





Abstract
Pseudomonas aeruginosa is a metabolically versatile bacterium that is found in a wide range of biotic and abiotic habitats. It is a major human opportunistic pathogen causing numerous acute and chronic infections. The critical traits contributing to the pathogenic potential of P. aeruginosa are the production of a myriad of virulence factors, formation of biofilms and antibiotic resistance. Expression of these traits is under stringent regulation, and it responds to largely unidentified environmental signals. This review is focused on providing a global picture of virulence gene regulation in P. aeruginosa. In addition to key regulatory pathways that control the transition from acute to chronic infection phenotypes, some regulators have been identified that modulate multiple virulence mechanisms. Despite of a propensity for chaotic behaviour, no chaotic motifs were readily observed in the P. aeruginosa virulence regulatory network. Having a ‘birds-eye’ view of the regulatory cascades provides the forum opportunities to pose questions, formulate hypotheses and evaluate theories in elucidating P. aeruginosa pathogenesis. Understanding the mechanisms involved in making P. aeruginosa a successful pathogen is essential in helping devise control strategies.
INTRODUCTION
Pseudomonas aeruginosa is a Gram-negative bacterium that has the ability to thrive in most natural and man-made environments. It is found in diverse habitats, including soil, water, plants and animals, and can infect multiple hosts (1,2). Pseudomonas aeruginosa causes a wide variety of acute (short duration, typically severe) and chronic (persisting for a long time, often refractory to treatment, severity varying with pathogen) human infections, including in patients with severe burn wounds, urinary tract infections, AIDS, lung cancer, chronic obstructive pulmonary disease, bronchiectasis and cystic fibrosis (CF) (3–6).
Metabolic versatility, intrinsic and acquired antibiotic resistance, biofilm formation and production of multiple virulence (disease-causing) factors make P. aeruginosa a formidable pathogen. The virulence machinery of P. aeruginosa comprises both cell-associated determinants (such as lipopolysaccharides, pili, flagella) and numerous secreted factors (such as elastases, proteases, exotoxins, pyocyanin, extracellular polysaccharides). One of the mechanisms by which P. aeruginosa senses external signals is using sensor proteins that, through phosphotransfer or phosphorelay, activate specific transcriptional regulators. These sensor–regulator protein pairs are called two-component systems (TCS). The P. aeruginosa PAO1 genome encodes ∼127 TCS members, compared with 60 in Escherichia coli (7) and 70 in Bacillus subtilis (8), reflecting the adaptability of P. aeruginosa. TCS and their modifications also feed into major regulatory pathways and play a critical role in allowing cells to modulate gene expression in response to environmental conditions (9,10). Many of the secreted virulence factors and phenotypes, such as biofilm formation, are under the control of a cell density recognition mechanism called quorum sensing (QS) that aids in the coordinated expression of genes (11,12). QS is a key to virulence gene expression in many bacteria and serves as an attractive target for antibacterial chemotherapy (13).
In humans, acute P. aeruginosa infections in specific sites, such as the CF lung, eventually lead to chronic inections. This is caused by adaptive modifications in the infecting clonal type, resulting in diverse morphotypes (14). Acute virulence factors include the Type 2 and Type 3 secretion systems, flagella, type IV pili and QS-regulated virulence factors (proteases, elastase, pyocyanin) (15). On establishing a chronic infection, P. aeruginosa overproduces extracellular polysaccharides, forms biofilms and small colony variants and upregulates the Type 6 secretion system (15–18). Antibiotic resistance plays a major role in both types of infection, although the cells display higher levels of resistance in chronic infections (18,19). The transition to a chronic infection phase is the result of numerous changes in cellular physiology in response to external stimuli (20). The changes include downregulation of acute virulence genes with a concomitant upregulation of chronic infection phenotypes and antibiotic resistance, facilitating recalcitrant infections (15,20). Host invasion, establishment of acute infection and the subsequent transition to the chronic phase involves tightly regulated expression of many genes associated with metabolism, virulence and antibiotic resistance. Several key players in these transition processes have been identified and include transcriptional and post-transcriptional regulators (21–23). Many of these will be discussed in subsequent sections of this review.
Gene regulation in P. aeruginosa is a complex process involving numerous transcriptional regulators, regulatory RNAs (rgRNA) and σ factors. The P. aeruginosa genome is >6 Mb (24), approaching that of lower eukaryotes. The genome is plastic and has acquired genes and undergone extensive rearrangements to adapt to specific niches (25). The large genome of P. aeruginosa supports a multitude of regulatory networks, with ∼8% of the total genome dedicated to the regulatory proteins (26). Pseudomonas aeruginosa PAO1 encodes 434 transcriptional regulators, 24 σ factors and 34 small RNAs, many of which remain to be characterized (24,27–29). Moreover, predicted regulatory networks indicate that there is an extensive crosstalk between the different transcriptional regulators (27,30). These networks, however, are based in part on in silico analyses, and their validity needs to be established. This review makes an effort to consolidate the empirically proven major virulence regulatory networks in P. aeruginosa with the hope of providing a framework for future studies to better understand pathogenic processes in P. aeruginosa and in related bacteria.
MAJOR VIRULENCE REGULATORY SYSTEMS IN P. AERUGINOSA
The experimentally established virulence regulatory network in P. aeruginosa is depicted in Figure 1. Our group and others have previously performed in silico analyses of the P. aeruginosa transcriptional regulatory network (27,30,31). Comparing those networks with the network depicted in Figure 1 clearly demonstrates the gap in knowledge between predicted networks and established ones. An important contributing factor to this discrepancy is the fact that the functions of the majority of the genes in the PAO1 genome remain unknown. Deep sequencing, transcriptome metaanalysis (32,33) and complementary studies will aid in assigning functions to the hypothetical genes and undoubtedly narrow this knowledge gap.
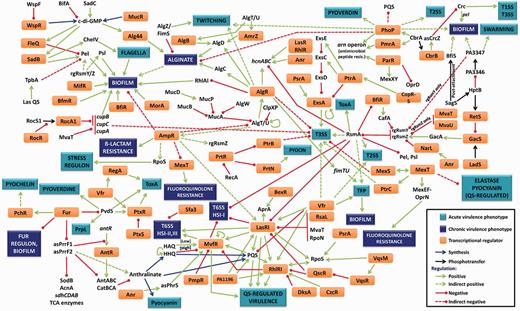
The P. aeruginosa virulence regulatory network. The pathogenic potential of P. aeruginosa is dictated by multiple virulence systems that are regulated transcriptionally, post-transcriptionally and post-translationally. The central mechanism for P. aeruginosa virulence regulation is QS, which controls expression of many virulence factors in a population density-dependent manner. Key activators of this system are LasR, RhlR, MvfR, VqsR, the cAMP receptor protein Vfr and the stationary phase σ factor RpoS. Las system repressors include RsaL, the H-NS protein MvaT, the σ factor RpoN and the sRNA-binding protein RsmA, whereas others like QscR repress both the Las and Rhl systems. Other regulators such as AmpR affect QS genes by an unknown mechanism. QS plays a role in regulating critical pathogenic mechanisms, including biofilm formation, secretion systems, production of numerous virulence factors, efflux pumps, antibiotic resistance and motility. Acute P. aeruginosa infections can lead to chronic infections in response to largely unidentified signals. A key regulatory pathway that controls this lifestyle switch is the RetS–LadS–GacSA–RsmA pathway. RetS and LadS are hybrid sensor proteins that, in response to external signals, either activate or repress the GacSA TCS. The GacA regulator then activates transcription of two rgRNAs, rgRsmZ and rgRsmY that sequester and inhibit activity of the sRNA-binding protein, RsmA. RsmA is a key activator/repressor that post-transcriptionally regulates numerous acute and chronic infection phenotypes, including multiple QS-regulated virulence factors, biofilm formation, Type 2, Type 3 and Type 6 secretion systems and motility. Another major phenotypic change associated with the switch from acute to chronic phases of infection is the formation of biofilm. This is associated with extensive changes in transcription. Three key TCS involved in activating biofilm formation are BfiSR, MifR and BfmSR. Cyclic-di-GMP is another major player influencing this process, whose levels are controlled by diguanylate cyclases and phosphodiesterases. QS and the cup genes enhance biofilms, whereas regulators like AmpR repress it. An important component of P. aeruginosa biofilms are extracellular polysaccharides, such as alginate, Pel and Psl. Alginate production is under the control of the master regulator ECF AlgT/U, whose activity is regulated transcriptionally by AmpR, post-translationally by MucA and MucB and by regulated intermembrane proteolysis involving MucP, AlgW, ClpXP and others. AlgT/U activates the alginate biosynthetic operon through AlgR, AlgB and AmrZ. In addition, biofilm formation is also affected by iron concentration, a process governed by the master repressor of iron uptake, Fur. Fur controls uptake of iron by regulating the σ factor PvdS, thereby modulating sidephore levels. Fur also modulates transcription of two key regulatory RNAs, asPrrF1 and asPrrF2. These two sRNAs are involved not only in regulating iron-uptake-related genes but also enzymes of the trichloroacetic acid cycle and genes involved in anthranilate synthesis. Anthralinate, a precursor for synthesis of PQS, is a key regulatory molecule of the PQS signalling system in P. aeruginosa, which is involved in expression of QS-regulated virulence factors. Details on the individual interactions and the appropriate references can be found in the text. Some of the interactions labelled as indirect are regulated by unknown mechanisms and warrant further investigation. In the figure, some regulators and phenotypes have been mentioned more than once.
Cis regulatory elements (CREs) form a critical part of transcription. CREs are non-coding DNA sequences present in or near a gene, and they often contain binding sites for transcription factors and/or other regulators of transcription (34). The two major CREs are promoters and enhancers (35,36). The promoters contain the binding sites for transcription factors and other regulatory molecules, such as σ factors and regulatory RNAs (37–39). Enhancers, once thought to be part of only eukaryotes, are found widely in prokaryotes also, and they function in conjunction with the σ54-RNA polymerase (40–42). The known P. aeruginosa transcription factor binding sites are listed in Table 1. This section will focus on the transcriptional and post-transcriptional regulation of critical pathways that determine P. aeruginosa pathogenesis.
Cis regulatory elements in P. aeruginosa transcriptional regulation
| Transcription factor | Cis regulatory element | Major virulence phenotype regulated | Reference |
|---|---|---|---|
| AlgR | ACCGTTCGTC | Alginate production, biofilm formation, T3SS | (43) |
| AlgZ | GGCCATTACCAGCC | Alginate production | (44) |
| Anr | TTGATN4ATCAA | Anaerobic regulator of QS | (45) |
| ArgR | TGTCGCN8AA | Carbon and nitrogen catabolism | (46) |
| ExsA | TNAAAANA | T3SS | (47) |
| FleQ | Box 1: CGCCTAAAAATTGACAGTT | Motility, biofilm formation | (48) |
| Box 2: CATTAGATTGACGTTAATC | |||
| Fur | GATAATGATAATCATTATC | Iron uptake | (49) |
| LasR (las box) | NHCTRNSNNDHNDKNNAGNB | QS | (50) |
| MexT | ATCAN5GTCGATN4ACYAT | Antibiotic resistance, T3SS, QS | (51) |
| MvfR | TTCGGACTCCGAA | QS | (52) |
| PsrA | G/CAAACN2-4GTTTG/C | Stress regulon (RpoS), T3SS | (53) |
| RcsBa | TTA-GAAACGTCCTAAA | Fimbriae | (54) |
| RhlR (lux box) | CCTGTGAAT/ATCC/TGGT/CAGTT | QS | (55) |
| Vfr | AATTGACTAATCGTTCACATTTG | QS | (56) |
| VqsR | TCGCCN8GGCGA | QS | (57) |
| Transcription factor | Cis regulatory element | Major virulence phenotype regulated | Reference |
|---|---|---|---|
| AlgR | ACCGTTCGTC | Alginate production, biofilm formation, T3SS | (43) |
| AlgZ | GGCCATTACCAGCC | Alginate production | (44) |
| Anr | TTGATN4ATCAA | Anaerobic regulator of QS | (45) |
| ArgR | TGTCGCN8AA | Carbon and nitrogen catabolism | (46) |
| ExsA | TNAAAANA | T3SS | (47) |
| FleQ | Box 1: CGCCTAAAAATTGACAGTT | Motility, biofilm formation | (48) |
| Box 2: CATTAGATTGACGTTAATC | |||
| Fur | GATAATGATAATCATTATC | Iron uptake | (49) |
| LasR (las box) | NHCTRNSNNDHNDKNNAGNB | QS | (50) |
| MexT | ATCAN5GTCGATN4ACYAT | Antibiotic resistance, T3SS, QS | (51) |
| MvfR | TTCGGACTCCGAA | QS | (52) |
| PsrA | G/CAAACN2-4GTTTG/C | Stress regulon (RpoS), T3SS | (53) |
| RcsBa | TTA-GAAACGTCCTAAA | Fimbriae | (54) |
| RhlR (lux box) | CCTGTGAAT/ATCC/TGGT/CAGTT | QS | (55) |
| Vfr | AATTGACTAATCGTTCACATTTG | QS | (56) |
| VqsR | TCGCCN8GGCGA | QS | (57) |
H = C/T/A; R = A/G; S = C/G; D = G/A/T; K = G/T; B = C/G/T; N = A/C/G/T.
aRcsB is found only in P. aeruginosa PA14, not in P. aeruginosa PAO1.
Cis regulatory elements in P. aeruginosa transcriptional regulation
| Transcription factor | Cis regulatory element | Major virulence phenotype regulated | Reference |
|---|---|---|---|
| AlgR | ACCGTTCGTC | Alginate production, biofilm formation, T3SS | (43) |
| AlgZ | GGCCATTACCAGCC | Alginate production | (44) |
| Anr | TTGATN4ATCAA | Anaerobic regulator of QS | (45) |
| ArgR | TGTCGCN8AA | Carbon and nitrogen catabolism | (46) |
| ExsA | TNAAAANA | T3SS | (47) |
| FleQ | Box 1: CGCCTAAAAATTGACAGTT | Motility, biofilm formation | (48) |
| Box 2: CATTAGATTGACGTTAATC | |||
| Fur | GATAATGATAATCATTATC | Iron uptake | (49) |
| LasR (las box) | NHCTRNSNNDHNDKNNAGNB | QS | (50) |
| MexT | ATCAN5GTCGATN4ACYAT | Antibiotic resistance, T3SS, QS | (51) |
| MvfR | TTCGGACTCCGAA | QS | (52) |
| PsrA | G/CAAACN2-4GTTTG/C | Stress regulon (RpoS), T3SS | (53) |
| RcsBa | TTA-GAAACGTCCTAAA | Fimbriae | (54) |
| RhlR (lux box) | CCTGTGAAT/ATCC/TGGT/CAGTT | QS | (55) |
| Vfr | AATTGACTAATCGTTCACATTTG | QS | (56) |
| VqsR | TCGCCN8GGCGA | QS | (57) |
| Transcription factor | Cis regulatory element | Major virulence phenotype regulated | Reference |
|---|---|---|---|
| AlgR | ACCGTTCGTC | Alginate production, biofilm formation, T3SS | (43) |
| AlgZ | GGCCATTACCAGCC | Alginate production | (44) |
| Anr | TTGATN4ATCAA | Anaerobic regulator of QS | (45) |
| ArgR | TGTCGCN8AA | Carbon and nitrogen catabolism | (46) |
| ExsA | TNAAAANA | T3SS | (47) |
| FleQ | Box 1: CGCCTAAAAATTGACAGTT | Motility, biofilm formation | (48) |
| Box 2: CATTAGATTGACGTTAATC | |||
| Fur | GATAATGATAATCATTATC | Iron uptake | (49) |
| LasR (las box) | NHCTRNSNNDHNDKNNAGNB | QS | (50) |
| MexT | ATCAN5GTCGATN4ACYAT | Antibiotic resistance, T3SS, QS | (51) |
| MvfR | TTCGGACTCCGAA | QS | (52) |
| PsrA | G/CAAACN2-4GTTTG/C | Stress regulon (RpoS), T3SS | (53) |
| RcsBa | TTA-GAAACGTCCTAAA | Fimbriae | (54) |
| RhlR (lux box) | CCTGTGAAT/ATCC/TGGT/CAGTT | QS | (55) |
| Vfr | AATTGACTAATCGTTCACATTTG | QS | (56) |
| VqsR | TCGCCN8GGCGA | QS | (57) |
H = C/T/A; R = A/G; S = C/G; D = G/A/T; K = G/T; B = C/G/T; N = A/C/G/T.
aRcsB is found only in P. aeruginosa PA14, not in P. aeruginosa PAO1.
QS
QS is a signalling mechanism that bacteria use to regulate gene expression in a population density-dependent manner, and it was first demonstrated in Vibrio fischeri (58). In QS, the bacteria produce and secrete small molecules called autoinducers or quoromones. When these molecules reach a concentration threshold, they diffuse back into the cell to elicit a coordinated response promoting group survival (59). Pseudomonas aeruginosa uses QS to regulate production of various virulence determinants, such as extracellular proteases, iron chelators, efflux pump expression, biofilm formation, motility and the response to host immune signals (60). This is achieved using two types of autoinducers, N-acyl-homoserine lactones (AHLs) and 2-alkyl-4 quinolones (AQs) (61).
AHL-mediated QS
Pseudomonas aeruginosa has two canonical AHL QS signalling pathways, the las and rhl systems. Together, these pathways affect expression of ∼10% of the P. aeruginosa transcriptome (62). The lasI (PA1432) and rhlI (PA3476) genes encode the N-3-oxododecanoylhomoserine lactone (3-oxo-C12-AHL) synthetase (63,64) and N-butyrylhomoserine lactone (C4-AHL) synthetase, respectively (65–68). The resulting AHLs then bind and activate their cognate LuxR family regulators, LasR (PA1430) (64) or RhlR (PA3477) (67). LasR and RhlR multimerize in the presence of their cognate AHL (69,70). In in vitro studies, LasR–DNA interaction is cooperative and non-cooperative in the presence or absence of a dyad symmetry in the binding sites, respectively (71). Rhl-regulated promoters have binding sites with a dyad symmetry (72).
AQ-mediated QS
Pseudomonas aeruginosa synthesizes two AQ QS signals, 2-heptyl-3-hydroxy-4-quinolone (PQS) and its precursor, 2-heptyl-4-quinolone (HHQ) (73). Both PQS and HHQ enhance in vitro binding of the LysR-type transcription regulator, MvfR (also known as PqsR, PA1003), to the promoter of the pqsABCDE operon (PA0996–PA1000), suggesting that they function as MvfR effectors (74). Microarray analysis identified 141 genes differentially expressed in an mvfR mutant strain, including lasR, algT/U (PA0762), rsmA (PA0905) and rsaL (PA1431) (75). PQS also acts independently of MvfR to induce expression of the Fur regulon through its ability to bind iron (73,76) and membrane vesicle formation by inducing membrane curvature (77,78). PmpR (PA0964), a YebC member, negatively regulates MvfR (Figure 1) (79).
QS regulation
The las, rhl and PQS/HHQ/MvfR systems exhibit positive feed-forward autoregulation (52,80). In addition, the P. aeruginosa AHL systems function in a hierarchical manner, as the 3-oxo-C12-AHL-LasR complex positively regulates rhlI, rhlR and mvfR expression as well as lasI (81–83). Exceptions to this have been noted. RhlR has been shown to regulate LasR-dependent genes in strains lacking lasR (84), and timing of lasI, lasR, rhlI and rhlR expression can vary drastically depending on growth conditions (85).
Many global regulators have been shown to modulate QS-dependent genes. RpoS (PA3622), the stationary phase σ factor affects ∼40% of the QS regulon (72,86). RpoS binding sites have been identified in several of the QS-dependent promoters. RpoS also affects lasR and rhlR expression, and LasR binding sites have been identified in promoters of other transcriptional regulators in the QS regulon, including PA2588, PA4778, pvdS (PA2426), vqsR (PA2591) and rsaL (87). Chromatin immunoprecipitation studies have shown occupancy by histone-like silencers MvaT (PA4315) and MvaU (PA2667) on lasI, lasR, mvfR, rpoS and rsaL (88). RsaL plays an important role in las signalling homeostasis, by binding to the lasI promoter and preventing LasR-mediated activation (89). In addition to affecting gene expression through las regulation, microarray analyses indicate that RsaL affects expression of 130 genes, including direct regulation of pyocyanin and hydrogen cyanide genes (89). RsaL also seems to be important in regulating the transition from planktonic to a sessile state, as rsaL mutants exhibit increased swarming motility and fail to form biofilms (90). RsaL expression is under the control of LysR-type regulator OxyR (PA5344) (91). The lasI promoter region has also been shown to be bound by CzcR (PA2523), which is required for expression of rhlI and rhlR in addition to lasI (Figure 1) (92). CzcR is part of the CzcRS TCS, which is shown to be involved in carbapenem and heavy metal resistance (93).
VqsR (PA2591), which is induced by H2O2 or human serum (94) and is under LasR regulation (95), regulates QS through inhibition of the LuxR-type regulator, QscR (PA1898, Figure 1) (96). Although QscR binds to 3-oxo-C12-AHL, its specificity is not as stringent as LasR (97). The QscR regulon partially overlaps that ascribed to the las and rhl systems, but also has unique targets (98). In the absence of AHL, QscR can multimerize and form heterodimers with LasR and RhlR (99). QscR also plays a role in LasI homeostasis, as mutations in qscR result in premature lasI expression (100). An AraC family member VqsM (PA2227) regulates VqsR in addition to numerous genes involved in QS, including RsaL, PprB (PA4296), MvfR, RpoS as well as AlgT/U and MexR (PA0424) (101).
Additionally, pqsH (PA2587), which encodes the enzyme responsible for oxidation of HHQ to form PQS, is positively regulated by the las system (102,103) and is negatively regulated by the rhl system (52). PQS is derived from anthranilate, which is synthesized by the kynurenine pathway (104). Kynurenine pathway anthranilate is also required for N-decanoyl-homoserine lactone (C10-AHL) dependent signalling, which is independent of las, rhl and qscR (104). The receptor for this signalling is yet to be identified (105). Besides potential heterodimerization with QscR, additional post-transcriptional regulation of QS has been described. In one such mechanism, QteE (PA2593) destabilizes LasR and RhlR, and in the absence of qteE, the quorum threshold-requirement for activation of QS-dependent genes is lost (106). RsmA negatively regulates rhl and las signalling, resulting in reduced AHL levels (107). Moreover, it has been shown the RNA chaperone Hfq (PA4944) positively regulates rhlI translation through rsmY and RsmA (108).
Recently, our laboratory has established a role for the ß-lactamase regulator AmpR (PA4109) in activating QS-regulated genes (23). The production of QS-regulated secreted virulence factors, such as LasA (PA1871) and LasB (PA3724) proteases, and pyocyanin production is significantly impaired in AmpR-deficient strains. Further, loss of ampR reduced virulence in the Caenorhabditis elegans toxicity assay (23,109). In addition, AmpR regulates non–ß-lactam resistance by repressing activity of the MexEF–OprN (PA2493–PA2495) efflux pump, the alginate master regulator AlgT/U (110) and biofilm formation (23), suggesting that it plays a role in maintaining the acute mode of infection.
TCS
TCS are sophisticated signalling mechanisms marked by a highly modular design that have been adapted and integrated into a wide variety of cellular signalling circuits. The archetypical TCS is composed of a membrane integrated sensory histidine kinase (HK) and a cytoplasmic response regulator (RR) (111). The HK contains a periplasmic N-terminal domain that detects specific stimuli (sensing domain) and a C-terminal cytoplasmic transmitter domain that comprises a dimerization domain, a conserved histidine and an adenosine triphosphate catalytic domain (112). HKs can have two or more transmembrane domains with little or no periplasmic domain, whereas others are completely cytoplasmic. The cognate RR contains a conserved receiver domain and a variable output domain (113). On receiving a signal, two HK monomers dimerize and cross-phosphorylate at the conserved histidine residue, and the phosphate is subsequently transferred to an aspartate residue in the receiver domain of the cognate RR (114). The phosphotransfer is catalysed by the receiver domain, and it results in a conformational change that activates the output domain, which often binds DNA and modulates gene expression or enzymatic activity (9,113,115). Variations to this model occur in phosphorelays, where a sensor kinase first transfers the phosphoryl group to an RR that has no output domain. This P∼RR then transfers the phosphoryl group to a histidine-containing phosphotransfer protein, and this in turn serves as a phosphate donor to a terminal RR, which has an output domain mediating a cellular response (10). In other cases, the sensor kinase and the RR lacking an output domain are fused into one protein (hybrid sensor kinase) (116). Other variations include the TCS connectors, a group of proteins that modulate the phosphorylation state and activity of sensor HK and RR and establish regulatory links between otherwise independent signal transduction pathways (117).
Pseudomonas aeruginosa, equipped with 55 HKs, 89 RRs and 14 HK–RR hybrids, possesses one of the largest pool of TCS proteins identified in any microorganism analysed thus far (24). This provides the bacterium with a sophisticated capability to regulate diverse metabolic adaptations, virulence and antibiotic resistance processes that are hallmark of P. aeruginosa infections. One of the critical TCSs is GacSA (GacS-PA0928, GacA-PA2586), which is central to expression of virulence factors, secondary metabolites, biofilm formation and QS (107,118) and is the switch between acute and chronic infections (1,119). GacS is a hybrid sensor HK that contains an HK domain, an RR domain and a histidine phosphotransfer (Hpt) domain (21,120). GacS phosphorylation is under the control of two hybrid sensor kinases, RetS (PA4856) (21) and LadS (PA3974) (22) (Figure 1). RetS can directly interact with GacS and prevent GacS phosphorylation (22,121), whereas LadS phosphorylates GacS (22). Phosphorylated GacA positively regulates the transcription of two small regulatory RNAs, rgRsmZ (PA3621.1) and rgRsmY (PA0527.1), which block the negative regulator RNA-binding protein RsmA (PA0905). RsmA positively regulates genes of the Type 3 secretion system, type IV pili formation and iron homeostasis while repressing QS, Type 6 secretion and potentially other transcription factors (122–124). The GacSA TCS is also involved in antibiotic resistance to three different families of antibiotics, tobramycin, ciprofloxacin and tetracycline (125), apparently through RsmA/rgRsmZ.
In P. aeruginosa, PhoPQ (PA1179–PA1180) together with PmrAB (PA4776–PA4777) are two TCSs that respond to limiting concentrations of cations, and regulate resistance to polymyxin B and cationic antimicrobial peptides through the regulation of the arnBCADTEF-pmrE (PA3552–PA3559) LPS modification operon (126,127). PhoQ is involved in swarming and twitching motility as well as in biofilm formation and is required for virulence without affecting the T3SS or QS systems (Figure 1) (128). The HK PhoQ activates the RR PmrA independently of PmrB, suggesting an interaction between these TCSs (129). In addition, increased resistance to antibiotics, including polymyxin B, aminoglycosides and quinolones in phoQ mutants suggests crosstalk between PhoPQ and other TCSs (130,131).
Formation of biofilms
Biofilms are surface-associated multicellular bacterial communities encapsulated in a self-produced extracellular matrix composed of polysaccharides, proteins and nucleic acids that mediate cell-to-cell and cell-to-surface interactions (132). Pseudomonas aeruginosa biofilms, typically associated with poor patient prognosis, signify the switch from an acute to a chronic infection. Biofilms can be formed on abiotic (environment) or biotic (wounds, surgical implants, CF lung) surfaces (133). Biofilm formation and maintenance is tightly regulated in response to environmental cues, conferring enhanced resistance against antimicrobial agents and immune defence mechanisms on the biofilm bacteria (12). Formation of biofilms is a multi-stage process that is initiated by the surface attachment of planktonic bacteria to form a monolayer, clonal growth/aggregation leading to the formation of microcolonies, maturation to form mushroom-shaped structures and dispersal (134–136). As can be imagined, this complex transition in the bacterial lifestyle is accompanied by drastic changes in gene regulation.
Surface attachment by P. aeruginosa to form microcolonies has been attributed to type IV pili, flagella, free DNA, alginate and Pel and Psl polysaccharides, although pili, alginate and flagella mutants also form biofilms (136,137). Attachment is a reversible process, and the commitment to form biofilms is partly under positive SadB (PA5346) regulation (138). SadB upregulates both Pel polysaccharide production and the chemotaxis-like cluster CheIV (PA0408–PA0417), which is thought to regulate flagellar motion by an unknown mechanism (139).
The Cup fimbriae, encoded by three distinct gene clusters cupA (PA2128–PA2133), cupB (PA4081–PA4086) and cupC (PA0992, PA0993, PA0994) in P. aeruginosa PAO1, have been demonstrated to play a role in different stages of biofilm formation on biotic and abiotic surfaces (140). Regulation of the cup genes is complex, involving a phase variation-dependent repression of cupA expression by an H-NS member MvaT (141,142). MvaT also regulates the cupB and cupC loci to a lesser extent (141). The cupB and cupC clusters are under the primary regulation of the RocS1–RocR–RocA1 (PA3946–PA3948) three-component system (143). This system is similar to the Bordetella pertussis BvgASR system (144) and consists of the hybrid sensor kinase RocS1, the response regulator RocA1 and the RocA1-repressor RocR (143,145). RocR has been hypothesized to bind to c-di-GMP through its EAL (diguanylate phosphodiestrerase) domain and prevents phosphotransfer from RocS1 to RocA1, thus preventing RocA1 activation (143,145). Pseudomonas aeruginosa PA14 has a fourth cup cluster (cupD) on the pathogenicity island PAPI-I, which is controlled positively by the response regulator RcsB (PA4080) and negatively by the EAL-domain containing response regulator PvrR (146). In addition, diguanylate cyclases and phosphodiesterases of the wsp gene cluster (PA3702–PA3708) (147,148) MorA (PA4601) (149) and TpbA–TpbB (PA3885, PA1120) (150) modulate intracellular levels of c-di-GMP to exert a regulatory effect on the cup gene clusters (Figure 1).
The main components of the extracellular polymeric substance matrix of biofilms are Pel and Psl polysaccharides, alginate and free DNA (12,136). Both pel and psl gene loci are post-transcriptionally regulated by the RetS–LadS (PA4856 and PA3974, respectively) system through rsmY and rzmZ (21,22,121) and by c-di-GMP levels, either directly (147) or by binding the transcriptional regulator FleQ (PA1097) (151). The pel operon is also repressed by the las QS system through the tyrosine phosphatase TpbA (PA3885) (150). A membrane-bound sensor, PpyR (PA2663) enhances biofilm formation through the psl operon and virulence through modulating QS (152). Although alginate is a major component of biofilms and affects biofilm structure, it is not essential for biofilm formation (136). Alginate regulation is discussed in a separate section (see later in the text). QS regulates cell lysis in biofilms (153–155), thereby controlling the release of extracellular DNA, a major component of the biofilm matrix (156,157). The QS system also regulates rhamnolipid production (67) that promotes motility and, hence, formation of the cap in the mushroom structure of mature biofilms (158), and maintenance of biofilm channels (159). BfiRS (PA4196–PA4197), BfmRS (PA4101–PA4102) and MifR (PA5511) are TCSs shown to regulate biofilm development and maturation by sequential phosphorylation (160). They activate biofilm formation at different transition stages, reversible to irreversible attachment (BfiRS), irreversible attachment to maturation stage-1 (BfmRS) and maturation stage-1 to mushroom structure formation (MifR) (Figure 1) (160). The BfiRS system may function in conjunction with the GacSA TCS and feed into the Rsm loop of regulation to control biofilm formation (160). SagS (PA2824), the cognate sensor of HptB (PA3345), modulates biofilm development (by controlling BifS phosphorylation) and other virulence phenotypes (by modulating rgsRmZ levels) depending on whether the cells are in the planktonic or biofilm phase (161).
Analyses of clinical isolates reveal a positive correlation between expression of lasR, rhlR and acute virulence factors (162,163), suggesting that QS is required for virulence in vivo. QS is also important when P. aeruginosa grows as biofilms in the CF lung (164). In vivo studies show that lasI and rhlI mutants produce milder chronic lung infections compared with their wild-type counterparts (165) and form more susceptible biofilms (166). However, in some in vitro studies, there was no apparent difference in the biofilms formed by the QS mutants and the wild-type strains (167,168). This discrepancy in the requirement of QS for biofilm formation and establishment of a successful chronic infection is probably not surprising, as QS regulates many different functions. Further, it has been demonstrated that the CF environment selects for strains with lasR mutations, although the rhl system is intact (169). Although lasR is higher up in the QS hierarchy, studies have shown that secondary mutations can re-establish rhl expression in las mutants (170). This suggests that in CF biofilms, the rhl system is more important, and lasR inactivation serves to downregulate the acute virulence factors (171).
A major cause of antibiotic resistance in biofilms has recently been attributed to the phenomenon of persistence. Persister cells are small subpopulations of antibiotic-sensitive cells that have acquired transient antibiotic tolerance (172). When the antibiotic levels drop, the persisters grow into a population of sensitive cells, again with a small sub-population of persisters (173,174). Many genes involved in the formation of persisters have been identified in P. aeruginosa PA14, including two transcriptional regulators AlgR (PA5261) and PilH (PA0409) (175). However, this topic is outside the scope of this review but has been extensively reviewed elsewhere (176,177).
Alginate production
In the lungs, especially in patients with CF, P. aeruginosa can convert from a non-mucoid to an alginate-overproducing mucoid phenotype signalling chronic infection (178). Chronic P. aeruginosa infection seems to be localized to foci within the anaerobic mucus environment in the lung’s respiratory zone (179–182). These foci lead to tissue damage decreasing lung function, and the appearance of the mucoid phenotype correlates with poor patient prognosis (183,184). The exopolysaccharide alginate is a linear polymer of β-d-mannuronic acid and α-l-guluronic acid (185), which stimulates production of IgG and IgA antibodies (186). Although production of alginate is metabolically taxing, it protects the bacteria from phagocytosis and antibodies, thus conferring a survival advantage (187,188). Conversion to mucoidy occurs when biofilms are treated with activated polymorphonuclear leucocytes (189), hydrogen peroxide (189), antibiotics (190) and nutrient starvation (191,192).
A complex regulatory pathway controls alginate biosynthesis. The central player is the σE family extracytoplasmic function σ factor AlgT/U (PA0762) (193,194), whose activity is inhibited post-transcriptionally by the anti-σ factor MucA (PA0763) and by MucB (PA0764) (195–197). Loss of function mutations in mucA or mucB result in a mucoid phenotype (195,198,199) because of release of AlgT/U from MucA by a regulated intramembrane proteolytic pathway [reviewed in (200,201)]. It was recently demonstrated that MucA proteolysis is regulated not only by AlgW (PA4446) but also by MucD (PA0766) by activating the MucP protease (PA3649) (202). In addition, AmpR links alginate production with antibiotic resistance and QS by negatively regulating algT/U expression (Figure 1) (110). AlgT/U regulates alginate production at least in part by autoregulation (193), controlling expression of the transcriptional regulators algR (203,204), algB (198,204), amrZ (205) and the algD (PA3540) alginate biosynthetic operon (204,206,207). AlgB (208), AlgR (43,209,210) and AmrZ (211) directly bind to the algD operon to activate transcription. The alternative σ factor RpoN (PA4462) is also required for high levels of algT/U and algD expression (212).
The algB (PA5483) and algR (PA5261) genes encode NtrC and LytR subfamily, respectively, of TCS RRs (213). Interestingly, aspartic acid phosphorylation in the regulatory domain is not essential for alginate production (214). Transcriptome analysis of a PAOmucA22 mucoid strain (PDO300) (196) identified seven predicted transcriptional regulators, PA1235, PA1261, PA1637 (KdpE), PA2881, PA3420, PA3771 and PA5431, and one sensor kinase, EraS (PA1979), whose expression was downregulated in an algB mutant but not in a strain containing a mutation in its cognate TCS sensor, KinB (PA5484) (208). In addition to regulating the algD operon, AlgR directly activates transcription of algC (PA5322), which encodes a phosphomannomutase/phosphoglucomutase essential for Psl, alginate and rhamnolipid synthesis (Figure 1) (215–218). AlgR also is important for mature biofilm formation, possibly by directly repressing rhl-QS (219), type IV pilus formation by binding to the fimTU–pilVWXY1Y2E promoter (220,221) and hydrogen cyanide production by binding to the hcnA (PA2193) promoter (222). Interestingly, in contrast to alginate production, the phosphorylation site is required for regulating cyanide production and twitching motility (220,223). AlgR has also been shown to indirectly regulate the cyclic AMP/Vfr-dependent pathway (224). The AlgR regulon has been characterized by several transcriptome studies (219,222,225). Two other regulators of alginate production in P. aeruginosa are Alg44 (PA3542) (226) and a diguanylate cyclase, MucR (PA1727) (227). MucR produces a pool of c-di-GMP in the vicinity of the PilZ domain of Alg44 (PA3542), which then positively regulates alginate production (Figure 1) (226–228).
Regulation of iron uptake
Iron is critical for growth of all organisms, and P. aeruginosa is no exception. Transcriptome studies reveal that a large number of genes are regulated in response to iron (229,230). Biologically useful iron (Fe2+) in the environment is scarce and is available mostly in the insoluble Fe3+ form. To help scavenge this free iron, bacteria produce siderophores that bind extracellular iron and transport them back into the cell through TonB-dependent receptors on the cell surface (231). Pseudomonas aeruginosa produces two siderophores, pyoverdine and pyochelin, and can also subvert siderophores produced by other organisms to take up haem (232,233). However, excess free iron in the cell leads to formation of toxic reactive oxygen species, and, therefore, cells tightly regulate the uptake (234). The ferric uptake regulator (Fur, PA4769) is a conserved protein in P. aeruginosa and other Gram-negative bacteria, and it is a major iron acquisition regulator (235). Fur dimerizes rapidly after synthesis, and it takes a minimum of two dimers to bind promoters of genes under Fur regulation in P. aeruginosa (236). Fur controls the iron regulon directly by binding the Fur box (237) and indirectly by modulating expression of other regulators, including the pyochelin uptake regulator PchR (PA4227), ECF σ factors like PvdS, TCS regulators and small regulatory RNAs [asPrrF1 (PA4704.1), asPrrF2 (PA4704.2); Figure 1] (237–239).
Iron concentrations in the cell also influence expression of virulence factors in P. aeruginosa. PvdS, for example, is critical in linking iron and virulence by controlling the production of pyoverdine, an outer membrane pyoverdine receptor [FpvA (PA2398)] and two important extracellular virulence factors [PrpL (PA4175) and exotoxin A (PA1148); Figure 1] (240–242). Also, pvdS mutants showed reduced virulence in a rabbit endocarditis model (243). Human lactoferrin inhibits P. aeruginosa biofilm formation, indicating a role for iron in the process (244). Iron chelation by lactoferrin induces twitching motility in P. aeruginosa negating colonization and ultimately, biofilm formation (244,245). Intracellular iron concentrations are one of the signals for biofilm development in a process involving Fur but not the iron uptake regulatory RNAs asPrrF1 and asPrrF2 (246). Further, high levels of iron suppress the PQS system, release of extracellular DNA and biofilm formation (247).
The link between iron and QS systems in P. aeruginosa is complex. QS systems are enhanced under limiting iron concentrations (85,248,249), and major QS regulators are also involved in regulating iron responsive genes (75,250,251). MvfR, for example, has been demonstrated to control transcription of iron-related genes, and it has an iron-starvation box in its promoter, a site recognized by PvdS (PA2426) to turn on transcription under low-iron concentrations (229,252). It was recently demonstrated that iron levels affect activity of the MvfR signalling molecule HAQ, adding another layer of complexity to the role of iron in QS (253). Another major QS and virulence regulator, VqsR regulates phenazine production by modulating phnAB expression (94). Moreover, the small regulatory RNAs asPrrF1 and asPrrF2, which are negatively regulated by Fur, positively regulate PQS production (Figure 1) (254). PQS has been shown to accumulate in the outer membrane and in membrane vesicles (77). PQS chelates iron, and this facilitates pyochelin and pyoverdin in scavenging iron (73,76).
Thus, iron uptake regulation in P. aeruginosa is a complex affair and involves multiple regulators that affect expression of numerous genes either by themselves or through other regulators. In addition, the interconnections between iron uptake mechanisms and other virulence systems, such as QS and biofilm formation, demonstrate the versatility of this bacterium in being able to pragmatically read environmental signals to accordingly modulate gene expression.
Toxins and exoproteins
Exoproteins are an important component of bacterial survival not only because they allow the bacteria to interact with their immediate environment and other organisms in the vicinity but also because they play a critical role in virulence. P. aeruginosa has a large complement of secreted proteins and five (type I, II, III, V and VI) of the seven secretion systems characterized in bacteria (255). A majority of the secreted proteins are toxins that aid in P. aeruginosa virulence, most of which, including LasA, LasB, PrpL, ToxA and phospholipases [PlcH (PA0844), PlcN (PA3319), PlcB (PA0026)], is secreted through the Xcp type II secretion system (T2SS) (255). Effector molecules that are crucial for evading the host phagocytic response are secreted through a dedicated T3SS (256), whereas the type I system (T1SS) secretes the alkaline protease AprA (257,258) and the haemophore HasAp (PA3407) (259). Substrates of the recently identified T6SS are just being discovered (260). In addition, c-di-GMP levels, modulated by the diguanylate cyclase WspR (PA3702) is involved in the switch between T3SS and T6SS independent of RetS but is dependent on rgRsmY and rgRsmZ (Figure 1) (261).
HasAp, a T1SS-secreted haem-uptake protein in P. aeruginosa, is under QS control (250). QS is also known to regulate PrpL that targets the human lactoferrin (see previous section) (240). Thus, QS in P. aeruginosa not only regulates enzymes to degrade the human lactoferrin but also produces proteins to retrieve the iron from the degraded lactoferrin. The other known T1SS substrate, AprA, is regulated by a novel LTTR named BexR (PA2432) that controls bistability in P. aeruginosa (Figure 1) (262). Inactivation of QS has been demonstrated to reduce expression of T2SS-secreted proteases, chitinases and lipases (63,263) because of downregulation of the Xcp T2SS (264,265). The TCS PhoBR (PA5360–PA5361) regulates other T2SS-secreted exoproteins, such as PlcH, PlcC, PlcN and the Hxc T2SS secreted alkaline phosphatase LapA (266,267). Microarray analysis revealed that a novel cell-surface signalling system PUMA3 regulates Hxc T2SS genes (268). The three T6SS systems (HSI-I, HSI-II and HSI-III) in P. aeruginosa are differentially regulated by the QS systems (269). Although LasR and MvfR negatively regulate the HSI-I system, they positively regulate expression of the functionally redundant HSI-II and HSI-III (Figure 1) (269). In addition, a putative regulator Sfa3 (SfnR) in P. aeruginosa PA14 (an orthologue of PA2359 in PAO1) potentially regulates the HSI-III cluster (269). HSI-I expression is also regulated by RetS through RsmA (270).
Pseudomonas aeruginosa T3SS is regulated in a complex and multi-tiered process, and it is probably the most well understood (271). Expression of T3SS is regulated transcriptionally and post-transcriptionally in response to host cell contact and environmental Ca2+ levels (272,273). ExsA (PA1713), an AraC member, regulates expression of the 43 genes that form the T3SS in P. aeruginosa by binding as a monomer to an A-rich 8-bp region upstream of the −35 in the promoter of genes under its regulation (272,47). ExsA autoregulates its own expression and is also activated by PsrA (PA3006), a member of the TetR family (274,275). Two anti-activators [ExsD (PA1714) and PtrA (PA2808)] also regulate ExsA-mediated activation (Figure 1). T3SS transcription is coupled to secretion and involves the anti-activator ExsD and the anti–anti-activator ExsC (PA1710) that regulate ExsA function. Under non-inducing conditions (high Ca2+), ExsE (PA1711) binds the anti–anti-activator ExsC, allowing the anti-activator ExsD to bind ExsA and inhibit transcription. Under Ca2+ delimiting conditions, ExsE is secreted, freeing ExsC to bind ExsD. Free ExsA then activates T3SS expression (Figure 1) (276). Although this is the primary mode of control, T3SS can also be triggered by stress because of DNA damage (RecA-mediated activation of PtrB) (277), high salt (278,279), metabolic stress (123,279,280), alginate regulators AlgT/U, AlgR and MucA (281), the MexEF–OprN efflux pump regulator MexT (PA2492) through PtrC (282) and the RetS/LadS/Gac-Rsm TCSs (21,22,283). Under low oxygen conditions, the anaerobic regulator Anr activates the response regulator NarL (PA3879), which in turn represses rgRsmY and rgRsmZ expression, allowing RsmA to activate T3SS (284). Expression of T3SS, however, happens only in a subset of the population even under inducing conditions (279,285). Multiple levels of control allow fine-tuning of T3SS expression, allowing P. aeruginosa to sense various environmental conditions and regulate expression in conjunction with other virulence factors.
Regulatory RNAs in P. aeruginosa virulence
RNAs other than messenger RNAs, transfer RNAs or ribosomal RNAs are termed small RNAs (sRNAs), and they affect all steps in gene expression pathways in both prokaryotes and eukaryotes (286). In general, sRNA-mediated regulation occurs in one of two ways, base pairing with DNA or mRNA, or by affecting the activity of a protein or protein complex (286). Not surprisingly, virulence gene expression in P. aeruginosa also relies on small rgRNA-mediated post-transcriptional regulation. The importance of rgRNAs in regulation of bacterial virulence is well established (39,287,288). Most of the P. aeruginosa rgRNAs that have been characterized play a role in virulence gene regulation (discussed later in the text). A recent study identified ∼150 novel sRNAs using sRNA-Seq in P. aeruginosa PAO1 and PA14, which includes both strain-specific and shared ones (289).
Perhaps the most well characterized system involves the rgRNAs, rgRsmY and rgRsmZ (whose roles in virulence regulation have been discussed in the ‘TCS’ and ‘Toxins and Exoproteins’ sections). These are two functionally redundant rgRNAs in P. aeruginosa that play a critical role in the switch between acute and chronic infections (21,290). GacA of the GacSA TCS positively regulates expression of rgRsmY and rgRsmZ, which then bind to and sequester the sRNA-binding protein RsmA through the GGA motif (291–293), leading to derepression of the genes that RsmA represses (107,124,294,295). The consequences of RsmA sequestration result in dysregulation of the expression of numerous virulence factors, as discussed in the ‘TCS’ section. Given the importance of this regulatory process in P. aeruginosa pathogenesis, regulation of expression of rgRsmY and rgRsmZ is multi-tiered. The histidine phosphotransfer protein HptB is phosphorylated by a phosphorelay involving the three sensor kinases PA2824, PA1611 and PA1976 (296). Phosphorylated HptB then transfers the phosphate to an anti–anti-σ factor PA3374, to negatively regulate expression of rsmY (296,297). In another mode of regulation, the BfiSR TCS activates expression of the ribonuclease CafA (PA4477), which specifically targets rgRsmZ (298). Further regulation is achieved by the global regulators of the H-NS family of proteins MvaT and MvaU, which bind to AT-rich regions upstream of the rsmZ gene repressing their expression (299). In addition to all this, there is a negative autoregulatory feedback mechanism, the details of which have not been elucidated yet (300). On synthesis, rgRsmY is stabilized by Hfq binding, either alone or in conjunction with RsmA (108,301).
Another example of post-transcriptional regulation by sequestering a RNA-binding protein links virulence with metabolism. Pseudomonas aeruginosa Crc (PA5332) is a RNA-binding protein that recognizes CA-motifs around the ribosome binding sites of the mRNA of carbon compound catabolism genes. Crc thus represses genes whose products help utilize less preferred carbon sources (302–304). When less preferred substrates, such as mannitol, have to be utilized, expression of catabolic genes is achieved by sequestration of Crc by the rgRNA, rgCrcZ (305). Expression of rgCrcZ is under the control of the TCS CbrAB (PA4725–PA4726), which in conjunction with Crc plays a role in carbon compound catabolism, biofilm formation, antibiotic resistance, secretion systems and swarming (306–312).
Pseudomonas aeruginosa antisense sRNAs (asRNAs) can also act by base pairing with target mRNAs, thus inhibiting translation (313). One such example is asPhrS (PA3305.1), which plays a role in PQS and pyocyanin expression (314). Transcriptome studies indicate an extensive overlap between the genes that are positively regulated by the transcriptional regulator PqsR (also known as MvfR, PA1003) and asPhrS, suggesting that asPhrS regulates pqsR mRNA (75,314). Interestingly, it was shown that asPhrS specifically targets a region in the RBS of a small ORF (uof), which is present upstream of PqsR (314). As translation of pqsR and uof are coupled, asPhrS regulates pqsR translation by modulating translation of uof (314). Expression of asPhrS is under the control of the oxygen responsive regulator Anr (314). Hfq controls asPhrS expression indirectly by regulating Anr expression, whose mechanism of action is yet to be elucidated (314,315).
Small asRNAs also play a role in regulation of iron uptake and involve base pairing by the sRNAs asPrrF1 and asPrrF2, which are the P. aeruginosa orthologues of E. coli RyhB (237,316). Expression of asPrrF1 and asPrrF2 is repressed by Fur when iron concentrations are high (237). Under iron-starvation conditions, asPrrF1 and asPrrF2 are expressed and base pair with the mRNA of target genes, which include the superoxide dismutase sodB (PA4366), genes involved in the trichloroacetic acid cycle and anthranilate and cathechol degradation (317). Thus, asPrrF1 and asPrrF2 link carbon metabolism, iron uptake and QS-mediated virulence. Another asRNA gene asPrrH is located in the same locus as asPrrF1 and asPrrF2. The asPrrH asRNA (at 325 nt) is longer than asPrrF1 (116 nt) and asPrrF2 (114 nt), and the coding region of asPrrH overlaps with the asPrrF1 terminator, the intergenic region between asPrrF1 and asPrrF2 and the 5′-end of the asPrrF2 ORF (318). The expression of asPrrH is maximal in the stationary phase of growth, similar to asPrrF1 and asPrrF2, and under iron-deplete conditions (318). Haem represses asPrrH expression, and this involves the outer membrane haem receptors PhuR (PA4710) and HasR (PA3408) (318). Interestingly, under conditions of haem starvation, asPrrH expression leads to the repression of achAB and sdhCDAB, which are also targets of the PrrF asRNAs (318). In addition to these targets, asPrrH also represses NirL, a protein involved in biosynthesis of haem, under haem and iron limitation (318).
CONCLUSIONS AND PERSPECTIVES
Pseudomonas aeruginosa is a versatile bacterium that can thrive in a wide range of habitats. This is achieved by an intricately interlinked regulatory system of transcriptional regulators, σ factors, sRNAs and their regulons. The exquisite control of gene expression is exemplified in the virulence regulatory network (Figure 1), which demonstrates that none of the virulence mechanisms are isolated. Expression of individual virulence networks is under transcriptional and post-transcriptional regulation of multiple-regulatory systems, either directly or indirectly. Furthermore, some signalling cascades inversely regulate the acute and chronic virulence phenotypes depending on the signals sensed. The next critical phase of research should focus on the signals that the bacteria recognizes to achieve gene regulation.
The extent of cross-regulation between the transcriptional regulators highlights the global nature of the regulation, where individual subnetworks (such as the QS network, alginate network and so forth) are interlinked to form a hyperconnected network (Figure 1). Given the complexity of the connections, one can expect the response of a cell to be elaborate even when faced with a simple stress condition. A fundamental point in a network setting is that one should evaluate the role of individual players (such as a regulator) not in isolation, but with the knowledge that the entire network will react to what it does. In other words, local changes can have global effects. This, in turn, results in subtle cause–effect relationships. Studying the functions of a regulator by generating deletion or overexpression strains is often performed under the assumption that other regulators will remain static. In reality, however, such modifications can lead to aberrant changes across the network in ways that were initially unintended. Moreover, there is a possibility that such changes can occur because the network connections might not always be obvious. This can be attributed, in part, to as yet uneludicated implicit players in the network that dictate or otherwise influence cellular response. This is a likely explanation for the many ‘global’ regulators in P. aeruginosa and in similar bacteria that have complex regulatory networks. In such cases, many of the phenotypes observed with single regulator mutant strains can be part of a ripple effect that propagates through the network affecting disparate phenotypes.
A simplified model of gene regulatory network treats genes as being on or off, that is, taking binary values. It is, therefore, no surprise that Boolean networks (discrete dynamical network models) have been used to model and study gene regulatory circuits (319–321). Probabilistic Boolean networks, which take into account molecular and genetic noise (322,323), and stochastic Boolean networks, which permit the modelling of gene perturbations (324), provide important insights into the dynamical behaviour of the system. Although they are computationally complex, they are a valuable addition to the numerous other programs that are available to analyse gene regulatory networks (31,325,326).
Dynamical systems theory helps us to analyse the behaviour of complex systems that can frequently be expressed by time-differential equations. When the behaviour of a dynamical system depends sensitively on small changes in initial conditions, then the system is said to be chaotic, that is, capable of exhibiting chaotic behaviour. Researchers have investigated whether regulatory networks can have subsystems that are capable of exhibiting chaotic behaviour (327). It has been shown that competition between two or more subnetworks of comparable importance can lead to chaos (328–330). In fact, chaos has been shown to be possible in biochemical systems with only two feedback loops, and positive feedback is known to be necessary for chaotic behaviour (331). So, one would expect chaotic subsystems in a regulatory network as complex as the one that controls P. aeruginosa virulence (Figure 1). Despite of this predisposition, gene regulatory networks seldom exhibit chaotic behaviour. This could be because the competitions between opposing nodes are not strong enough (332) or that chaotic behaviours are short-lived because of triggering of other pathways, such as cell–cell communication (333). Another possibility is that the natural random variability of biochemical systems masks the chaotic behaviour (332). However, maintaining a low level of chaos in such a complex network is probably a combination of the aforementioned and, potentially, as yet unknown factors.
In gene regulatory networks, a particular dynamical system is characterized by time-evolving variables (chemical concentrations, gene expression and so forth) and by parameters (temperature, ambient chemical concentrations and so forth). A network can exhibit chaotic or non-chaotic behaviour depending on the parameters that influence it (334). Environmental factors, such as the temperature or the nutritional status of the cells, parameterize the relationship between transcription factors and the genes that they regulate. Although it is understood that some choices of parameters can induce chaotic behaviours, the parameter may not even be achievable, such as high temperatures (334). Mutations can also alter relationships in regulatory networks by causing changes in existing links or forming new ones. In a dynamically robust (non-chaotic) system, small finite changes in the parameters lead to only qualitative changes in the dynamical behaviour. However, there are boundaries in the parameter space where the behaviour of the system changes qualitatively and may include the possibility of chaotic dynamics. Predicting whether a network will be stable or chaos-prone under some conditions has been proven to be difficult and remains poorly characterized. Recent work has identified the minimum number, types and interactions among three and four nodes/subnetworks that can lead to chaos in a gene regulatory network (332). Such minimal subnetworks have been termed ‘chaotic motifs’, and networks with these motifs can exhibit chaotic behaviour under the right parameters (332). Analysis of the network in Figure 1 does not readily show such chaotic motifs. This could be because the network is incomplete (lack of data on the interactions among the P. aeruginosa regulators) or because of errors in the inferred interactions. Although P. aeruginosa virulence regulation has been extensively studied, there is yet much to learn. Thus, absence of empirical evidence does not preclude a propensity to chaos and is worth further investigation.
Depending on an elaborate network to achieve gene regulation is likely an adaptive mechanism by P. aeruginosa. Possessing alternate pathways to regulate the same phenotype ensures a rapid response to stimuli even if one of the pathways is affected, thus enhancing survival. Such examples can be seen throughout the network. As discussed in the ‘Toxins and Exoproteins’ section, expression of T3SS genes can be regulated at multiple levels, in response to various different signals and stress conditions, and it is not entirely dependent on any one signal. However, the extent of contributions of the individual regulators and, consequently, the fine balance that exists in some regulatory cascades within the network, are sometimes not easily apparent. Network dependence is also a probable reason for regulator genes being non-essential, in the sense that deleting a transcriptional regulator gene typically does not affect cell viability because of the presence of alternate regulatory mechanisms. Having key regulators modulate different related phenotypes has the added advantage in allowing the cells to adapt to external signals by modulating one or a few regulators instead of individually regulating different virulence systems. A case in point is AmpR that positively regulates acute virulence factors while downregulating chronic infection phenotypes (23). Also of importance is the co-regulation of metabolism and virulence. Studies have identified regulators like CbrB that, with its cognate sensor CbrA, not only regulate carbon metabolism but also virulence phenotypes through the rgRNA, rgCrcZ and the RNA-binding protein Crc (306,307). Moreover, there is crosstalk between CbrA and regulators other than CbrB, highlighting the complexity of the system (306).
The plethora of transcriptome studies using microarrays or deep sequencing will add to the database of genes that are differentially expressed in response to regulator mutations or specific growth conditions. Differentiating the direct effect of a change from a ripple effect can, at least partly, be achieved by meta-analysis studies that look at multiple transcriptomes, identifying effects unique to each condition and differentiating them from the so-called ripple (32,33). Network analyses can help us understand the relationship between different regulators, group them based on function and, more importantly, help identify critical nodes and prominent players. This can serve as a means of target identification in attempting to deal with P. aeruginosa infections. In Figure 1, we see that some parts of the network are more densely connected than others, with central cores containing most of the links. A case in point is LasR of the QS subnetwork. It is well known that QS is central to virulence regulation in P. aeruginosa and targeting key regulators will have a better chance of therapeutic success. Recently, inhibitors of a key QS regulator were shown to reduce pathogenicity in Vibrio cholera (335).
With the extensive use of high-throughput transcriptomics, gene regulation studies are now focusing on the role of non-coding RNAs in bacteria. rgRNAs have been shown to be extensively involved in gene regulation in P. aeruginosa and other bacteria (124,237,299,305,314,336). Techniques such as RNA-seq allow for the entire transcriptome to be sequenced, giving us an unprecedented insight into non-coding RNAs, asRNAs and sRNAs involved in regulation. Preliminary studies using prediction software and complementary experiments have already advanced our understanding (29,108,124,299,314,337). Given the many different ways in which small RNAs can modulate gene expression (313) and potentially undiscovered ones, we can look forward to exciting new discoveries in bacterial gene regulation in the coming years.
FUNDING
National Institutes of Health-Minority Medical Research Support SCORE [S06 GM08205 and 5SC1AI081376 to K.M.]; FIU Research Assistantship (Herbert Werthiem College of Medicine to D.B.). Funding for open access charge: National Institutes of Health-Minority Medical Research Support SCORE [5SC1AI081376 to K.M.].
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors are extremely grateful to Giri Narasimhan (Florida International University) and Edward Celarier (NASA Goddard) for extensive discussions and critical comments on chaos theory.

{kind=link}
Comments