5 g of chlortetracycline/kg, Alpharma, Bridgewater, NJ) referred to whether all cattle in a pen received chlortetracycline treatment top-dressed on feed, subsequent to the CCFA treatment. The study day (Day) was considered as a third factor in the statistical models. This accounted for the effect of day of trial on the quantities of resistance genes detected in feces from the steers.
On Day 0, a single dose of CCFA treatment was injected subcutaneously at the base of the ear in all steers in eight of the 16 pens at a dose rate of 6.6 mg/kg. Subsequently, all animals in four of these eight pens received chlortetracycline treatment at the labeled dose rate of 22 mg/kg body weight administered during three separate 5-day periods (with a one-day break in between). Top-dressing occurred after the morning feed was delivered starting on Day 4. In the remaining eight pens, only one steer among 11 pen-mates within the pen received CCFA treatment. Subsequent chlortetracycline treatment among these eight pens was likewise given to all animals in just four of the pens (Table 1).
Fecal grab samples were collected perrectum every other day from Day 0 to Day 26, after restraining the animals in a squeeze chute; in total, 2,424 samples were collected. For each fecal sample, two 5 ml vials were filled to the 4 ml level with fresh feces. These fecal samples were stored at −70°C for further genotypic analysis.
Community DNA extraction
Total community DNA was extracted from 200 mg feces via the QIAamp DNA Stool Mini Kit™ (Qiagen, Valencia, CA) according to the manufacturer's instructions. The extraction was performed in the QIAcube robot™ (Qiagen, Valencia, CA). The quality and concentration of DNA samples were determined via NanoDrop® ND-1000 UV-Vis Spectrophotometer (NanoDrop Technologies, Wilmington, DE). These DNA samples were used for further genotypic quantification.
Genotypic quantification of target genes in community DNA
Gene copies/gram wet feces of blaCMY-2, blaCTX-M, tet(A), tet(B) and 16S rRNA genes were determined in fecal community DNA using quantitative real time PCR (qPCR). All four resistance gene copy numbers were standardized to the 16S rRNA gene, used as the reference gene in this study. The PCR plates were set up using an automated robot (QIAgility™, Qiagen, Valencia, CA). The fecal community DNA was used directly as a template in qPCR reactions for quantification of the genes using a Stratagene Mx3005P™ real-time qPCR system (Agilent Technologies, La Jolla, CA). The real time qPCR reactions for blaCMY-2 and 16S rRNA genes were adapted from a technique previously described by Alali et al. (2009)32; qPCR reactions for tet(A) and tet(B) genes were adapted from Fan et al. (2007)45; and qPCR reactions for blaCTX-M were adapted from Cottell et al. (2013)25. These blaCTX-M primers targeted all five major CTX-M groups (1, 2, 8, 9 and 25). The variants considered for blaCTX-M primer design were derived from Novais et al. (2010)46. All 2,424 samples were quantified for blaCMY-2 and 16S rRNA genes per gram; however, only samples from Days 0, 4, 12 and 26 from both replicates were chosen for tet(A), tet(B) and blaCTX-M gene quantification. All community DNA qPCR assays were run in triplicate. Primers set details are listed in Table 2.
Controls
An E. coli strain (M1) was used as the positive control for the blaCMY-2 and 16S rRNA gene. It was earlier obtained from the University of Illinois, Chicago47. XL1-Blue E. coli strain (Agilent Technologies, Santa Clara, CA) and E. coli ATCC 47042 were used as the positive controls for tet(A) and tet(B) genes, respectively. The blaCTX-M positive control was an E. coli isolate internal control strain obtained from this study, which was positive on PCR with the specific primers. The PCR product from this strain was sequenced for confirmation of the blaCTX-M-32 gene and has been published elsewhere25. These controls were also used for the generation of template for the standard curve. Two negative controls were run with each 96-well PCR plate. One was the no-template control and the other had DNA-free water as a template.
Standard curve template generation
Template for the standard curve for two genes (blaCMY-2 and 16S rRNA) was generated with a PCR reaction utilizing the broad range primers in a total volume of 22 μl. Each reaction contained 12.5 μl of 2 × Brilliant II SYBR® Green QPCR Master Mix (Agilent Technologies, Santa Clara, CA), 4.75 μl of nuclease free water (Qiagen, Valencia, CA), 1 μl of each primer (135 nM and 185 nM for blaCMY-2 and 16S rRNA genes, respectively), 0.75 μl of reference dye (30 nM) and 2 μl of the DNA extracted from M1 strain (positive control). The thermal profile for amplification was similar to as described by Alali et al. (2009)32. Both tet(A) and tet(B) gene PCR reactions were run in a total volume of 20 μl. Each reaction contained 10 μl of 2 × Brilliant II SYBR® Green QPCR Master Mix, 6.25 μl of nuclease free water, 0.5 μl of each primer (556 nM), 0.75 μl of reference dye (30 nM) and 2 μl of the positive control DNA [XL1-Blue E. coli strain-tet(A) and E. coli ATCC 47042-tet(B)]. The thermal profile for amplification for both tet genes was 95°C for 10 minutes, 45 cycles of 95°C for 15 seconds, 60°C for 30 seconds and 72°C for 28 seconds. The blaCTX-M gene PCR reactions were run in a total volume of 25 μl. Each reaction contained 12.5 μl of 2 × Brilliant SYBR® Green II QPCR Master Mix, 9.5 μl of nuclease free water, 0.625 μl of each primer (260 nM), 0.75 μl of reference dye (30 nM) and 1 μl of the positive control DNA. The thermal profile of these reactions was 94°C for 5 minutes, 40 cycles of 94°C for 1 minute, 57°C for 1 minute and 72°C for 1 minute. Thermal profiles for all gene targets ended with one last cycle to obtain a dissociation curve, which consisted of 95°C for 1 minute, 55°C for 30 seconds and 95°C for 30 seconds.
These PCR products were purified using a DNA purification kit (Promega Corporation, Madison, WI). This purified DNA was used as the template for generating standard curves, after determining its concentration via NanoDrop® ND-1000 UV-Vis Spectrophotometer (NanoDrop Technologies, Wilmington, DE) at wavelengths of 260 and 280 nm. The quantities of target gene copies μl−1 for standard curve generation were determined under consideration of the molecular weight of the amplicons, weight of one mole of base pairs and concentration of PCR product.
Quantification of target genes in unknown samples
Narrow range primers for blaCMY-2 and 16S rRNA genes and the same set of primers for the tet(A), tet(B) and blaCTX-M resistance genes were used to quantify the gene copies in the unknown samples along with the standard curve. Standard curves used the 10 fold serial dilution of the 2 μl purified PCR products described above (with known number of gene copies) to 18 μl of nuclease-free water to a minimum concentration of 100 for blaCMY-2, blaCTX-M and both tetracycline genes and a minimum concentration of 103 for 16S rRNA gene. The lowest five concentrations were used to build each standard curve. All reactions, including standards, were run in triplicate. The reaction mixture composition was same as that used for the standard curve template generation for all respective genes. The thermal profile for amplification for the blaCMY-2 gene was 95°C for 10 minutes, 40 cycles of 95°C for 10 seconds, 52°C for 30 seconds and 79°C for 17 seconds. Likewise, the thermal profile for the16S rRNA gene was 94°C for 10 minutes, 40 cycles of 94°C for 2 minutes, 60°C for 1 minute and 72°C for 17 seconds. Thermal profiles for the tet(A), tet(B) and blaCTX-M resistance genes were the same as for the standard curve template generation. Dissociation curves were run with every plate to examine the specificity of the amplifications.
Statistical analysis
The gene copy number estimates were derived from either 1 or 2 μl of the sample community DNA, depending on the gene being quantified. The gene quantities per microliter were back calculated to estimate gene copies per gram wet feces. Non-standardized blaCMY-2, blaCTX-M, tet(A) and tet(B) gene copies per gram wet feces and the quantities obtained by standardizing them in a ratio with the 16S rRNA gene were used for the outcomes. All standardized and non-standardized quantities were logarithmically transformed to log base 10 for use as a dependent variable in both multiple imputation and mixed (fixed and random effect) linear regression models.
A missing value was recorded in instances where qPCR amplification was below the quantification limit of the assay, or else was estimated below the threshold limit determined by the assay. These observations were considered missing; however, they were initially assigned an arbitrary very low non-zero value so that logarithmic transformation was possible for graphical purposes. Many of these observations were expected to have values more than zero but below the limit of quantification of the assay utilized. Estimates of missing values were imputed using a multiple imputation procedure in STATA® SE Release 12.1 (STATA Corp., College Station, TX). A multiple regression method was utilized for the imputation process. Our imputation model was based on the unique animal id (repeats within animal), sampling day and the number of observations missing among the triplicates from each sample (i.e., 0, 1, 2 or all 3 triplicates were missing). Twenty imputations were performed to reduce the sampling variability from the imputation process41,48. The descriptive statistics (supplementary Table S1) from the first five imputed datasets (out of 20 datasets) were compared to the non-imputed dataset.
Before deciding to utilize the linear regression-based imputation methods, two other imputation techniques (truncated regression and interval censored regression, with upper limit cutoff values of 3 log10 and 7 log10 gene copies per gram, respectively) were explored. The distribution of the estimates of the imputed values for blaCTX-M gene and the comparative descriptive statistics for blaCTX-M and tet(B) from all three imputation techniques are provided as supplementary Figure S1 and supplementary Table S2, respectively. Additionally, results of the main linear regression models, both before and after imputation, were compared to examine and rule out any biases that might introduced during the imputation procedure. Supplementary Figure S2 represents the graphical output of marginal predicted means obtained from the non-imputed dataset, where missing observations were substituted with low non-zero values for each of the 5 genes (blaCMY-2, blaCTX-M, tet(A), tet(B) and 16s rRNA genes). Supplementary Table S3 facilitates direct comparison of the predicted marginal means obtained from regression analyses using non-imputed and imputed data.
To determine the effect of treatments on gene quantities, a multi-level mixed linear model was utilized on the datasets following imputation. The three explanatory variables tested in the model were pen-level CCFA treatment (ratio at which CCFA was administered within a pen), pen-level CTC administration and the sampling day (period). The relationships among the outcome and the explanatory variables were assessed using the mixed linear regression model with three-way full factorial design of the three explanatory variables (fixed effects) in STATA® SE Release 12.1 (STATA Corp., College Station, TX). The models accounted for pen level dependencies as random effects; however, due to convergence issues the animal level dependencies could not be accounted for within the models. Because animals are nested within pens/treatments, this is perhaps not surprising.
References
U. S. Food and Drug Administration. Summary report on antimicrobials sold or distributed for use in food-producing animals. (2011). Available at: http://www.fda.gov/downloads/ForIndustry/UserFees/AnimalDrugUserFeeActADUFA/UCM338170.pdf, accessed February 24, 2014.
World Health Organization (WHO). Critically important antimicrobials for human medicine (3rd revision). (2012). http://www.who.int/foodborne_disease/resistance/cia/en/, accessed February 24, 2014.
Mann, S., Siler, J., Jordan, D. & Warnick, L. Antimicrobial susceptibility of fecal Escherichia coli isolates in dairy cows following systemic treatment with ceftiofur or penicillin. Foodborne Pathog. Dis. 8, 861–867 (2011).
Lamb, H. M., Ormrod, D., Scott, L. J. & Figgitt, D. P. Ceftriaxone: an update of its use in the management of community-acquired and nosocomial infections. Drugs 62, 1041–1089 (2002).
Tragesser, L. A., Wittum, T. E., Funk, J. A., Winokur, P. L. & Rajala-Schultz, P. J. Association between ceftiofur use and isolation of Escherichia coli with reduced susceptibility to ceftriaxone from fecal samples of dairy cows. Am. J. Vet. Res. 67, 1696–1700 (2006).
Fey, P. D. et al. Ceftriaxone-resistant Salmonella infection acquired by a child from cattle. N. Engl. J. Med. 342, 1242–1249 (2000).
Dunne, E. F. et al. Emergence of domestically acquired ceftriaxone-resistant Salmonella infections associated with AmpC beta-lactamase. JAMA 284, 3151–3156 (2000).
Zhao, S. et al. Identification and expression of cephamycinase bla(CMY) genes in Escherichia coli and Salmonella isolates from food animals and ground meat. Antimicrob. Agents Chemother. 45, 3647–3650 (2001).
Centers for Disease Control and Prevention. National Antimicrobial Resistance Monitoring System for Enteric Bacteria (NARMS): Human isolates final report, 2010. Atlanta, Georgia: U.S. Department of Health and Human Services, CDC (2012). Available at: http://www.cdc.gov/narms/pdf/2010-annual-report-narms.pdf, accessed February 20, 2014.
United States Department of Agriculture. National Antimicrobial Resistance Monitoring System (NARMS): 2010 Executive report. Washington D.C. (2011). Available at: http://www.fda.gov/AnimalVeterinary/SafetyHealth/AntimicrobialResistance/NationalAntimicrobialResistanceMonitoringSystem/ucm312356.htm, accessed February 20, 2014.
U. S. Food and Drug Administration. FDA to protect important class of antimicrobial drugs for treating human illness. (2012). Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm285704.htm, accessed February 24, 2014.
Hayes, D. J. & Jensen, H. H. Lessons from the Danish ban on feed-grade antibiotics. Briefing Paper 03-BP 41. Center for Agricultural and Rural Development, Iowa State University, Ames, IA. (2003). Available at: http://www.card.iastate.edu/publications/dbs/pdffiles/03bp41.pdf, accessed February 20, 2014.
Phillips, I. et al. Does the use of antibiotics in food animals pose a risk to human health? A critical review of published data. J. Antimicrob. Chemother. 53, 28–52 (2004).
Volkova, V. V., Lanzas, C., Lu, Z. & Grohn, Y. T. Mathematical model of plasmid-mediated resistance to ceftiofur in commensal enteric Escherichia coli of cattle. PLoS ONE 7, e36738 (2012).
Ayscue, P., Lanzas, C., Ivanek, R. & Grohn, Y. T. Modeling on-farm Escherichia coli O157:H7 population dynamics. Foodborne Pathog. Dis. 6, 461–470 (2009).
Lipsitch, M., Bergstrom, C. T. & Levin, B. R. The epidemiology of antibiotic resistance in hospitals: paradoxes and prescriptions. Proc. Natl. Acad. Sci. U.S.A. 97, 1938–1943 (2000).
Sun, Z. et al. Antibiotic resistance in Pseudomonas Aeruginosa is associated with decreased fitness. Cell. Physiol. Biochem. 31, 347–354 (2013).
Schrag, S. J. & Perrot, V. Reducing antibiotic resistance. Nature 381, 120–121 (1996).
Subbiah, M., Top, E. M., Shah, D. H. & Call, D. R. Selection pressure required for long-term persistence of blaCMY-2-positive IncA/C plasmids. Appl. Environ. Microbiol. 77, 4486–4493 (2011).
Cottell, J. L., Webber, M. A. & Piddock, L. J. Persistence of transferable extended-spectrum-beta-lactamase resistance in the absence of antibiotic pressure. Antimicrob. Agents Chemother. 56, 4703–4706 (2012).
Kaye, K. S., Cosgrove, S., Harris, A., Eliopoulos, G. M. & Carmeli, Y. Risk factors for emergence of resistance to broad-spectrum cephalosporins among Enterobacter spp. Antimicrob. Agents Chemother. 45, 2628–2630 (2001).
Schwaber, M. J., Cosgrove, S. E., Gold, H. S., Kaye, K. S. & Carmeli, Y. Fluoroquinolones protective against cephalosporin resistance in gram-negative nosocomial pathogens. Emerg. Infect. Dis. 10, 94–99 (2004).
Platt, T. et al. Antimicrobial susceptibility of enteric bacteria recovered from feedlot cattle administered chlortetracycline in feed. Am. J. Vet. Res. 69, 988–996 (2008).
Kanwar, N. et al. Effects of ceftiofur and chlortetracycline treatment strategies on antimicrobial susceptibility and on tet(A), tet(B) and blaCMY-2 resistance genes among E. coli isolated from the feces of feedlot cattle. PLoS ONE 8, e80575 (2013).
Cottell, J. L. et al. blaCTX-M-32 on an IncN plasmid in Escherichia coli from beef cattle in the United States. Antimicrob. Agents Chemother. 57, 1096–1097 (2013).
Cottell, J. L. et al. Characterization of E. coli antibiotic resistance mechanisms and plasmids recovered from US beef cattle exposed to antibiotic agents. Paper presented at 3rd ASM conference on antimicrobial resistance in zoonotic bacteria and foodborne pathogens in animals, humans and the environment, Aix-en-Provence, France. Washington: American Society of Microbiology. (2012 June 26–29).
Alexander, T. W. et al. Longitudinal characterization of resistant Escherichia coli in fecal deposits from cattle fed subtherapeutic levels of antimicrobials. Appl. Environ. Microbiol. 75, 7125–7134 (2009).
Boerlin, P. et al. Antimicrobial resistance and virulence genes of Escherichia coli isolates from swine in Ontario. Appl. Environ. Microbiol. 71, 6753–6761 (2005).
Gow, S. P., Waldner, C. L., Harel, J. & Boerlin, P. Associations between antimicrobial resistance genes in fecal generic Escherichia coli isolates from cow-calf herds in western Canada. Appl. Environ. Microbiol. 74, 3658–3666 (2008).
Poppe, C. et al. Characterization of antimicrobial resistance of Salmonella Newport isolated from animals, the environment and animal food products in Canada. Can. J. Vet. Res. 70, 105–114 (2006).
Winokur, P. L., Canton, R., Casellas, J. M. & Legakis, N. Variations in the prevalence of strains expressing an extended-spectrum β-Lactamase phenotype and characterization of isolates from Europe, the Americas and the Western Pacific Region. Clin. Infect. Dis. 32, S94–S103 (2001).
Alali, W. Q., Scott, H. M., Norby, B., Gebreyes, W. & Loneragan, G. H. Quantification of the bla(CMY-2) in feces from beef feedlot cattle administered three different doses of ceftiofur in a longitudinal controlled field trial. Foodborne Pathog. Dis. 6, 917–924 (2009).
Boyer, T. C. & Singer, R. S. Quantitative measurement of bla(CMY-2) in a longitudinal observational study of dairy cattle treated with ceftiofur. Foodborne Pathog. Dis. 9, 1022–1027 (2012).
Lowrance, T. C. et al. Changes in antimicrobial susceptibility in a population of Escherichia coli isolated from feedlot cattle administered ceftiofur crystalline-free acid. Am. J. Vet. Res. 68, 501–507 (2007).
Doyle, M., Loneragan, G. H., Scott, H. M. & Singer, R. S. Antimicrobial resistance: challenges and perspectives. Compr. Rev. Food Sci. F. 12, 234–248 (2013).
Welch, T. J. et al. Multiple antimicrobial resistance in plague: an emerging public health risk. PLoS ONE 2, e309 (2007).
Smith, M. A. & Bidochka, M. J. Bacterial fitness and plasmid loss: the importance of culture conditions and plasmid size. Can. J. Microbiol. 44, 351–355 (1998).
Fogel, G. B., Collins, C. R., Li, J. & Brunk, C. F. Prokaryotic Genome Size and SSU rDNA Copy Number: Estimation of Microbial Relative Abundance from a Mixed Population. Microb. Ecol. 38, 93–113 (1999).
Sterne, J. A. et al. Multiple imputation for missing data in epidemiological and clinical research: potential and pitfalls. Br. Med. J. 339, 157 (2009).
Greenland, S. & Finkle, W. D. A critical look at methods for handling missing covariates in epidemiologic regression analyses. Am. J.Epidemiol. 142, 1255–1264 (1995).
Schafer, J. L. & Graham, J. W. Missing data: Our view of the state of the art. Psychol. Methods 7, 147–177 (2002).
Peters, S. A. et al. Multiple imputation of missing repeated outcome measurements did not add to linear mixed-effects models. J. Clin. Epidemiol. 65, 686–695 (2012).
Burton, A., Billingham, L. J. & Bryan, S. Cost-effectiveness in clinical trials: using multiple imputation to deal with incomplete cost data. Clin. Trials 4, 154–161 (2007).
Boyer, T. C., Hanson, T. & Singer, R. S. Estimation of low quantity genes: a hierarchical model for analyzing censored quantitative real-time PCR data. PLoS ONE 8, e64900 (2013).
Fan, W., Hamilton, T., Webster-Sesay, S., Nikolich, M. P. & Lindler, L. E. Multiplex real-time SYBR Green I PCR assay for detection of tetracycline efflux genes of Gram-negative bacteria. Mol. and Cell. Probes 21, 245–256 (2007).
Novais, A. et al. Evolutionary trajectories of beta-lactamase CTX-M-1 cluster enzymes: predicting antibiotic resistance. PLOS Pathogens 6, e1000735 (2010).
Odeh, R. et al. Broad resistance due to plasmid-mediated AmpC β-Lactamases in clinical isolates of Escherichia coli. Clin. Infect. Dis. 35, 140–145 (2002).
Horton, N. J. & Lipsitz, S. R. Multiple imputation in practice: Comparison of software packages for regression models with missing variables. Am. Stat. 55, 244–254 (2001).
Ng, L. K., Martin, I., Alfa, M. & Mulvey, M. Multiplex PCR for the detection of tetracycline resistant genes. Mol. and Cell. Probes 15, 209–215 (2001).
Steinman, C. R. et al. Domain-directed polymerase chain reaction capable of distinguishing bacterial from host DNA at the single-cell level: characterization of a systematic method to investigate putative bacterial infection in idiopathic disease. Anal. Biochem. 244, 328–339 (1997).
Acknowledgements
We are grateful to Ms. Mal Hoover for her assistance with preparing the figures. The field trial was funded by USDA-NIFA-AFRI (formerly CSREES; grant numbers 2008-35201-30235 and 2008-35201-04682) entitled: “Novel pre-harvest interventions to protect antimicrobials of critical importance in human and veterinary medicine”. The subsequent phenotypic and genotypic analysis of E. coli isolates was funded by USDA-NIFA-NIFSI (grant number 2010-51110-21083) entitled: “Practical interventions to effectively manage antibiotic resistance in beef and dairy cattle systems: a fully integrated approach.”
Author information
Authors and Affiliations
Contributions
H.M.S., B.N., G.H.L. and P.B. conceived and designed the study. N.K., H.M.S., B.N., G.H.L., J.V., P.B., J.L.C. and G.C. performed the experiments. N.K., H.M.S., B.N., G.H.L., P.B., M.M.C. and J.B. analyzed and interpreted data. N.K. and H.M.S. drafted the manuscript. All authors revised manuscript for critically important intellectual content and approved the final version to be published.
Ethics declarations
Competing interests
Dr. Guy H. Loneragan has provided scientific consulting services to Zoetis Inc. (manufacturer of the CCFA product). This does not alter the authors' adherence to all the journal policies on sharing data and materials.
Electronic supplementary material
Supplementary Information
supplementary material
Supplementary Information
Supplementary Dataset 1
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Kanwar, N., Scott, H., Norby, B. et al. Impact of treatment strategies on cephalosporin and tetracycline resistance gene quantities in the bovine fecal metagenome. Sci Rep 4, 5100 (2014). https://doi.org/10.1038/srep05100
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05100
This article is cited by
-
Age influences the temporal dynamics of microbiome and antimicrobial resistance genes among fecal bacteria in a cohort of production pigs
Animal Microbiome (2023)
-
Ruminal resistome of dairy cattle is individualized and the resistotypes are associated with milking traits
Animal Microbiome (2021)
-
In vitro synergy of sertraline and tetracycline cannot be reproduced in pigs orally challenged with a tetracycline resistant Escherichia coli
BMC Microbiology (2019)
-
Antimicrobial use and antimicrobial resistance in food animals
Environmental Science and Pollution Research (2018)
-
Antimicrobial usage and resistance in beef production
Journal of Animal Science and Biotechnology (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
