1Servio de Endocrinologia, Hospital Central do Funchal, Portugal; 2Servicio de Pediatrίa, Hospital Clίnico de Ourense, Spain; 3Servio de Pediatria, Hospital Pedro Hispano, Matosinhos, Portugal; 4Fundaciόn Pública Galega de Medicina Xenόmica, 5Departamento de Pediatrίa, Hospital Clίnico Universitario de Santiago de Compostela, 6Unidad de Endocrinologίa Pediátrica y Crecimiento, Departamento de Pediatrίa, Hospital Clίnico Universitario de Santiago de Compostela, Universidad de Santiago de Compostela; Santiago de Compostela, Spain
Autoimmune polyglandular syndrome type 2 (type 2 APS), or Schmidt’s syndrome, is defined by the presence of Addison’s disease in combination with type 1 diabetes and/or autoimmune thyroid disease. The estimated prevalence of this syndrome is 1.4-4.5 per 100,000 inhabitants and it is more frequent in middle-aged females, whilst it is quite rare in children. Type 2 APS, which shows a pattern of autosomal dominant inheritance with low penetrance, has been associated with HLA specific DR3/DQ2 and DR4/DQ8 haplotypes. However, it has been hypothesized that genetic variability in the AIRE gene, which causing type 1 APS, may play a role in more common organ-specific autoimmune conditions like type 1 diabetes, Hashimoto’s disease and type 2 APS, among others. Here we present the case of an 8-year-old girl, with a past medical history of type 1 diabetes diagnosed at the age of 3. She was taken to the Emergency Department because she complained of abdominal pain, nausea and vomiting, and her blood analysis revealed a severe hyponatremia. She also had seizures as a consequence of the hyponatremia and frequent hypoglycemia. She was ultimately found to be suffering from autoimmune primary adrenal insufficiency. The combination of both mentioned conditions, type 1 diabetes and Addison’s disease, in the absence of chronic mucocutaneous candidiasis, made a diagnosis of type 2 APS plausible in this girl. The genetic study showed two heterozygous variants: NM_000383.2:C.1411C>T (p. Arg471Cys) in exon 12 and IVS9+6G>A in intron 9 of the AIRE gene. The description of an uncommon case of type 2 APS with precocious presentation associated with an AIRE mutation in a very young girl could help to clarify the role of AIRE in the development of autoimmune diseases.
AIRE gene, Autoimmune polyglandular syndrome, Autoimmune Regulator gene, Child, Mutation, Schmidt’s syndrome, Type 2 APS
INTRODUCTION
The combined occurrence of Addison’s disease and chronic lymphocytic thyroiditis was first reported by Schmidt in 1926,1 while Carpenter noticed the frequent occurrence of diabetes in patients with Schmidt’s syndrome.2 Addison’s disease may occur as a component of two autoimmune polyglandular syndromes: type 1 APS and type 2 APS. Type 1 APS is defined by the presence of chronic mucocutaneous candidiasis, hypoparathyroidism and Addison’s disease with an incidence lower than 1 in 100,000 inhabitants. Type 2 APS, or Schmidt’s syndrome, is characterized by the obligatory occurrence of Addison’s disease in combination with autoimmune thyroid dysfunction and/or type 1 diabetes. In addition, it can also be associated, inter alia, with other autoimmune disorders like pernicious anemia, premature ovarian failure, vitiligo, alopecia and myasthenia gravis. Type 2 APS is a rare syndrome with an estimated prevalence of 1.4–4.5 per 100,000 inhabitants. Females are three times more frequently affected than males and it is commonly found in middle age, while it is quite rare in children. On the other hand, APS type 1 usually appears in childhood or teenage years.2-5
Other autoimmune polyglandular syndromes are type 3 APS (thyroid autoimmune diseases associated with other autoimmune diseases, excluding Addison’s disease and/or hypoparathyroidism) and type 4 APS (combinations of organ-specific autoimmune diseases not included in the previous group).6
Type 1 APS is a monogenic autosomal recessive disease caused by mutations in the Autoimmune Regulator gene (AIRE).7,8 By contrast, type 2 APS is more complex and the pattern of inheritance appears to be autosomal dominant with incomplete penetrance.2 It has been associated with class II HLA alleles, in particular with DR2/DQ2 and DR4/DQ8.9,10 However, it has been hypothesized that genetic variability in the AIRE gene may play a role in more common organ-specific autoimmune conditions like type 1 diabetes, Hashimoto’s disease and type 2 APS.3,8,11
CASE REPORT
An 8-year-old girl, with a history of type 1 diabetes diagnosed when she was 3 years old, was taken to the Emergency Department complaining of abdominal pain, nausea and vomiting. She reported no diarrhea, fever or dysuria. She had carried a portable continuous insulin perfusion system since the age of 6 years old because of her frequent episodes of hypoglycemia; in the last two months her insulin needs had been reduced due to more frequent hypoglycemia. Her physical examination showed her height to be 117 cm (-2.14 SDS), her weight 22 Kg (-0.81 SDS), her blood pressure 96/55 mmHg, her heart rate 139 bpm and her auricular temperature 37°C. She was lethargic and dehydrated. The rest of her physical examination was unremarkable. Her blood analysis revealed high glucose (330 mg/dl) and hyponatremia of 111mEq/L (Table 1). The venous arterial blood gases showed a pH of 7.32, pO2 of 26.9 mmHg, pCO2 of 41 mmHg, HCO3- of 21 mmol/L and lactates of 2.3 mmol/L. The urinalysis showed a discrete elevation of ketones (10 mg/dl).
She was started on an
infusion of hypertonic saline (2% NaCl) and regular insulin 0.1 U/Kg/h. The
patient subsequently had seizures, which were attributed to the hyponatremia.
The lumbar puncture and the cerebral computed tomography (CT) revealed no
alterations. She had seizures again, this time in the context of an attack of
hypoglycemia, thus the insulin perfusion was stopped and 5% dextrose perfusion
was initiated. When the clinical situation stabilized, she was admitted to the
Pediatrics Department and started again on hypertonic saline; her sodium level subsequently
increased (Table 2) and the perfusion of insulin stabilized at 0.3 U/Kg/h;
however, some episodes of hypoglycemia persisted (Figure 1). At that point a
cosyntropin (Synachten®) stimulation test was performed that
revealed an immeasurable level of cortisol (Table 3). The adrenocorticotrophic
hormone (ACTH) was extremely elevated (>1,250 µg/dl). The remainder of her
hormonal profile showed an immeasurable plasmatic aldosterone and
11-deoxycortisol and elevated renine (Table 4).

Figure 1.
She was diagnosed with primary adrenal insufficiency and was started on hydrocortisone: 5mg/Kg every 6 hours on the first day, which was then gradually reduced until attaining the maintenance dose (15 mg/m2/day divided in 3 doses). She was also started on fludrocortisone (0.1 mg/day). The antibodies against adrenal cortex were positive, confirming the diagnosis of Addison’s disease. The patient had no history of chronic mucocutaneous candidiasis and her thyroid function, parathyroid hormone and calcium levels were all normal; antimicrosomal antibodies were negative. Antibodies against intrinsic factor, antithyroid, anti-transglutaminase and anti-endomysium were all negative.
Genetic analysis
For the genetic analysis, DNA of the patient was obtained from peripheral leukocytes by chemagic DNA Blood100 Kit (Perkin Elmer Chemagen). All the AIRE coding region as well as intron exon boundaries were subjected to PCR amplification and direct sequencing using the BigDye terminator kit and were run in the 3730 xl DNA Analyzer (Applied Biosystems, Foster City, CA U.S.A.).
This analysis showed
two heterozygous variants: NM_000383.2:C.1411C>T (p. Arg471Cys) in exon 12
(Figure 2) and IVS9+6G>A in intron 9 of the AIRE gene. No other
mutation was found.
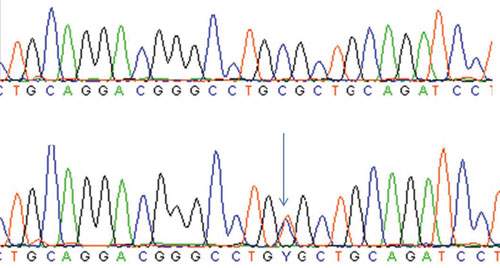
Figure 2.
DISCUSSION
In the present case report, we describe a young patient with a diagnosis of type 1 diabetes who was evaluated due to the occurrence of frequent hypoglycemia, this determining the need for a continuous insulin perfusion system, and the occurrence of unexplained hyponatremia. These conditions constituted the main reasons for consideration of a diagnosis of Addison’s disease, which was confirmed by an immeasurable level of plasmatic cortisol after the cosyntropin (Synacthen®) stimulation test. Interestingly, the patient did not have skin hyperpigmentation, although her ACTH level was high. The combination of both mentioned conditions, type 1 diabetes and Addison’s disease, in the absence of chronic mucocutaneous candidiasis, made a diagnosis of type 2 APS plausible.
In 1981, Neufeld12 reported that, in the vast majority of the population studied, this syndrome began after 20 years of age, with an increased prevalence in middle-aged women. Thyroid autoimmune diseases were present in 69% and type 1 diabetes in 52% of the cases. Here we present a singular case occurring in an 8-year-old girl with a combination of type 1 diabetes and Addison’s disease with 5 years’ lapse of presentation between the two autoimmune diseases.
Type 2 APS, which shows a pattern of autosomal dominant inheritance with low penetrance, has been associated with HLA specific DR3/DQ2 and DR4/DQ8 haplotypes. On the other hand, type 1 APS, or autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), is an autosomal recessive monogenic disease caused by AIRE gene mutations. However, it has been hypothesized that mutations in the AIRE gene may play a role in more common organ-specific autoimmune conditions like type 1 diabetes, Hashimoto’s disease and type 2 APS, among others.10,13
Taking into account this hypothesis and the precocious presentation of the disease in the patient, we decided to study the AIRE gene. This study showed that the patient was a double heterozygote for the transition c.1411C>T and IVS9+6G>A. It is not possible to exclude completely the presence of a second mutation in the regulatory or intronic regions of the AIRE gene, which were not analyzed; nevertheless, no mutation in these regions has been described so far.
The c.1411C>T would result in the substitution of arginine at position 471 by cysteine at the protein level (p.Arg471Cys). This variant is registered in the single nucleotide polymorphism (SNP) database as the rs74203920 with a minor allele frequency (T) ranging from 0.001 to 0.015 depending on the population studied, which does not necessarily indicate that it is a functional polymorphism. In fact, the in silico predictions of the effect of the aminoacid substitution in the protein function (Align GVGD, SIFT, MutationTaster and Polyphen) suggest that this variant is a deleterious or disease-causing one. These prediction methods are based on the degree of conservation of aminoacid residues in multiple sequence alignments from closely related sequences and the biophysical characteristics of the aminoacids. Moreover, this mutation was described in heterozygosis in a patient with chronic hypoparathyroidism, transient diabetes insipidus, chronic thyroiditis and evidence of circulating parietal cell antibodies.8,11,14 The compound heterozygous state of IVS9+6G>A/c.1411C>T was found in another patient with adrenal failure and autoimmune thyroiditis.3,8,11 It is important to note that our patient is also heterozygous for the IVS9+6G>A (c.1095+6G>A, nomenclature following the Human Genome Variation Society recommendations), although we consider this irrelevant because this is a genetic polymorphism (rs1800525) with a minor allele frequency (A) among European-American populations of 8.49% and the in silico splicing predictions (Alamut Module 2.4) indicates that it has no effect on normal splicing. In fact, Toth et al3 also consider this variant an innocent polymorphism. These results suggest that the c.1411C>T variant must be an important factor for the development of the disease. Further studies are needed to discover what other genetic or environmental factors are responsible for the different genotypes among the carriers of this mutation and for the uncommon early presentation in our patient.
In summary, we present an uncommon case of type 2 APS in a child associated with an AIRE mutation. This study could contribute to elucidation of the role of AIRE in the development of autoimmune diseases.
CONFLICT OF INTEREST
The authors have nothing to declare.
REFERENCES
2. A Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L, 2013 Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens) 12: 39-45.
3. Toth B, Wolff ASB, Halasz Z, et al, 2010 Novel sequence variation of AIRE and detection of interferon – omega antibodies in early infancy. Clin Endocrinol 72: 641-647.
4. Betterle C, Dal Pra C, Mantero F, Zanchetta R, 2002 Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocrinol Rev 23: 327-364.
5. Zak T, Noczynska A, Wasikowa R, Zaleska-Dorobisz U, Golenko A, 2005 Chronic autoimmune thyroid disease in children and adolescents in the years 1999 – 2004 in Lower Silesia, Poland. Hormones (Athens) 4: 45-48.
6. Eisenbarth GS, Gottlieb PA, 2004 Autoimmune polyendocrine syndromes. N Engl J Med 350: 2068-2079.
7. Nagamine K, Peterson P, Scott HS, et al, 1997 Positional cloning of the APECED gene. Nat Genet 17: 393-398.
8. Palma A, Gianchecchi E, Palombi M, et al, 2013 Analysis of the autoimmune regulator gene in patients with autoimmune non-APECED polyendocrinopathies. Genomics 102: 163-168.
9. Fourati H, Mahfoudh N, Abida O, et al, 2011 HLA-DRB1/DQB1 susceptibility for autoimmune polyglandular syndrome type II and III in south of Tunisia. Ann Endocrinol (Paris) 72: 232-238.
10. Cutolo M, 2014 Autoimmune polyendocrine syndromes. Autoimmun Rev 13: 85-89.
11. Fierabracci A , 2011 The role of heterozygous mutations of the autoimmune regulator gene (AIRE) in non-APECED autoimmunity: a comment on recent findings. Clin Endocrinol (Oxf) 74: 532-553.
12. Neufeld M, Maclaren NK, Blizzard RM, 1981 Two types of autoimmune Addison’s disease associated with different polyglandular autoimmune (PGA) syndromes. Medicine (Baltimore) 60: 355-362.
13. Maurer A, Schwarting A, Kahaly GJ, 2011 Polyglandular autoimmune syndromes. Z Rheumatol 70: 752-759.
14. Cervato S, Morlin L, Albergoni MP, et al, 2010 AIRE gene mutations and autoantibodies to interferon omega in patients with chronic hypoparathyroidism without APECED. Clin Endocrinol 73: 630-636.
Address for correspondence:
Eduarda Resende, Avenida do Amparo, Edifício Monumental Residence, Bloco 1 B – AA, 9000 – 783, Funchal, Portugal, Tel.: +351919837755, E-mail: edu.bene84@gmail.com
Received: 26-06-2014, Accepted: 02-07-2014