Article Text

Abstract
Background Spinocerebellar ataxia type 28 (SCA28) is a dominantly inherited neurodegenerative disease caused by pathogenic variants in AFG3L2. The AFG3L2 protein is a subunit of mitochondrial m-AAA complexes involved in protein quality control. Objective of this study was to determine the molecular mechanisms of SCA28, which has eluded characterisation to date.
Methods We derived SCA28 patient fibroblasts carrying different pathogenic variants in the AFG3L2 proteolytic domain (missense: the newly identified p.F664S and p.M666T, p.G671R, p.Y689H and a truncating frameshift p.L556fs) and analysed multiple aspects of mitochondrial physiology. As reference of residual m-AAA activity, we included SPAX5 patient fibroblasts with homozygous p.Y616C pathogenic variant, AFG3L2+/− HEK293 T cells by CRISPR/Cas9-genome editing and Afg3l2 −/− murine fibroblasts.
Results We found that SCA28 cells carrying missense changes have normal levels of assembled m-AAA complexes, while the cells with a truncating pathogenic variant had only half of this amount. We disclosed inefficient mitochondrial fusion in SCA28 cells caused by increased OPA1 processing operated by hyperactivated OMA1. Notably, we found altered mitochondrial proteostasis to be the trigger of OMA1 activation in SCA28 cells, with pharmacological attenuation of mitochondrial protein synthesis resulting in stabilised levels of OMA1 and OPA1 long forms, which rescued mitochondrial fusion efficiency. Secondary to altered mitochondrial morphology, mitochondrial calcium uptake resulted decreased in SCA28 cells.
Conclusion Our data identify the earliest events in SCA28 pathogenesis and open new perspectives for therapy. By identifying similar mitochondrial phenotypes between SCA28 cells and AFG3L2+/− cells, our results support haploinsufficiency as the mechanism for the studied pathogenic variants.
- genetics
- molecular genetics
- movement disorders (other than parkinsons)
- mitochondria
- cell biology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Spinocerebellar ataxias (SCAs) are a clinically and genetically heterogeneous group of autosomal dominant neurodegenerative disorders primarily characterised by progressive gait ataxia, imbalance and dysarthria. Among the 46 different SCA subtypes identified to date (http://neuromuscular.wustl.edu/ataxia/domatax.html), SCA type 28 (SCA28; MIM #610246) is the only one caused by pathogenic variants in a resident mitochondrial protein, AFG3L2.1 The clinical spectrum of SCA28 comprises slowly progressive gait and limb ataxia, and oculomotor abnormalities (eg, ophthalmoparesis and ptosis) as typical signs.2
AFG3L2 belongs to the AAA-protease subfamily (ATPases associated with various cellular activities) and assembles into homo-oligomers and hetero-oligomers with paraplegin in the inner mitochondrial membrane (IMM), named m-AAA proteases. AFG3L1 is a third m-AAA paralogue subunit that is expressed in mouse, but not in humans due to a pseudogenisation event.3 AFG3L1 can also form homo-oligomers or hetero-oligomers, while paraplegin can only form hetero-oligomers. Based on the expression levels of each of the m-AAA subunits, different tissues will possess a peculiar cohort of these homo-oligomeric or hetero-oligomeric complexes.4
The m-AAA proteases are crucial component of the quality control system of the IMM and mediate the selective degradation of non-assembled and damaged proteins.5 Furthermore, they exert additional regulatory functions to promote mitochondrial protein synthesis,6 assembly of respiratory chain complexes,7 8 mitochondrial dynamics9 10 and mitochondrial calcium homeostasis.11 12
Most of the reported SCA28 pathogenic mutations are heterozygous missense variants within the key proteolytic domain encoded by exons 15 and 16 of AFG3L2.1 13–17 Even though the genetic variants are similar in type and location within AFG3L2, the clinical phenotype of SCA28 patients is associated with a wide range of symptoms severity and age of onset.2 Other variants have included one missense in exon 4,18 one missense in exon 10,1 a truncating frameshift in exon 1519 and a deletion spanning exons 14–16.20
In addition, a homozygous missense pathogenic variant in AFG3L2 in two brothers of a consanguineous family has been shown to cause a different and more severe disease, spastic ataxia type 5 (SPAX5), characterised by early-onset spastic ataxia, neuropathy, ptosis, oculomotor apraxia, dystonia, cerebellar atrophy and progressive myoclonic epilepsy.21 Recently, the clinical phenotype associated to AFG3L2 pathogenic variants has been further expanded by two reports of different homozygous pathogenic variants associated with progressive myoclonus epilepsy22 and refractory epilepsy with progressive microcephaly,23 respectively.
We provided evidence that Afg3l2+/− haploinsufficient mouse recapitulates most features of SCA28 patients, displaying defects in motor coordination and balance due to dark cell degeneration (DCD) of Purkinje cells (PCs).24 In this SCA28 model, DCD originates from increased cytoplasmic calcium concentrations ([Ca2+]c) because of inefficient transport of mitochondria in PC dendrites.11 Similarities between human and mouse models of AFG3L2-related disorders also extend to Afg3l2−/− mice. These mice resemble patients with homozygous pathogenic variants by having a severe neurological syndrome associated with ataxia and spasticity and early lethality at P16.8
These findings suggest that loss of function of the mutated protein is behind the pathogenesis of the human AFG3L2 variants (heterozygous for SCA28 and homozygous for SPAX5). However, since in Afg3l2+/− mouse the protein is reduced in amount rather than mutated, this model does not fully explain whether the reduced m-AAA complex activity in SCA28 is caused by haploinsufficiency or by a dominant negative effect exerted by the mutated protein on the wild-type (wt). Furthermore, the presence of the additional m-AAA protease subunit AFG3L1 in the mouse may be compensating for some of effects caused by the haploinsufficiency/absence of AFG3L2.
In order to clarify the pathogenic mechanism of SCA28, in this work, we characterised patient fibroblasts carrying different heterozygous missense or truncating frameshift pathogenic variants (the newly identified p.F664S, p.M666T, p.G671R, p.Y689H and p.L556fs). We found that the missense changes are similar to controls with regards to the total amount and ability of AFG3L2 to assemble into m-AAA complexes, while the truncating frameshift variant is associated with half of this amount. Common phenotypic hallmarks of SCA28 cells were inefficient mitochondrial fusion and reduced mitochondrial calcium buffering, while mitochondrial membrane potential (ΔΨm) was unaffected. The altered mitochondrial morphology is the result of OMA1 hyperactivation causing increased processing and turnover of the long forms of OPA1. Of note, we demonstrated that the trigger for OMA1 hyperactivation in condition of reduced AFG3L2 activity is proteostatic stress due to the accumulation of mitochondria-encoded polypeptides. In agreement with these findings, we found that pharmacological attenuation of mitochondrial protein synthesis stabilised OMA1 activity with restoration of OPA1 long forms and mitochondrial fusion in human SCA28 cells and Afg3l2 −/−murine embryonic fibroblasts (MEFs). These results were replicated in AFG3L2+/− HEK 293 T cells generated by targeted genome editing. Taken together, our data provide new insights into the pathogenic cascade of SCA28 and identify a potential upstream target for therapy. Finally, different and complementary experimental findings indicate haploinsufficiency as the primary mechanism of these SCA28 pathogenic variants.
Materials and methods
Ethics statement
All patients or their legal representatives signed informed consent before enrolment.
Fibroblast derivation from skin biopsies
Patient-derived skin fibroblasts were obtained following standard procedures. Fibroblasts were cultured in Dulbecco’s Modified Eagle Medium (DMEM) medium supplemented with 20% Fetal Bovine Serum (FBS), 1 mM sodium piruvate, 2 mM L-glutamine and 100 U/mL penicillin-streptomycin. All cell culture reagents were from Thermo Fisher Scientific (Waltham, Massachusetts, USA). Seven SCA28 patient-derived fibroblast lines were used that included several previously reported SCA28 variants (p.M666T, p.G671R13 and p.Y689H15), and a partial deletion of exons 14–16.20 A novel SCA28 variant (p.F664S) from a recently identified Italian family was also used, along with fibroblasts from a SPAX5 subject with a homozygous AFG3L2 variant (p.Y616C).21
Antibodies, drugs and reagents
Commercially available antibodies were used in western blots (WBs), for the detection of OPA1 and TIM44 (BD Transduction Laboratories, Franklin Lakes, New Jersey, USA), OMA1 (Novus Biologicals, Littleton, Colorado, USA), tubulin (DSHB, University of Colorado, Colorado, USA), HSP60 (Enzo Life Science, Farmingdale, New York, USA), MTCO1 and VDAC1 (Abcam, Cambridge, UK). Anti-AFG3L2 N-terminal (OriGene, Rockville, Maryland, USA) and an anti-AFG3L2 C-terminal antibody (previously generated in the lab)7 were used to detect AFG3L2. Secondary antibodies used included Horseradish Peroxidase (HRP)-conjugated antimouse and antirabbit IgG (Amersham Bioscience, Buckinghamshire, UK). Chloramphenicol succinate sodium salt (Merck, KGaA, Billerica, Massachusetts, USA) was used at a concentration of 200 µg/mL for 24 hours. Carbonyl cyanide-4 -(trifluoromethoxy)phenylhydrazone (FCCP) (Merck) was used at a concentration of 10 µM for 10 min.
Mitochondria isolation and Blu-Native PAGE
Mitochondrial enrichments and Blu-Native PAGE (BN-PAGE) were performed as previously described8 followed by standard immunoblotting procedures.
Analysis of mitochondrial morphology
Primary skin fibroblasts (n=20 000) were plated on a bottom-glass culture dishes (MatTek Corporation, Ashland, Massachusetts, USA) and infected with lentivirus expressing mitoDsRed2 (Clontech, Mountain View, California, USA) (1:200 from 4.67*108 U/mL). Seventy-two hours after infection, the mitochondrial network morphology was evaluated with live imaging using an Axio Observer.Z1 inverted microscope (Zeiss). On average, at least 100 cells/cell line were evaluated per experiment. Three independent operators performed blinded analysis of the collected images.
Generation of monoclonal AFG3L2+/− HEK 293 T cells by CRISPR/Cas9-based technology
A 20 bp targeting sequence for spCas9 was selected in the coding sequence of exon 1 of AFG3L2 by using NCBI BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi.). Candidate sequences with off-targets with up to two mismatches were excluded (GRCh38/hg38 was used as assembly reference). Complementary oligonucleotides (AFG3L2 FWD 5′ gatcgCTCCTCGTGCCTGGCGGCGTg 3′ and AFG3L2 REV 5′ aaaacACGCCGCCAGGCACGAGGAGc 3′) for gRNA were annealed and cloned into BamHI and BsmBI sites of pCas-Guide-EF1a-GFP CRISPR/Cas9 All-in-One vector (GE10018, OriGene). HEK 293 T cells were transfected with the pCas-Guide-EF1a-GFP vector containing the AFG3L2 targeting sequence, and 48 hours after transfection, Green Fluorescent Protein (GFP)-positive cells were sorted through fluorescence-activated cell sorting. Sorting was performed with FACSAria Fusion instrument and FACSDiva 8.0 software (BD). Positive cells were collected into a 96 multiwell plate. After 24 hours, cells were resuspended, and single cells were isolated by serial dilutions. Wells with single colonies were checked at the microscope, and monoclonal cell lines were expanded. Genomic DNA from monoclonal cells was amplified by PCR (primers: FWD 5′CCGCGTTGAGAGCTTGGG3′ and REV 5′ACCTTGACGTCCGCTCTC3′), and products were Sanger-sequenced to identify AFG3L2 heterozygous clones. WB analysis was then performed to verify the reduction of AFG3L2 protein by 50%.
The clones used in this study are: clone #1 (allele1: p.R28fs; allele 2: p.G26del) and clone #2 (allele1: p.A14fs; allele 2: p.G27_V28del). The p.G26del and p.G27_V28del in frame deletions are located upstream of the leader peptide-cleavage site by mitochondrial matrix peptidase and therefore do not affect the mature AFG3L2.
Mitochondrial membrane potential evaluation
ΔΨm was measured using the mitochondrial potentiometric dye tetramethylrhodamine methyl ester (TMRM) (Invitrogen) and cytofluorimetric analysis as previously described.10 Physical parameters settings were specific for each cell line and data were reported as ratio between TMRM/(TMRM+FCCP).
In vitro pulse labelling of mitochondrial translation products
Cells were incubated for 3.5 hours in methionine-free and cysteine-free DMEM supplemented with 250 µg/mL of the cytoplasmic translation inhibitor cycloheximide and 10% dialysed FBS. They were then labelled with 35S-methionine/cysteine (0.22 mCi/plate) for 30 min and then harvested or chased (DMEM supplemented with 10% FBS) for 2 or 4 hours. For each condition, 30 µg of proteins were electrophoresed through a 10%–20% denaturing gel. Proteins were transferred to nitrocellulose membranes before autoradiography and sequentially probed with antibodies (anti-OMA1 and anti-VDAC1). To evaluate the efficacy of chloramphenicol in stabilising OMA1 activity, cells were incubated for 24 hours with chloramphenicol before pulse labelling.
Cytosolic and mitochondrial calcium evaluation
Measurement of [Ca2+]c was performed as previously described.25 To quantify mitochondrial calcium responses ([Ca2+]m), fibroblasts were transfected with plasmid 4mtD126 and imaged 48 hours after transfection as reported.11
Statistical analysis
Continuous variables were summarised by their mean values and SEM; categorical variables were summarised by means of frequencies and percentages.
To evaluate possible differences in protein levels between patients and controls, densitometric analysis of WB bands from at least three independent experiments was performed using Image J (imagej.nih.gov/ij/index.html), followed by Student’s t-test analysis. Differences in mitochondrial network morphology were calculated by χ2 analysis, using two df. To investigate differences in ΔΨm between patients and controls, and thus differences in the mean values of [(TMRM-Basal)]/[(TMRM+FCCP)-Basal] among the groups (=‘kindof’), we fitted a generalised linear model with nested random effect by means of penalised quasi-likelihood method taking into account the dependencies between measurements coming from the triplicates of the same cell line.27
Results
Missense AFG3L2 variants do not alter AFG3L2 monomer stability or m-AAA assembly
We derived primary fibroblasts from skin biopsies from seven different SCA28 patients. Some of the analysed pathogenic variants were already reported (p.M666T, p.G671R,13 a partial deletion of exons 14–16,20 p.Y689H15), while we identified p.F664S as a new SCA28-causing pathogenic variant in an Italian family. We also included in the study the only living SPAX5 patient, carrying the homozygous AFG3L2 p.Y616C pathogenic variant.21
We first sequenced the cDNA of SCA28 patients carrying the C-terminal deletion, and we defined the pathogenic variant as a frameshift (p.L556fs) occurring in the end of exon 13, resulting in a premature stop codon (online supplementary figure S1A and S1B).
The clinical features of the analysed SCA28 patients are reported in table 1. The primary phenotype consisted of gait–limb ataxia and dysarthria for all patients, with variability in the severity of symptoms and age of onset. The patient with the p.Y689H pathogenic variant has a milder phenotype with less severe gait–limb ataxia and no oculomotor or extrapyramidal signs.
The analysed pathogenic variants are all localised in the key metallopeptidase domain of AFG3L2 (online supplementary figure S2A). In silico analysis (PolyPhen-228 and SIFT29 tools) showed that all the SCA28 missense pathogenic variants analysed in this study are predicted to damage AFG3L2 function. Moreover, multiple species alignment shows that mutated residues are highly conserved through evolution (online supplementary figure S2B).
Clinical features of SCA28 patients analysed in this study
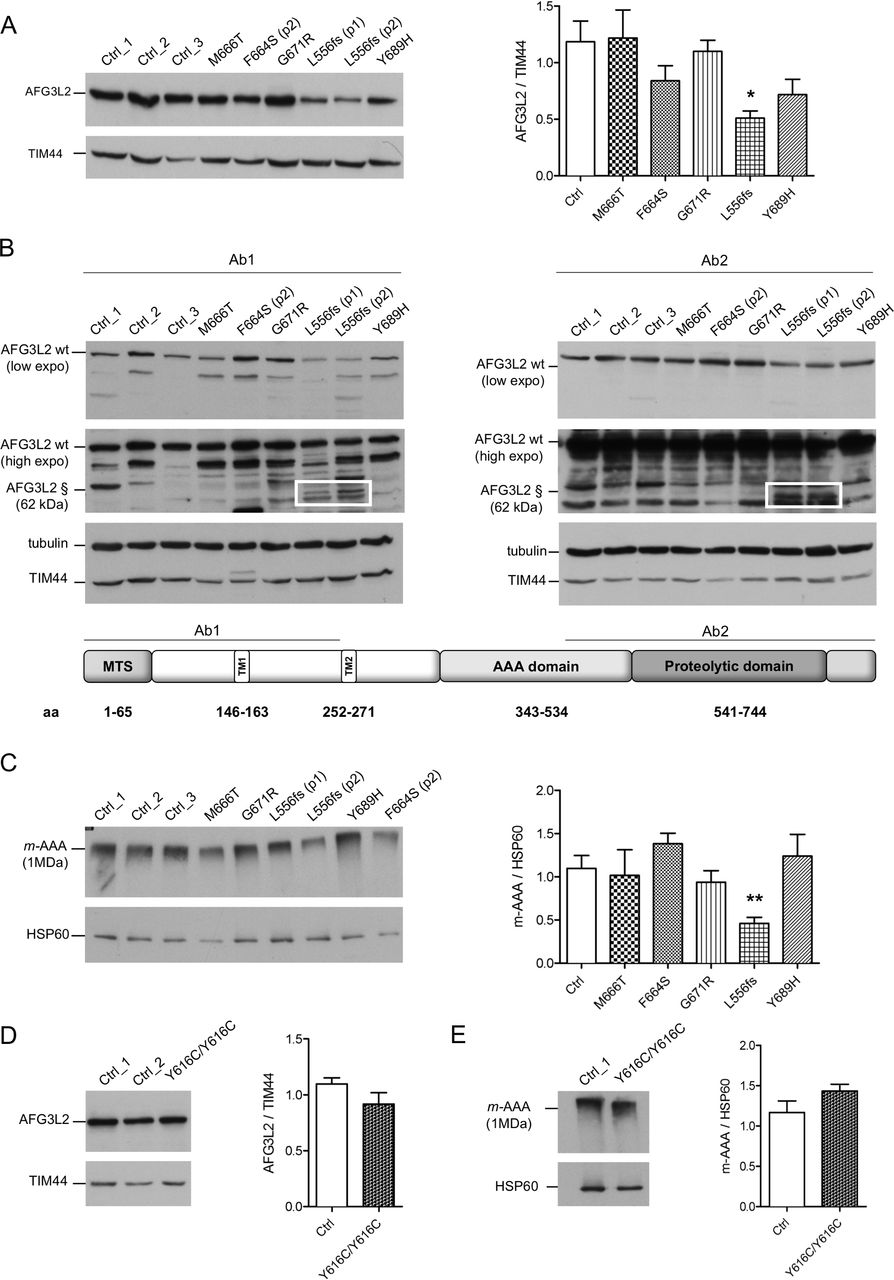
We analysed by WB the effect of the pathogenic variants on the stability of AFG3L2 monomer. The heterozygous missense pathogenic variants expressed similar total amount of full-length AFG3L2 protein as compared with controls (figure 1A). In contrast, this amount was decreased by ~50% in the p.L556fs fibroblasts (figure 1A). In these cells, the mutated allele is predicted to encode for a truncated protein of approximately 62 kDa. In order to quantify the amount AFG3L2-p.L556fs protein relative to the full-length protein, we employed two different antibodies recognising either N-terminal or C-terminal epitopes of AFG3L2. In experiments with both antibodies, we observed that the truncated AFG3L2-p.L556fs has dramatically reduced expression compared with the wt protein (almost 20% relative to the full-length) (figure 1B and online supplementary figure S2C). Treatment with MG132 for the inhibition of proteasomal activity did not rescue truncated AFG3L2-p.L556fs protein in patient cells (online supplementary figure S2D), suggesting that this mutant is either degraded inside the mitochondria after being imported or its mRNA is unstable.
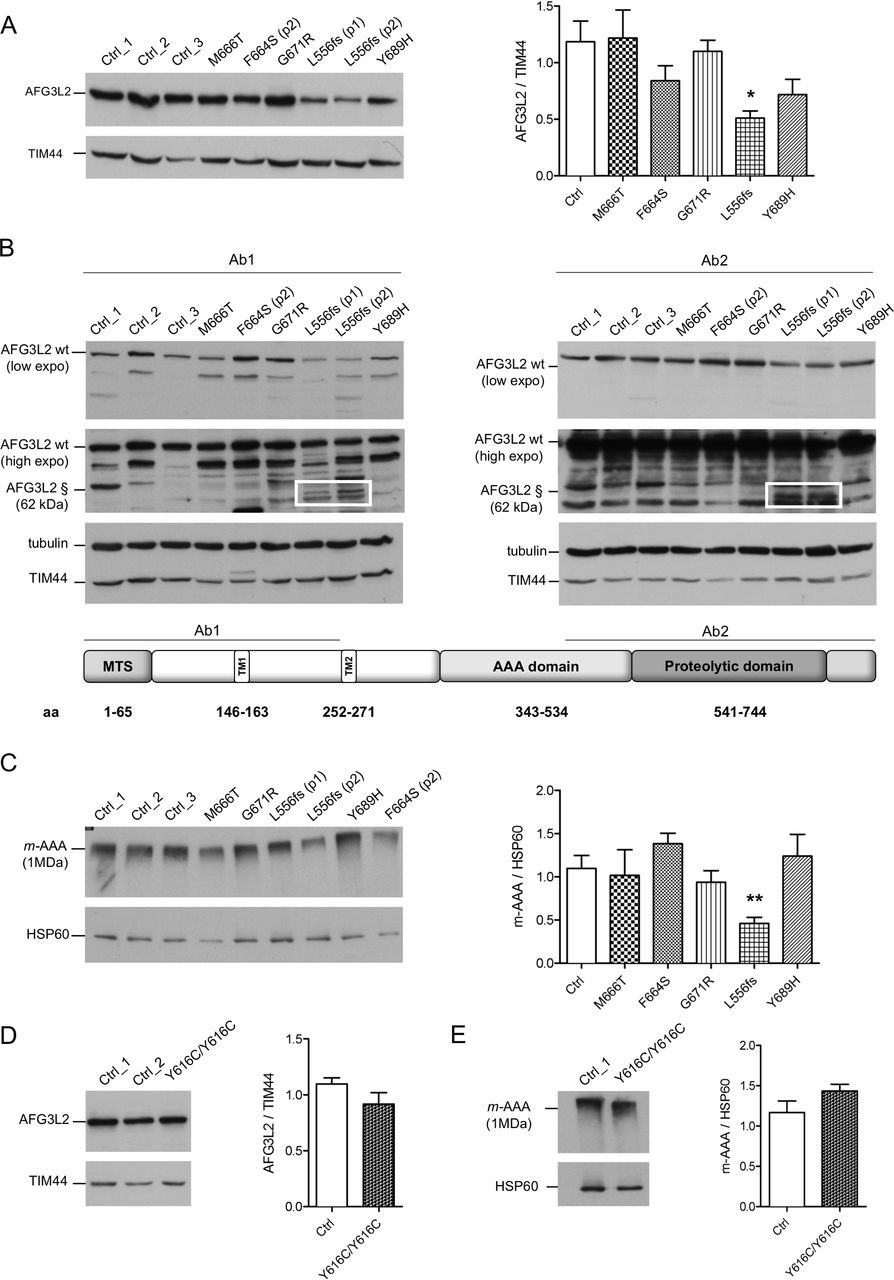
Missense pathogenic AFG3L2 variants do not alter the amount of m-AAA complexes. (A) WB analysis of fibroblast cell lysates showing levels of AFG3L2 in controls and patients with relative quantification. Bars represent means±SEM of three independent experiments. Student’s t-test: *p<0.05. (B) WB analysis showing residual levels of p.L556fs AFG3L2 (§) encoded by the mutant allele (boxed) using antibodies recognising an epitope on the N-terminus (left) or on the C-terminus of AFG3L2 (right), respectively. (C) BN-PAGE of mitochondrial extracts from SCA28 fibroblasts and controls stained with anti-AFG3L2 and anti-HSP60 as loading control. Bars represent means±SEM of four independent experiments. Student’s t-test: **p<0.01. (D) WB analysis of fibroblast cell lysates from SPAX5 patient and controls showing levels of AFG3L2 monomer with relative quantification. Bars represent means±SEM of three independent experiments. (E) BN-PAGE on mitochondrial extracts from SPAX5 fibroblasts and controls revealed with anti-AFG3L2 and anti-HSP60 as normaliser. Bars represent means±SEM of four independent experiments. WB, western blot.
To verify if AFG3L2 pathogenic variants impact on the ability of the encoded protein to assemble into m-AAA complexes, we performed BN-PAGE on mitochondrial extracts (figure 1C). Fibroblasts carrying missense pathogenic variants had similar amounts of assembled m-AAA complexes, while these levels were decreased by ~50% with p.L556fs cells, demonstrating that the p.L556fs pathogenic variant causes a functional haploinsufficiency. Indeed, BN-PAGE followed by second dimension SDS-PAGE identified only full-length AFG3L2 in assembled m-AAA complex, indicating that the truncated AFG3L2-p.L556fs protein is unable to oligomerise (online supplementary figure S3A and B). Cells with homozygous p.Y616C pathogenic variant were also shown to have similar amounts of AFG3L2 protein, as both a monomer and assembled m-AAA complexes compared with controls (figure 1D and E).
Altered mitochondrial morphology in SCA28 patient fibroblasts
We and others have reported increased mitochondrial fragmentation in Afg3l2 −/−MEFs and primary PCs.9–11 To evaluate if mutated AFG3L2 affects mitochondrial morphology, we analysed the mitochondrial network in primary SCA28 patient fibroblasts by live imaging microscopy. While control cells possessed a highly fused and interconnected mitochondrial network, most of the SCA28 cells (and SPAX5 cells) had shorter and less interconnected organelles with more numerous mitochondrial units (figure 2A). Only the p.Y689H fibroblasts were not statistically different compared with controls in this regard.

Mitochondrial fragmentation due to enhanced OPA1 processing and turnover in SCA28 fibroblasts. (A) Representative images of mitochondrial morphology in SCA28 and SPAX5 primary fibroblasts infected with mtDsRed2 and visualised by live imaging microscopy. The graph shows the morphometric analysis of mitochondrial morphology in SCA28 and SPAX5 primary fibroblasts. One hundred randomly selected cells were analysed on average in each experiment. Bars represent means±SEM of three independent experiments. χ2 test (2 df): controls (mean of 7 different lines) versus different pathogenic variants: ***p<0.0001. (B and C) WB of fibroblast cell lysates showing levels of OPA1 in controls and patients with relative quantifications (# highlights a non-specific band). Magnification of two independent anti-OPA1 immunoblots shows the OPA1-band pattern in SCA28-p.Y689H cells, with higher amount of OPA1 long forms and decreased OPA1 short forms (arrows) compared with SCA28-p.L556fs cells. Bars represent means±SEM of four independent experiments. Student’s t-test: *p<0.05 and **p<0.01. (D) WB of CRISPR/Cas9-engineered AFG3L2+/− HEK 293 T cells (clone #1) and controls showing levels of AFG3L2 and OPA1 with relative quantifications. AFG3L2 protein is reduced by 50% in AFG3L2+/− HEK 293 T cells and migrates at the same molecular weight compared with the wt controls, indicating that it is imported and undergoes correct maturation inside mitochondria. Bars represent means±SEM of four independent experiments. Student’s t-test: *p<0.05. SCA28, spinocerebellar ataxia type 28; wt, wild-type.
To understand the molecular basis of this mitochondrial morphology defect, we analysed by WB the total protein amount, as well as post-translational processing of the pro-fusion protein OPA1. At steady state, the eight splicing variants of OPA1 undergo constitutive processing at site 1 by OMA1-operated cleavage and/or at site 2 by YME1L-operated cleavage, leading to the accumulation of long (non-cleaved) and short forms. The OPA1 long forms have been demonstrated to be the active mediators of IMM fusion.30 31 We and others have previously shown that the absence of AFG3L2 destabilises OPA1 long forms compared with controls, leading to inhibition of mitochondrial fusion and mitochondrial network fragmentation.9 10 We observed SCA28 fibroblasts with mutated AFG3L2 have a significant reduction in the total amount of OPA1 (figure 2B and C). This phenotype is not the result of decreased mitochondrial mass or transcriptional downregulation (online supplementary figure S4A and B) but is likely due to faster processing and degradation of OPA1 in the presence of the AFG3L2 mutants. Of note, fibroblasts with the p.Y689H variant had reduced levels of OPA1 long forms but was not as significant compared with cells with other pathogenic variants (figure 2B). This could explain their unaltered mitochondrial morphology.
The comparable phenotypes (altered mitochondrial morphology and OPA1 processing) seen with p.L556fs fibroblasts and missense-carrying cells suggest haploinsufficiency as the cause of SCA28 instead of dominant negative mechanism. To prove it, we used CRISPR/Cas9-mediated editing to generate AFG3L2 haploinsufficient HEK 293 T cells. These AFG3L2+/− cells also had the same reduction of OPA1 protein amount observed in the SCA28 patient-derived cells (figure 2D: clone #1 and online supplementary figure S4D and E: clone #2), indicating that 50% reduction in m-AAA is capable of increasing OPA1 processing and alters mitochondrial morphology. Furthermore, the overexpression of the missense variants AFG3L2-p.F664S, AFG3L2-p.M666T or AFG3L2-p.G671R in Afg3l2 −/− MEFs did not recover mitochondrial fragmentation even partially, indicating that these variants have no residual activity. By contrast, the coexpression of wt AFG3L2 with AFG3L2-p.F664S, AFG3L2-p.M666T or AFG3L2-p.G671R restored mitochondrial tubulation in Afg3l2 −/− MEFs (online supplementary figure S4F), further indicating haploinsufficiency being the disease-causing mechanism for these variants.
Proteostatic stress in the IMM activates OMA1 in SCA28 cells
OMA1 has been shown to increase OPA1 cleavage in the absence of AFG3L2.9 In physiological conditions, OMA1 is a constitutively active protease, but it becomes hyperactivated in response to mitochondrial depolarisation and undergoes autocatalytic degradation.32 Although the link between the loss of AFG3L2 and OPA1 processing is well established,9 10 the OMA1-related mechanisms triggering the increased processing of OPA1 in this setting are unknown. Therefore, we measured endogenous OMA1 levels in patient-derived SCA28 and SPAX5 cells and found a significant reduction in OMA1 protein compared with controls (figure 3A). This result was not due to a change in OMA1 transcriptional levels (online supplementary figure S4C), which indicates that OMA1 becomes hyperactivated (and thus highly autocatalysed) when AFG3L2 activity is reduced, leading to excessive processing of OPA1 long forms. The collapse of ΔΨm is the major stimulus that induces OMA1-mediated processing of OPA1.32 However, ΔΨm was maintained in SCA28 and SPAX5 cells, as measured by cytofluorimetric evaluation of the steady-state loading of the potentiometric dye tetramethylrhodamine methyl ester (TMRM) (figure 3B). This suggests a different stimulus being responsible for OMA1 activation in this setting of reduced AFG3L2 activity. Recent data point to a role for AFG3L2 in regulating the stability of mitochondria-encoded proteins in the IMM.33–35 Therefore, we performed pulse-chase experiments, which revealed the accumulation of mitochondrial translation products in Afg3l2 −/− MEFs compared with controls (figure 3C). After 2 hours and 4 hours of chase, we observed specific mitochondria-encoded polypeptides having a higher stability in Afg3l2 −/− MEFs versus controls (ie, ND1, ND4, ND6 and ND4L/ATP8), with OMA1 levels decreasing proportionally with this stabilisation (figure 3C). To prove that the accumulation of mitochondria-encoded polypeptides was the stimulus for OMA1 activation, we treated p.L556fs cells, Afg3l2 −/− MEFs, AFG3L2+/− HEK 293 T cells and human fibroblasts knockdown for AFG3L2 with chloramphenicol, an inhibitor of mitochondrial protein synthesis. Interestingly, in all cases we observed a complete rescue of OPA1 long forms, which correlated with the stabilisation of OMA1 levels (figure 4A and online supplementary figure S5A–D). Of note, the restored OPA1 long form levels were associated with a recovery of mitochondrial network morphology in both SCA28 cells and Afg3l2 −/− MEFs (figure 4B). As further evidence of this mechanism, we treated Afg3l2 −/− MEFs for 24 hours with chloramphenicol and then performed pulse-chase experiments of mitochondrial translation products. Interestingly, in this setting, OMA1 protein levels were found to be stabilised immediately after pulse phase, but progressively decreased during the chase phase because of the reactivation of mitochondrial protein synthesis (figure 4C). In addition, we demonstrated that the efficacy of chloramphenicol in maintaining OPA1 long forms lasted for over 48 hours after treatment in Afg3l2 −/− MEFs (online supplementary figure S6A–C).

OMA1 is activated by accumulation of mitochondria-encoded polypeptides in AFG3L2-mutated and depleted cells. (A) WB of fibroblast cell lysates from control and AFG3L2 patients showing levels of OMA1 in controls and patients with relative quantifications. Bars represent means±SEM of three independent experiments. Student’s t-test: *p<0.05; **p<0.01. (B) Analysis of ΔΨm by cytofluorimetric evaluation of the potentiometric dye TMRM. Data are reported as the ratio between TMRM/(TMRM+FCCP). Bars represent means±SEM of at least three independent experiments. (C) Autoradiogram and WB analysis of the same membrane probed with antibodies directed against OMA1 and VDAC1 (loading control). In this experiment, Afg3l2−/− MEFs and controls were pulse-labelled with 35S-methionine/cysteine for 30 min and harvested or chased for 2 or 4 hours. WB, western blot.
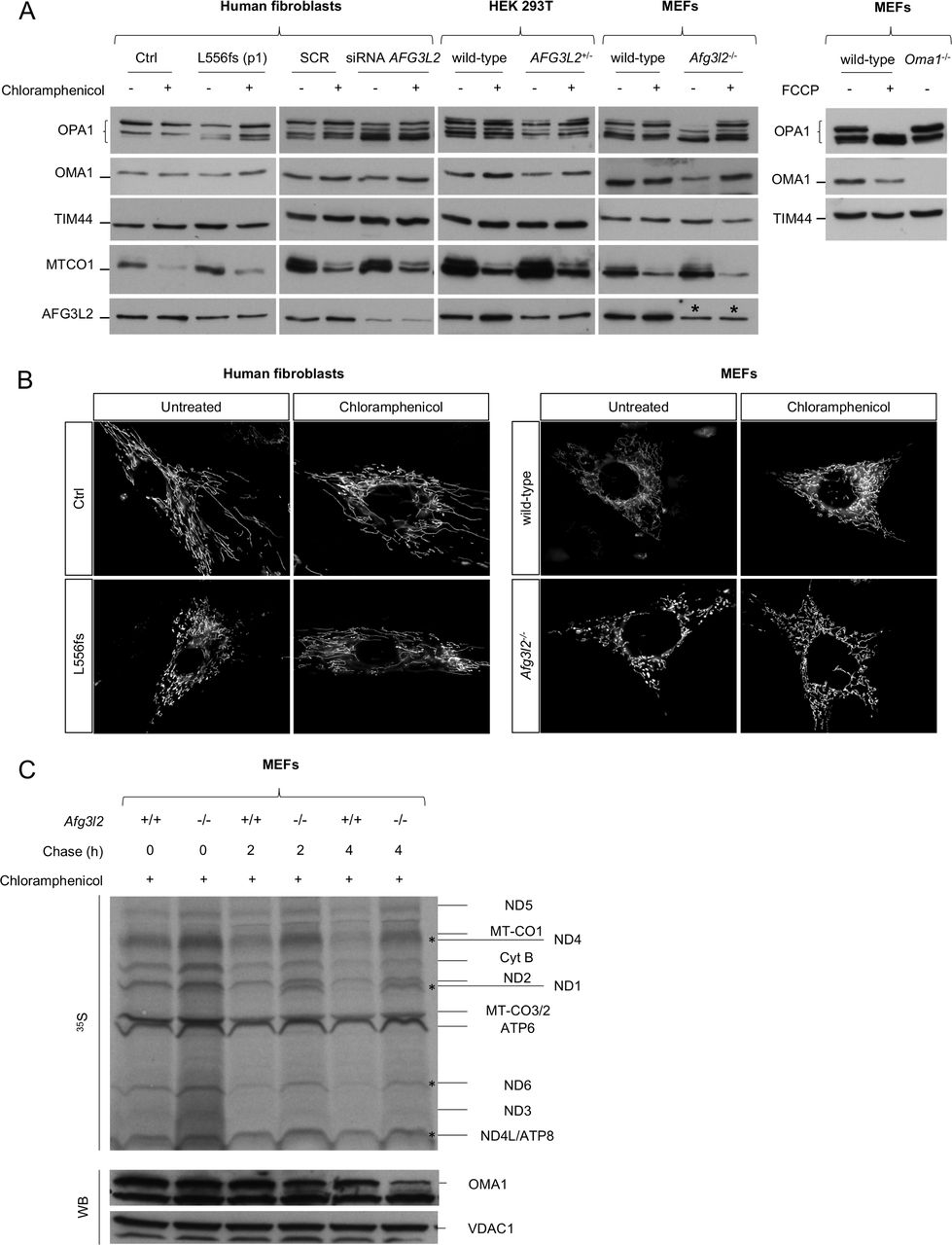
Rescue of OPA1 long forms and mitochondrial fragmentation by inhibition of mitochondrial protein synthesis in AFG3L2-mutated and depleted cells. (A) WB of fibroblast cell lysates showing levels of OPA1 and OMA1 in cells of the indicated genotype with or without chloramphenicol treatment. TIM44 was used to verify equal loading, and MTCO1 was used to verify inhibition of mitochondrial protein synthesis by chloramphenicol. FCCP was used to verify OMA1 activation. * represents the cross reactivity of anti-AFG3L2 antibody with murine AFG3L1 in mouse cells. (B) Representative pictures of mitochondrial morphology in SCA28-p.L556fs primary fibroblasts and Afg3l2 −/− MEFs in normal conditions and on chloramphenicol treatment. (C) Autoradiogram and WB analysis of the same membrane probed with antibodies directed against OMA1 and VDAC1 (loading control). In this experiment, Afg3l2−/− MEFs and controls were treated with chloramphenicol for 24 hours and then pulse-labelled with 35S-methionine/cysteine for 30 min and harvested or chased for 2 or 4 hours. MEFs, murine embryonic fibroblasts; SCA28, spinocerebellar ataxia type 28; WB, western blot.
Reduced mitochondrial calcium uptake in SCA28 patient fibroblasts
We previously reported that Afg3l2 −/− MEFs and primary PCs show a reduction in evoked mitochondrial calcium rises.10 11 Recently, this defect was reported also in HeLa cells lacking the m-AAA complex.12 In Afg3l2+/− mouse model, defective calcium handling was shown to alter calcium homeostasis in PCs, which led to DCD. This mechanism represents a crucial step in disease pathogenesis, therefore we assayed whether this defect was present in SCA28 and SPAX5 patient fibroblasts. To exclude differences in global cellular calcium signalling, [Ca2+]c was evaluated in patient cells and compared with controls. To this end, we performed fura-2 measurement of cytosolic calcium response after histamine stimulation, which leads to an inositol (1,4,5) triphosphate-triggered release of calcium from the ER stores. This analysis revealed no significant differences between SCA28 cells and controls (figure 5A). We then measured [Ca2+]m using a mitochondria-targeted version of the fluorescence resonance energy transfer-based calcium probe 4mtD1cpv.26On challenge with 100 µM histamine, mitochondrial calcium rises were significantly smaller in SCA28 patient cells as compared with controls (figure 5B,C).

Decreased mitochondrial calcium uptake in SCA28 fibroblasts. (A) Means±SEM of [Ca2+]c (peak responses) after histamine stimulation evaluated by the fura-2 fluorescence 340/380 ratio (fold increase above the basal value; n=3–5 for each cell line, an average of 15 traces analysed/experiment). (B) Means±SEM of [Ca2+]m (peak responses) after histamine stimulation (normalised increase in YFP/CFP ratio measured above the initial value; n=5, from 5 to 10 traces analysed/experiment). Student’s t-test: *p<0.05. (C) Representative traces of evoked mitochondrial calcium responses in SCA28 and SPAX5 fibroblasts expressing 4mtD1cpv. Traces from different cell lines have been shifted along the time-axis for clarity. SCA28, spinocerebellar ataxia type 28; SPAX5, spastic ataxia type 5.
Discussion
In this study, we examined the pathogenic mechanisms underlying SCA28 by analysing a panel of patient-derived primary fibroblasts with different AFG3L2 pathogenic variants. These included several missense (p.M666T, p.G671R, p.Y689H and the newly identified p.F664S) and frameshift (p.L556fs) variants. We assayed in these SCA28 fibroblasts all the functional aspects of mitochondrial physiology that so far have been linked to m-AAA activity (ie, mitochondrial morphology, OPA1 processing, OMA1 activation, mitochondrial protein synthesis, ΔΨm and calcium buffering). By comparing SCA28 fibroblasts with cells with reduced m-AAA activity (SPAX5 patient fibroblasts carrying the homozygous p.Y616C pathogenic variant, AFG3L2+/− HEK293 T cells generated by CRISPR/Cas9 genome editing and Afg3l2 −/− MEFs) in regard of these phenotypes, our data indicate haploinsufficiency as the primary pathogenic mechanism in these SCA28 cells as opposed to a dominant negative process. Also, our findings identified the earliest steps in SCA28 pathogenic cascade, which may provide valuable targets for future therapy.
We demonstrated that p.L556fs fibroblasts possessed half of the amount of assembled m-AAA complexes (1MDa) compared with controls, being therefore functionally haploinsufficient. In contrast, cells from subjects with missense variants had similar steady-state levels of AFG3L2 monomers and assembled m-AAA complexes compared with controls. Interestingly, almost all AFG3L2 pathogenic variants we studied had similar mitochondrial phenotypes regardless of the total amount of AFG3L2 monomers or assembled m-AAA complexes. These phenotypes included altered mitochondrial morphology associated with increased processing of OPA1 long forms by hyperactive OMA1. These results indicated that inefficient mitochondrial fusion is an important cause of AFG3L2-related pathogenesis. Of note, the only cells that did not have significantly reduced levels of OPA1 long forms and altered mitochondrial network morphology were derived from a subject (p.Y689H) with a milder clinical phenotype compared with the other SCA28 patients. These data may indicate that OPA1-mediated mitochondrial fragmentation could be a cellular phenotype that correlates with disease severity.
We also measured mitochondrial calcium uptake in SCA28 and SPAX5 cell lines because it was previously demonstrated to be the trigger of PC-DCD in the Afg3l2 haploinsufficient mouse.11 We found it reduced in SCA28 and SPAX5 cells, even though ΔΨm was unchanged. This result demonstrates that faulty mitochondrial calcium uptake could play a crucial role in SCA28 cellular pathogenesis and that it is likely a consequence of altered mitochondrial morphology, as cells with the p.Y689H pathogenic variants do not have any significant changes with these two parameters.
Mitochondrial abnormalities are seen to comparable extent in cells expressing missense changes or in the haploinsufficient ones, indicating that a dominant negative effect is unlikely. Indeed, if a dominant negative effect was the cause of the pathology, we would expect the overall activity of the m-AAA complexes to be more severely compromised than what we see with our missense and truncated mutants. Our data indicate that the missense variants we have studied are heterozygous loss of function variants that reduce m-AAA complexes functionality. Indeed, when overexpressed in a null Afg3l2 background, they failed to recover mitochondrial fragmentation, not even partially.
The extent of m-AAA complexes reduction is however hard to be precisely quantified. This is because it depends on the stoichiometry of individual subunit types in homo-oligomeric m-AAA complexes composed of AFG3L2 and in hetero-oligomeric m-AAA complexes composed of AFG3L2 and paraplegin. Studies have demonstrated that m-AAA proteases are organised in hexamers,36 but how AFG3L2 and paraplegin subunits assemble in the m-AAA complexes, in terms of relative amount and reciprocal disposition, has not been clarified yet, even though it is known to be influenced by the relative abundance of subunits in distinct tissue types.4 Therefore, depending on the ratio between wt and mutated AFG3L2 subunits in homo-oligomeric complexes, as well as between wt and mutated AFG3L2 subunits with paraplegin in hetero-oligomeric complexes, we hypothesise that a gradient of reduced activity of m-AAA complexes occurs in SCA28. Further evidence was provided with AFG3L2+/− HEK 293 T cells, wherein two different clones had AFG3L2 protein reduced by 50%. These cells also had decreased levels of OPA1 long forms and OMA1 as seen in patient cells. This result, together with AFG3L2 truncating and missense pathogenic variants expressing similar effects on mitochondrial physiology, indicate the overall residual activity of the m-AAA should be around 50% or more, consistent with haploinsufficient mechanism. Although strongly supported by our functional data, a haploinsufficient mechanism is not in agreement with the higher than expected frequency of AFG3L2 loss of function variants in ExAC database (http://exac.broadinstitute.org). Many explanations, such as bias in sequencing, CNVs, as well as the age of individuals (ie, sequencing was done prior to the onset of symptoms in the late-onset disorder) and clinical misdiagnosis may account for this discrepancy. Indeed, SCA28 is a mild and slowly progressing form of ataxia, frequently underdiagnosed. Of note, all the pathogenic variants that we analysed in this study are located within the proteolytic domain of AFG3L2; therefore, we can only comment on these types of mutations and cannot exclude that variants located in other AFG3L2 domains may act through a dominant negative effect. The p.Y689H variant had a milder cellular phenotype compared with the other missense variants. We may speculate that the p.Y689 residue, being located almost 20 aminoacids downstream the other studied variants, affects the proteolytic activity of AFG3L2 in some milder form.
We included in this study fibroblasts from a SPAX5 patient carrying the AFG3L2 homozygous recessive variant p.Y616C. At the mitochondrial level, the phenotype of SPAX5 cells were comparable with those of SCA28 and AFG3L2+/− HEK 293 T cells but markedly milder compared with Afg3l2 −/− MEFs. These data suggest that the p.Y616C mutant is a pathogenic hypomorph and is not a complete loss of function variant, and so requires both alleles to be affected to cause AFG3L2-related disease. This is also consistent with the asymptomatic status of both the heterozygous parents of this SPAX5 patient,21 although they may be presymptomatic. However, this hypothesis is in contrast with the very severe neurological phenotype of the SPAX5 patient.21 One explanation could be that in the different neuronal populations affected in SPAX5, the homozygous p.Y616C pathogenic variant causes a more severe phenotype compared with fibroblasts. We should also consider that other genes may contribute/modify the clinical phenotype in this consanguineous pedigree.
We found abnormal mitochondrial morphology as a unifying element of both SCA28 and SPAX5 pathogenesis. In both disorders, cells lose the highly tubulated and interconnected mitochondrial network as a result of decreased levels of OPA1 long forms, which is essential for efficient fusion of the IMM.30 31 While it has been previously shown that OMA1 is the protease mediating increased OPA1 processing in the absence of AFG3L2,9 the detection of activated OMA1 in condition of absence/pathogenic variant of AFG3L2 or the stimuli that trigger this activation remains elusive.
We found that OMA1 has increased activation in SCA28 and SPAX5 cells, as well as in AFG3L2+/− HEK 293 T cells, in human fibroblasts treated with AFG3L2 siRNA and in Afg3l2 −/− MEFs, to a higher extent. The trigger for OMA1 activation in SCA28 and SPAX5 cells was not a drop of ΔΨm, which we verified conserved in patient cells and we previously found unaltered in Afg3l2 −/− MEFs.10 Our data indicate that the primary stressor regulating OMA1 activation in SCA28 is the accumulation of mitochondria-encoded polypeptides. Several reports indicate that m-AAA proteases are important for preventing an overaccumulation of mitochondria-encoded polypeptides.33–35 Data form pulse-chase experiments indeed revealed the stabilisation of specific mitochondrial translation products in the absence of AFG3L2 over time, which correlated with decreased levels of OMA1. Chloramphenicol-treatment studies supported this mechanism, as we found that this drug stabilises OMA1 levels, OPA1 long forms and rescues mitochondrial elongation.
Our data reveal the fine titration of OPA1 turnover likely depends on the residual amount of functional m-AAA complexes. In SCA28 cells and AFG3L2+/− HEK 293 T cells, where there is at least 50% of residual functional m-AAA, there was no accumulation of short forms despite OPA1 processing being accelerated. In this context, it seems likely that other proteases in the intermembrane space may be working efficiently to prevent accumulation of soluble OPA1 short forms. However, OPA1 short forms accumulated in Afg3l2−/− MEFs, where residual function of m-AAA is below the critical level of 50%, indicating that mitochondria are more severely damaged and overall protein quality control is compromised, leading to a slower turnover of OPA1 short forms. Together, these data strengthen the concept that the amount of OPA1 long forms is the key mediator for efficient mitochondrial fusion. Consistent with this finding, OPA1+/− cells show increased mitochondrial fragmentation despite unaltered OPA1 processing.37 38
Another important finding of this work is that mitochondrial calcium uptake is reduced in SCA28 and SPAX5 fibroblasts. We had previously shown that primary murine Afg3l2 −/− PCs also have reduced mitochondrial calcium uptake, leading to disruption of calcium homeostasis and activation of PC-DCD. This mechanism was consistent with previous data where treatment with ceftriaxone, a drug capable of blocking calcium influx in PCs by reducing glutamatergic stimulation, was effective in rescuing the ataxic phenotype of the Afg3l2 +/− haploinsufficient mice.11 In the current work, we demonstrated similar findings, which indicate that ceftriaxone therapy may be beneficial for SCA28 or SPAX5 patients.
In conclusion, this study established the physiopathological cascade of events in SCA28 primary fibroblasts, which included: (1) proteostatic stress as the major trigger for OMA1 activation, leading to (2) OPA1 overprocessing, (3) mitochondrial fragmentation and (4) decreased calcium buffering (our model is summarised in figure 6). Because fibroblasts are not the primary site of affection in SCA28, future studies in primary neurons and preclinical models are required to reconfirm this proposed cascade of events and to determine if attenuation of mitochondrial synthesis may represent a new translational perspective for AFG3L2-associated neurodegeneration.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Model explaining the pathogenic cascade of events in SCA28 cells. In physiological conditions, the amount of mitochondria-encoded proteins is controlled, and OMA1 activity is limited, leading to normal OPA1 processing, efficient mitochondrial fusion and calcium buffering. In condition of decreased AFG3L2 activity, there is accumulation of mitochondria-encoded proteins, with proteostatic stress leading to OMA1 hyperactivation and autocatalysis, enhanced OPA1 processing, inefficient mitochondrial fusion and decreased calcium buffering. SCA28, spinocerebellar ataxias type 28.
Supplementary file 1
Supplementary file 2
Acknowledgments
We would like to thank SCA28 patients collaborating with this study. We are grateful to Laura Cassina and Luca Rampoldi for scientific discussion. We would like to thank Giancarlo Zerbini for providing primary fibroblasts from healthy subjects. We are grateful to Fabiana Longo, Daniele De Ritis and Maurizio De Fusco for technical assistance. Oma1-/- MEFs cells were kindly provided by Professor C Lopez Otin, University of Oviedo (Spain); 4mtD1 construct was kindly provided by Professor T Pozzan, University of Padova, Italy. This project was supported by Italian Ministry of Health #GR-2011-02351638 and FY2014 5x1000 funds (FM); Association Belge contre les Maladies Neuromusculaire – Aide à la Recherche ASBL #2017-2018/05 and Research Foundation – Flanders #1805016N (JB), Nando and Elsa Peretti Foundation (GC) and Cedars-Sinai Medical Center Institutional Funding and the Diana and Steve Marienhoff Fashion Industries Guild Endowed Fellowship in Pediatric Neuromuscular Disease and the Undiagnosed Disease Program (TMP).
References
Footnotes
ST and ADB contributed equally.
Contributors FM and GC conceived the study, interpreted the results and wrote the manuscript. TMP revised and edited the manuscript. ST and ADB performed the experiments, analysed data and helped writing the manuscript. VB, DM and FC performed the experiments. AA performed statistical analysis of data. DN, CG, TMP, CT, JB, TD, PD and PM provided primary fibroblasts or skin biopsies from patients. All authors critically discussed the data.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Ethics approval The local institutional review boards approved this study.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.