 Guillermo Blanco1,2,3
Guillermo Blanco1,2,3 Borja Sánchez2Florentino Fdez-Riverola1,3,4
Borja Sánchez2Florentino Fdez-Riverola1,3,4 Abelardo Margolles2
Abelardo Margolles2 Anália Lourenço1,3,4,5*
Anália Lourenço1,3,4,5*- 1ESEI – Department of Computer Science, University of Vigo, Ourense, Spain
- 2Department of Microbiology and Biochemistry of Dairy Products, Superior Council of Scientific Investigations, Institute of Dairy Products of Asturias, Villaviciosa, Spain
- 3SING Research Group, Galicia Sur Health Research Institute (IIS Galicia Sur), SERGAS-UVIGO, Vigo, Spain
- 4Centre for Biomedical Research, University of Vigo, Vigo, Spain
- 5Centre of Biological Engineering, University of Minho, Braga, Portugal
This work presents a novel in silico approach to the prediction and characterization of the glycolytic capacities of the beneficial intestinal bacterium Faecalibacterium prausnitzii. Available F. prausnitzii genomes were explored taking the glycolytic capacities of F. prausnitzii SL3/3 and F. prausnitzii L2-6 as reference. The comparison of the generated glycolytic profiles offered insights into the particular capabilities of F. prausnitzii SL3/3 and F. prausnitzii L2-6 as well as the potential of the rest of strains. Glycoside hydrolases were mostly detected in the pathways responsible for the starch and sucrose metabolism and the biosynthesis of secondary metabolites, but this analysis also identified some other potentially interesting, but still uncharacterized activities, such as several hexosyltransferases and some hydrolases. Gene neighborhood maps offered additional understanding of the genes coding for relevant glycoside hydrolases. Although information about the carbohydrate preferences of F. prausnitzii is scarce, the in silico metabolic predictions were consistent with previous knowledge about the impact of fermentable sugars on the growth promotion and metabolism of F. prausnitzii. So, while the predictions still need to be validated using culturing methods, the approach holds the potential to be reproduced and scaled to accommodate the analysis of other strains (or even families and genus) as well as other metabolic activities. This will allow the exploration of novel methodologies to design or obtain targeted probiotics for F. prausnitzii and other strains of interest.
Introduction
The high complexity of the human gut microbiota and the lack of know-how on all the individual players is a key challenge in gut microbiome research. Attempts have been made to define the microorganisms that constitute a healthy human gut microbiota but the scientific community has not yet reached a consensus to define the human beneficial intestinal microbial fingerprint. However, a number of observational studies have repeatedly shown a correlation between some bacterial populations and different physiological states, including those having an influence on human health. Specifically, some groups of strict anaerobes, such as Faecalibacterium prausnitzii, are found to be underrepresented in physiological conditions in which inflammation and oxidative stress are present (Joossens et al., 2011; Morgan et al., 2012; Gevers et al., 2014). Thus, during the last decade, F. prausnitzii research experienced a boosting, and this microorganism emerged as one of the most promising next-generation probiotics (O’Toole et al., 2017).
The species Fusobacterium prausnitzii was reclassified in 2002 by Stewart and co-workers, who proposed the new species F. prausnitzii (Stewart et al., 2002). F. prausnitzii is highly prevalent in the large intestine of humans and represents a significant proportion of the fecal anaerobes that produce major quantities of butyrate (Sokol et al., 2008; Arumugam et al., 2011). It is part of the autochthonous mucosal bacteria inhabiting the colon and its population has been found to be decreased in several diseases, particularly in those with mucosal inflammation, such as the inflammatory bowel disease or the type 2 diabetes (Ahmed et al., 2007; Sokol et al., 2008; Machiels et al., 2014; Tilg and Moschen, 2014; Rossi et al., 2016). In fact, it was demonstrated that F. prausnitzii has anti-inflammatory activity, likely due to its capacity to induce the secretion of the anti-inflammatory cytokine IL-10 in human immune cells and thus modulate T-cell responses (Rossi et al., 2016), as well as to produce microbial anti-inflammatory proteins able to inhibit the NF-κB pathway in intestinal epithelial cells (Quévrain et al., 2016), or extracellular polysaccharides able to attenuate clinical parameters of colitis (Rossi et al., 2015).
Microbiota studies using massive sequencing methodologies, either metataxonomics or metagenomics analyses, point to a high abundance of Faecalibacterium in the human gut (Arumugam et al., 2011). However, since F. prausnitzii is very sensitive to low oxygen concentrations and possesses a low technological robustness for industrial purposes, it is difficult to isolate from fecal samples and to grow using traditional cultivation methods. Also, although different phylogroups can be distinguished within the species (Benevides et al., 2017), the phenotypic characterization of strains has been restricted to a few isolates.
Metabolic maps have been generated using as the strain A2-165 (DSM17677T) as reference (El-Semman et al., 2014; Rowland et al., 2018), but the F. prausnitzii pangenome has never been explored to describe the metabolic capabilities of the genus and be able to develop novel microbial cultivation strategies. Therefore, the aim of the present work was to investigate the genome-based metabolic potential of F. prausnitzii, based on a deep prediction of the glycolytic capacities of this bacterium. The F. prausnitzii genomes currently deposited in the NCBI Genome Database were explored, taking the metabolisms of F. prausnitzii SL3/3 and F. prausnitzii L2-6 and data on all known glycoside hydrolases as references. To the best of our knowledge, this is the first time that a high throughput bioinformatics approach is carried out to predict the glycolytic capacities of F. prausnitzii.
Materials and Methods
The primary aim of the computational workflow developed in this work was to support the investigation of glycoside hydrolase activities in F. prausnitzii. Complimentarily, programmatic access to public data sources and the use of broad scope genomic and metabolic tools were sought as means to make this study completely reproducible as well as be able to apply this workflow to similar studies in the future. The proposed workflow (Figure 1) encompasses three main steps, i.e., data retrieval, data integration and data analysis. The primary data sources were the carbohydrate-active enzymes database (CAZy), the kyoto encyclopedia of genes and genomes (KEGG), and the Genome database of National center for biotechnology information (NCBI). Data integration was based on the matching of the glycoside hydrolase sequences against the sequences of the proteins encoded in the genomes of F. prausnitzii strains. As a rule of thumb, a threshold of 80% was used as baseline to identify most prominent activities in the different strains. Protein homology data were used to segregate clusters of glycoside hydrolases and strains for further pattern analysis (notably, based on the obtained identity percentage). Moreover, gene neighborhood maps were generated for the genes coding for some relevant glycoside hydrolases. Experts curated the results obtained throughout the execution of the workflow towards presenting a comprehensive portrait of current knowledge on glycoside hydrolases activities in F. prausnitzii as well as ensuring the validity of the overall process of analysis. The next sections describe the main steps in the workflow, including processing statistics, methods, and Supplementary Material produced during the analysis.
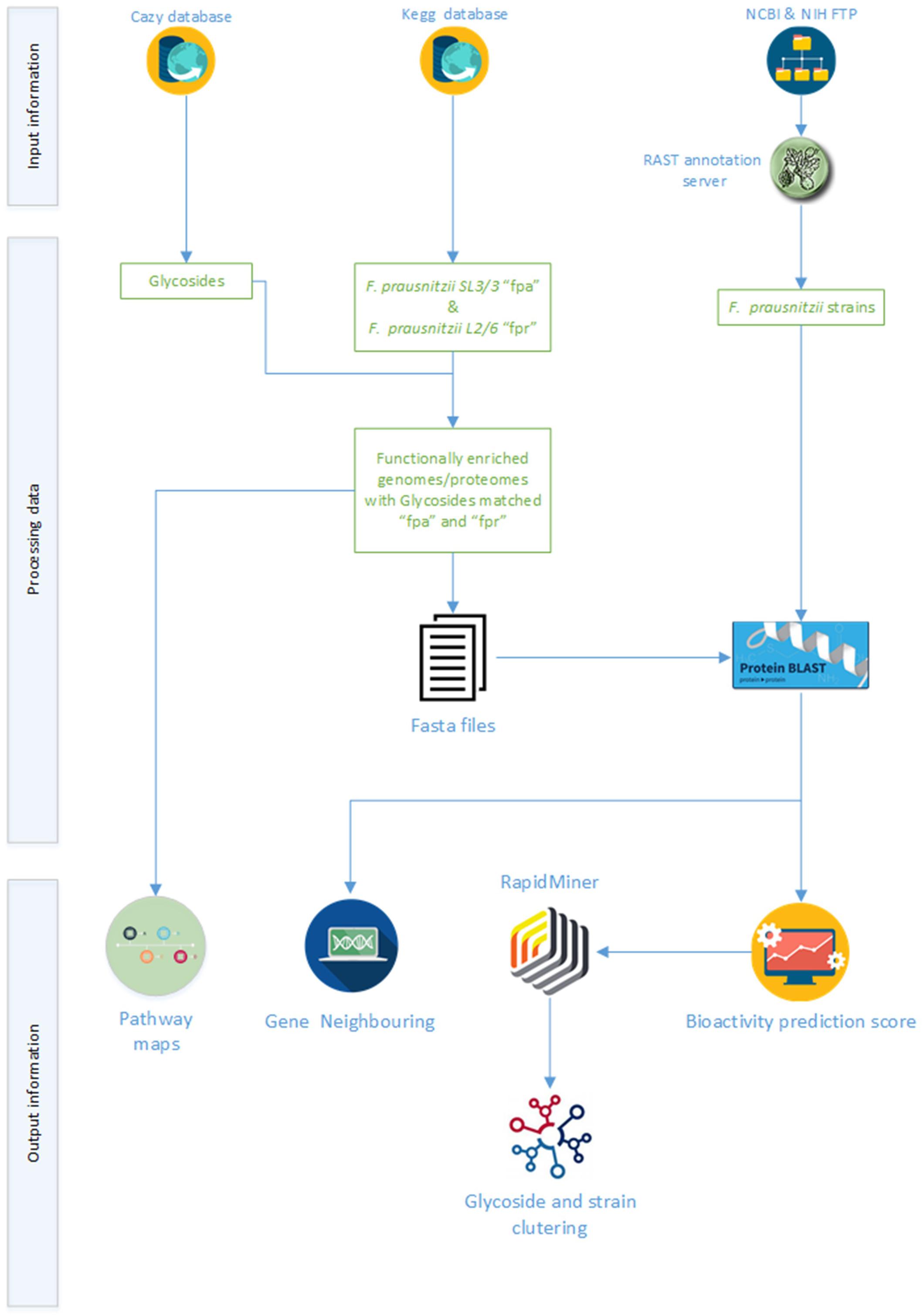
Figure 1. Schematic of the computational workflow supporting the analysis of the glycolytic potential of available F. prausnitzii genomes.
Glycoside Hydrolases and Reference Strains
Data on the glycoside hydrolase families were retrieved from the CAZy (Cantarel et al., 2009). From the families present in the CAZy database, only those that have associated fully specified Enzyme Commission numbers (EC number) (Tipton, 1994) were considered (i.e., enzyme codes including four numerical classes separated by periods).
Data on the metabolism of F. prausnitzii strains were retrieved from the KEGG (Kanehisa et al., 2017) using its programmatic API. Two strains of F. prausnitzii were used as reference in the present study, i.e., F. prausnitzii SL3/3 (identified hereinafter as “fpa”) and F. prausnitzii L2-6 (identified hereinafter as “fpr”). Protein and gene information was retrieved for all the previously selected glycoside hydrolases, including definition (i.e., reference name in GenBank database) (Benson et al., 2018), pathway, amino acid sequence and nucleotide sequence. The KEGG Mapper1 was applied to generate the pathway maps for both strains. The lists of amino acid sequences of glycoside hydrolases found in “fpa” and “fpr” were gathered in two fasta files (see Supplementary Table S1).
Publicly available genomes of F. prausnitzii strains were retrieved from the Genome database of the NCBI via FTP (Agarwala et al., 2018). From a total of 27 strains, 15 strains have been released in 2017 and the protein annotations are not yet available. Therefore, these genomes required further annotation. The rapid annotation using subsystem technology (RAST) platform was used for this purpose (Overbeek et al., 2014).
Homology Analysis
The basic local alignment search tool (BLAST), most notably the Blastp 2.6.0 tool, supported the search of glycoside hydrolase sequences in the compiled F. prausnitzii genomes (Altschul et al., 1997). A Matlab script was coded to generate the results, generally referred to as “fpa” (i.e., F. prausnitzii SL3-3 as reference) and “fpr” (i.e., F. prausnitzii L2-6 as reference), and obtain the homology data for each of the strains. Specifically, data on the percentage identity, the alignment length, the number of mismatches, gap opens, the start and end of alignment in query/subject, the expected value, and the bit score.
A heatmap-alike representation of the best homology values with associated hierarchical clustering (Euclidean distance and average linkage) was further implemented to support a more intuitive inspection of the results. Values of homology below 50% were suspected to represent non-orthologs enzymes. Conversely, homology values close to 1 were investigated as likely pointing to orthologs enzymes. Supplementary Table S2 details all these data.
Clustering F. prausnitzii Strains
The k-means partitioning clustering method was applied to further analysis of the homology results, most notably to identify groups of strains showing similar traits (Neyman, 1967). This analysis included the clustering of glycoside hydrolases activities according to their prevalence in the analyzed strains as well as the clustering of strains according to the predicted glycoside hydrolases activities.
Distance similarity was based on the Euclidean metric. Several values of k were analyzed for each scenario and the best number of clusters was determined based on the rule of thumb:
where n being the number of instances to cluster, and the Davies-Bouldin index, which evaluates intra-cluster similarity (i.e., the members of a cluster should present rather similar traits), and inter-cluster differences (i.e., clusters should be reasonably different) (Davies and Bouldin, 1979). Specifically, the number of clusters (k) was initially set to 4 and then fine-tuned using the Davies-Bouldin index (in general, the lowest the better). See Supplementary Table S3 for details on this analysis.
Rapid Miner 8.2 software platform was used to conduct the clustering experiments (Hofmann and Klinkenberg, 2013). The graphical representation of the results was produced using a Python script, based on the graphical libraries NumPy (van der Walt et al., 2011) and Matplotlib (Hunter, 2007).
Gene Neighboring
The SEED genome annotation viewer2 was used to investigate the gene neighborhoods related to glycoside hydrolase activities. SEED had publicly available annotations for 5 of 27 strains under analysis, i.e., the strains A2-165, M21/2, SL3/3, L2-6, and KLE1255. In-house experts, via the RAST server3, annotated the other 22 genomes (managed as a private annotation project in SEED). Next, the SEED Viewer tool4 was used to create a graphical representation of the gene neighborhood for the genes encoding glycoside hydrolases in F. prausnitzii or related strains.
Supplementary Table S4 lists the SEED URLs for public inspection. While it is not possible to supply public links for the genomes annotated privately, this document explains how to reproduce the analysis at its full extent.
Results
Metabolic Portrait of F. prausnitzii
Currently, KEGG database describes the metabolism of F. prausnitzii SL3/3 (identified as “fpa,” and documenting 2820 genes and 2756 proteins) and F. prausnitzii L2-6 (identified as “fpr,” and documenting 2816 genes and 2746 proteins). In particular, KEGG describes 90 metabolic pathways in these organisms. Glycoside hydrolases are annotated in 17 pathways of F. prausnitzii SL3/3 and 14 pathways of F. prausnitzii L2-6, respectively (Table 1). For the most part, glycoside hydrolases are located in the pathways responsible for the starch and sucrose metabolism and the biosynthesis of secondary metabolites.
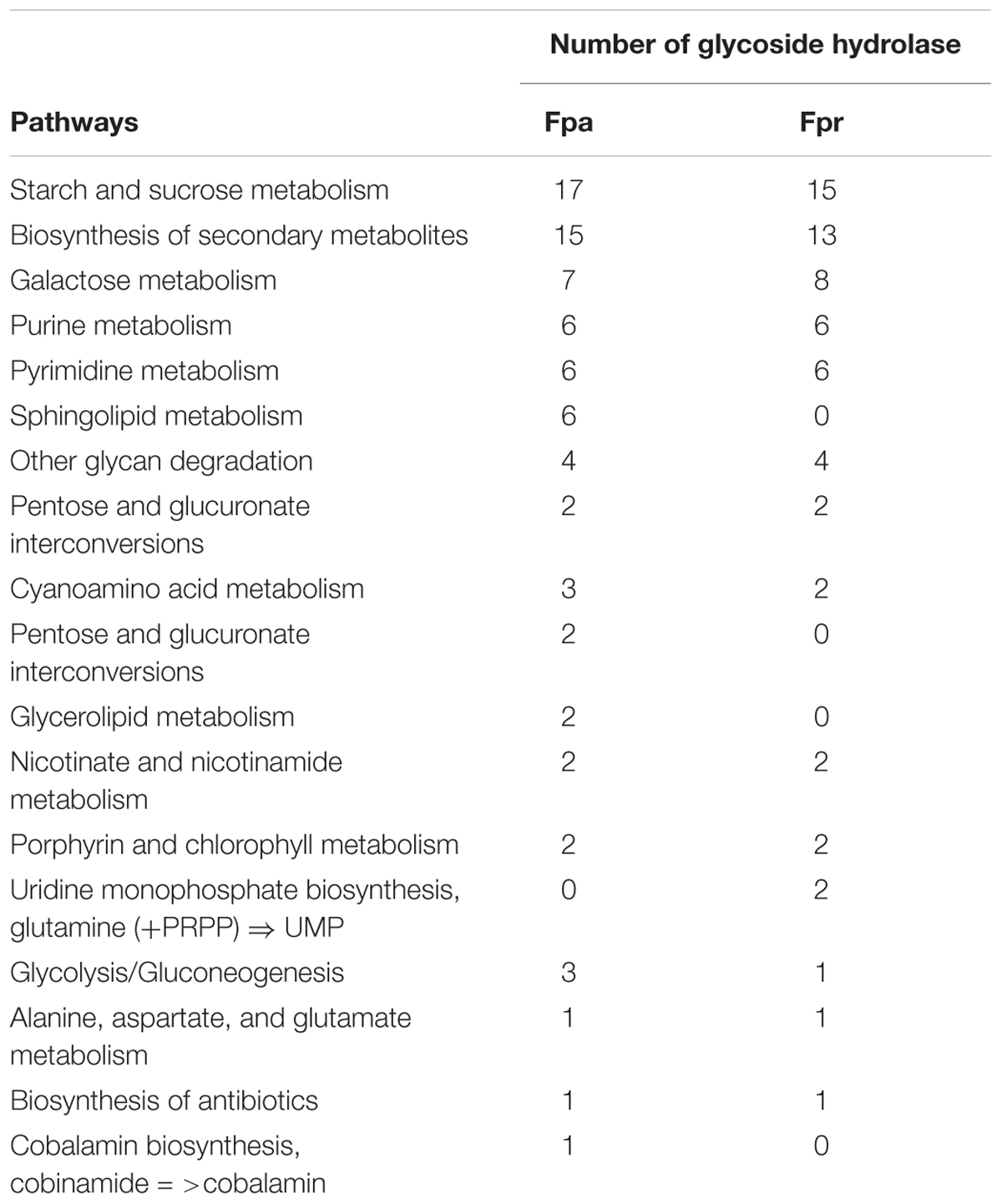
Table 1. Pathways of the metabolisms of F. prausnitzii SL3/3 and F. prausnitzii L2-6 containing glycoside hydrolases.
Figure 2 illustrates the gene orthology and the glycolytic activities in the two strains, as described in KEGG. F. prausnitzii SL/3 has 44 glycoside coding genes and F. prausnitzii L2-6 has 37 of such genes. 27 of these genes are orthologs, whilst 8 of the genes are specific of F. prausnitzii SL/3 and 3 are specific of F. prausnitzii L2-6. As a result, the two strains have in common 24 glycoside hydrolases. In terms of specific activity in F. prausnitzii SL3/3, this can be pin pointed to the pathways related with sphingolipid metabolism, the pentose and glucuronate interconversions, the glycerolipid metabolism, and the cobalamin biosynthesis (i.e., the glycoside hydrolases 2.4.1.20, 2.4.1.281, 2.4.1.319, 2.4.1.320, 3.2.1.22, 3.2.1.40, and 3.2.1.122). Likewise, the glycoside hydrolases 2.4.1.4, 3.2.1.10, and 3.2.1.26 show activity only in the uridine monophosphate biosynthesis of F. prausnitzii L2-6.
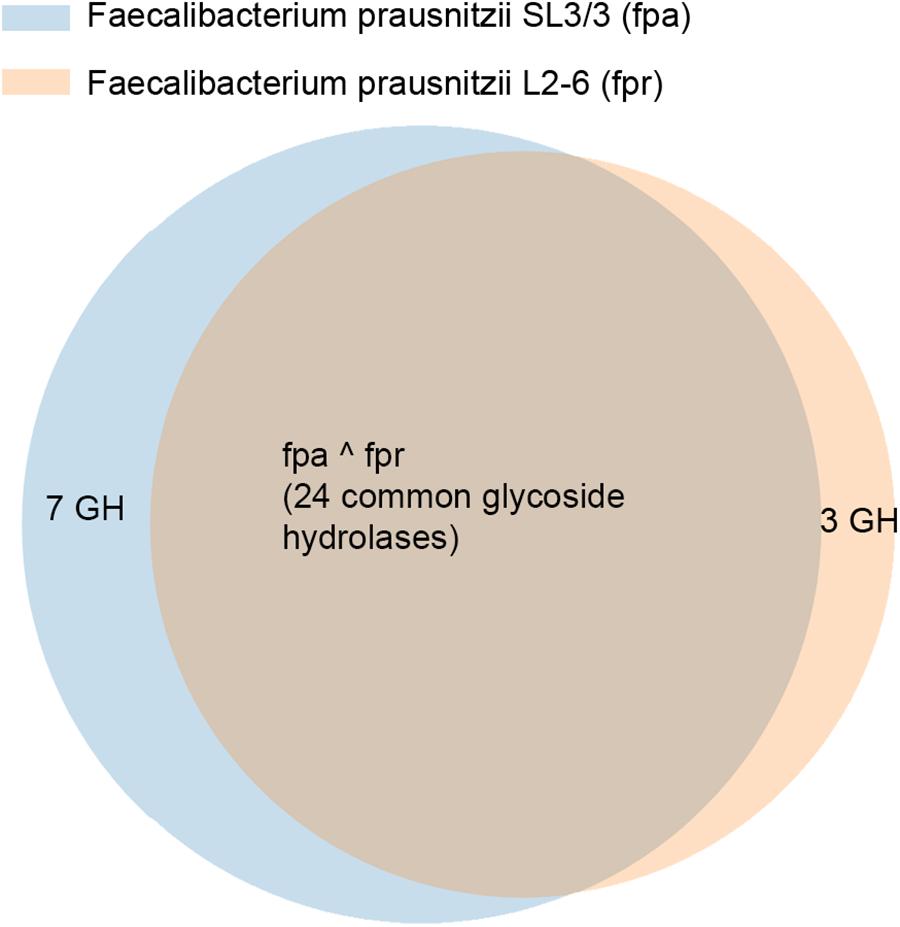
Figure 2. Relation of distribution of glycoside hydrolases in F. prausnitzii SL3/3 and F. prausnitzii L2-6.
As an example, Figure 3 details the enzymatic activities found in the starch and sucrose metabolism of F. prausnitzii SL3/3. The program implemented in-house to generate the pathway maps using the KEGG Mapper depicts the enzymatic activities of interest as well as organism-specific activity. That is, organism-specific reactions are colored in green and glycoside hydrolases are colored in red. All objects have links to the corresponding KEGG entries.
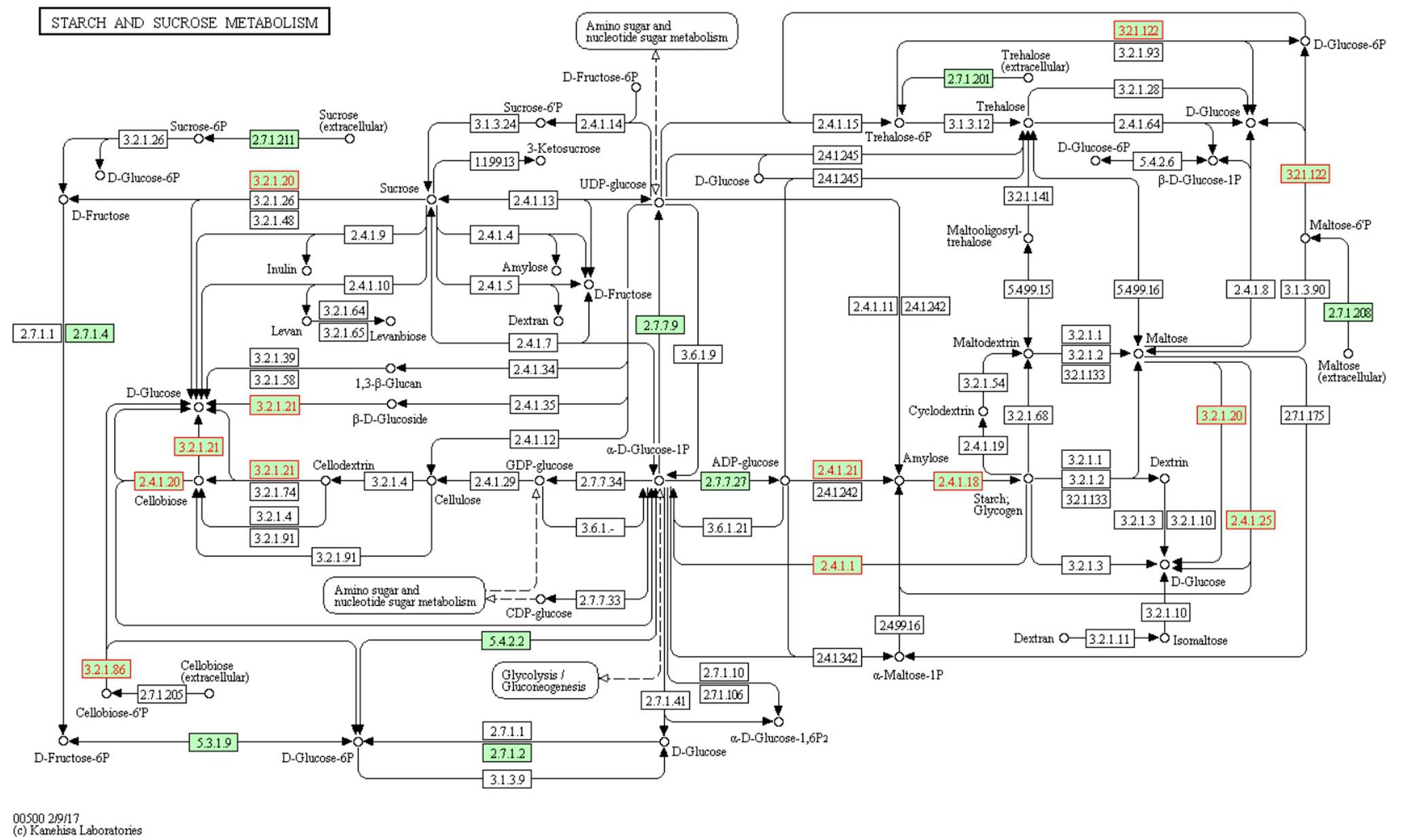
Figure 3. Metabolic map of the starch and sucrose metabolism in F. prausnitzii SL3/3. The EC numbers colored in green are organism-specific and red colored font highlights glycoside hydrolase activities
Homology Analysis
The results of the sequence similarity analysis, in particular the percentage identity data, were further analyzed. Figure 4 illustrates the results obtained having F. prausnitzii SL3/3 (fpa) as reference. Table 2 describes the glycoside hydrolase activities that have broader, acceptable, and lower probability of being present in the F. prausnitzii strains analyzed. A threshold of 50% of homology guided the analysis of gene orthology, i.e., the likelihood of the same gene being present across multiple strains.
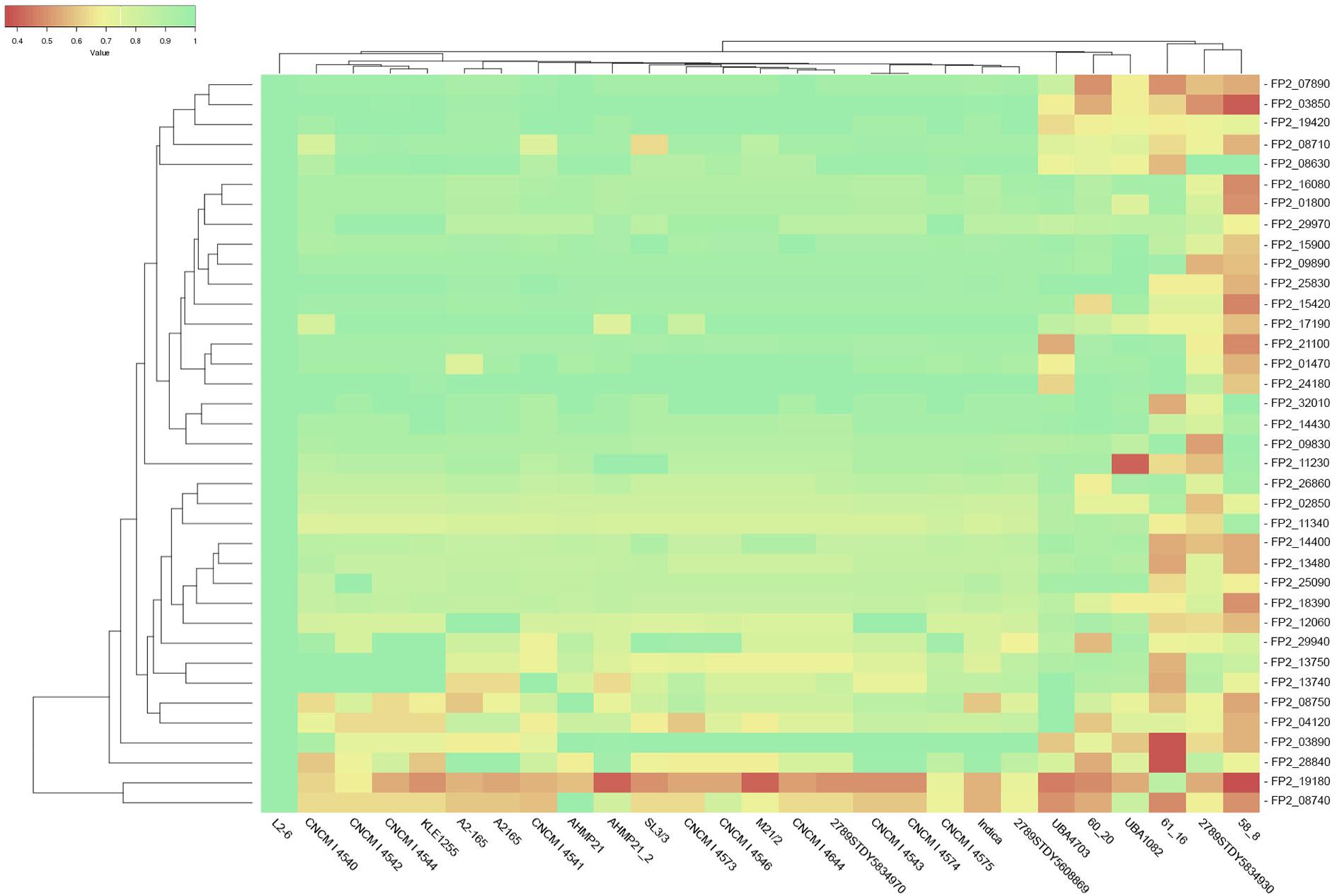
Figure 4. Extract of the heatmap representation of the homology results for F. prausnitzii SL3/3. The rows describe glycoside hydrolases and the columns depict the F. prausnitzii strains under analysis. The color hue of the cells represents the magnitude of the obtained homology score (red for 0% to green for 100%).
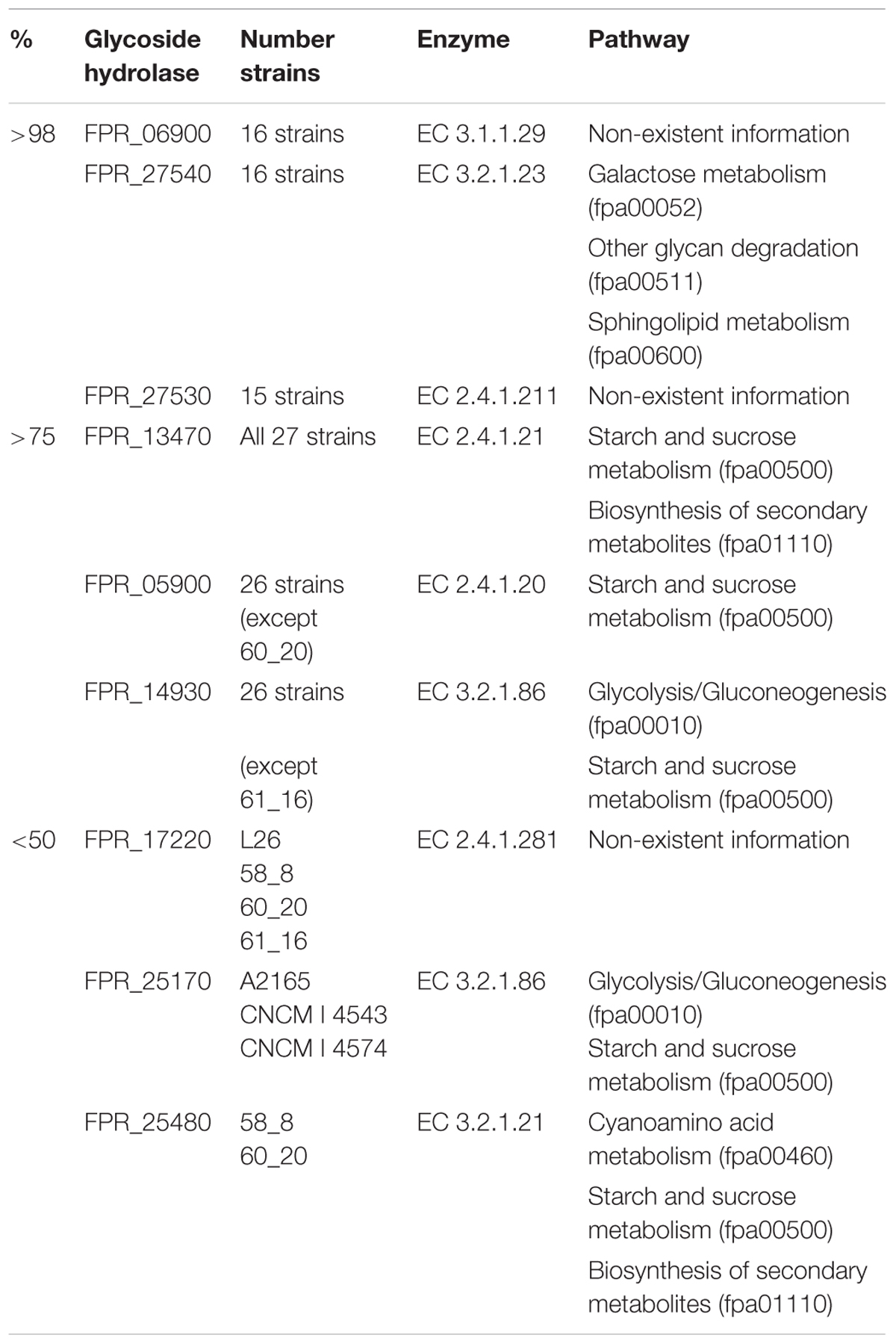
Table 2. Glycoside hydrolases showing very high (>98%), acceptable (>75%), and low values of homology (<50%) having the F. prausnitzii SL3/3 strain as reference.
Three glycoside hydrolases showed a perfect homology score (98%) for almost all the strains namely: a beta-galactosidase/beta-glucuronidase (FPR_27540) that participates in the galactose and sphingolipid metabolisms (fpa00052 and fpa00600, respectively) as well as in glycan degradation (fpa00511); an uncharacterized peptidyl-tRNA hydrolase (FPR_06900); and, a conserved hypothetical 1,3-beta-galactosyl-N-acetylhexosamine phosphorylase (FPR_27530).
Various other glycoside hydrolases showed homology scores above 75% for (almost) all strains. Notably, a starch synthase (FPR_13470), a cellobiose phosphorylase (FPR_05900), and a 6-phospho-beta-glucosidase (FPR_14930) are highlighted as being potentially present in practically all the studied strains (Table 2). The starch and sucrose metabolism (fpa00500) is again in evidence, but these enzymes are also present pathways such as the glycolysis/gluconeogenesis (fpa00010) and the biosynthesis of secondary metabolites (fpa01110).
A predicted 4-O-beta-D-mannosyl-D-glucose phosphorylase (FPR_17220), a 6-phospho-beta-glucosidase (FPR_25170), and a beta-glucosidase (FPR_25480) were among those glycoside hydrolases that showed values of homology below 50%. FPR_17220 is a predicted glycoside hydrolase that was found in only 4 of the strains with a score of no more than 48%. FPR_25170 is a glycoside hydrolase located in the glycolysis/gluconeogenesis metabolism (fpa00010) and the starch and sucrose metabolism (fpa00500), and did not achieve any result above 45%. Finally, FPR_25480 is an aryl-beta-glucosidase, which is present in the starch and sucrose metabolism (fpa00500), the cyanoamino acid metabolism (fpa00460) and the biosynthesis of secondary metabolites (fpa01110). The prediction scores of these glycoside hydrolases were below 45%. Also noteworthy, the aryl-beta-glucosidase (FPR_25480) is the glycoside hydrolase predicted in fewer strains, i.e., 2 out of the 27 strains, even though homology scores are greater than 50%.
The strains CNCM I 4546 and M21/2 showed the most overall homology with respect to the reference strain (SL3/3). They show homology values above 75% for all glycoside hydrolases, with the exception of the 6-phospho-beta-glucosidase (i.e., FPR_25170) in M21/2. Conversely the strain 58_8 showed the lowest overall homology results, i.e., 32 out of the 44 glycoside hydrolases have homology results below 70%. Supplementary Table S2 details these data.
Clustering of F. prausnitzii Strains
Previous works proposed possible divisions of the Faecalibacterium group based on phylogenetic analyses (Benevides et al., 2017; Fitzgerald et al., 2018). In this study, the clustering of the studied strains was more specialized, i.e., it was based on the homology results for the glycoside hydrolases. Therefore, here, the strains clustered together have similar homology profiles for the glycoside hydrolases (i.e., glycolytic traits) whereas the clusters represent different, potentially meaningful sets/combinations of glycolytic activities.
Figure 5 describes the clustering of the F. prausnitzii strains based on the glycolytic traits as well as phylogeny. The strains present in the glycolytic study are also in the reference phylogenetic studies, with the exception of the strains 2789STDY5834930, 58_8 and 61_16. Color notation helps to compare the present homology-based clustering to two phylogenetic groupings. Overall, most of the groupings are in accordance to one another. The most noticeable difference is that the strain CNCM I 4541 is grouped apart from the rest of the strains showing similar glycolytic traits (i.e., cluster 4, light green colored) in both phylogenetic studies. This glycolytic group showed a high prevalence of glycosides participating in the glycolysis/gluconeogenesis as well as the metabolism of starch and sucrose.
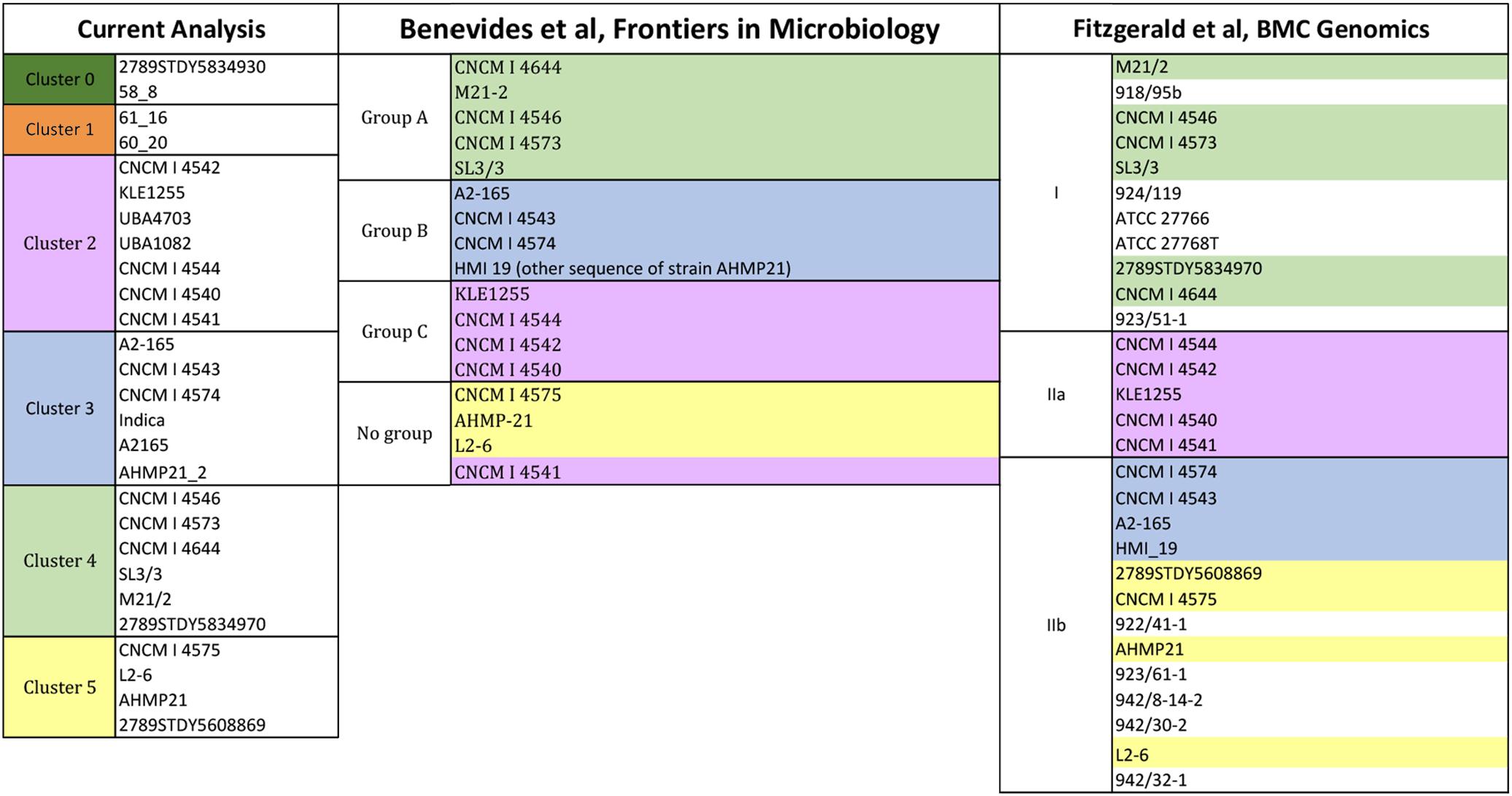
Figure 5. Clustering of strains activities having F. prausnitzii SL3-3 (fpa) as reference. The reference strains are located in cluster 4. Strain per cluster: cluster 0 (58_8, 2789STDY5834930), in cluster 1 (60_20 and 61_16), in cluster 2 (UBA4703, UBA1080, CNCM | 4544, KLE1255, CNCM | 4542, CNCM | 4540, and CNCM | 4541), in cluster 3 (A2-165, Indica, CNCM | 4574, A2165, CNCM | 4543, and AHMP21-2), in cluster 4 (M21/2, CNCM | 4573, 2789STDY5834970, SL3/3, CNCM | 4546, and CNCM | 4644), and in cluster 5 (CNCM | 4575, L2-6, AHMP21, and 2789STDY5608869).
The strains in the cluster 1 (orange colored) and the cluster 2 (pink colored) showed a high frequency of some glycosyltransferases and presence in the pyrimidine metabolism. The biggest difference between these clusters is that cluster 1 shows a low presence of some hexosyltransferases (i.e., FPR_17220 and FPR_17240) whereas cluster 2 is characterized by a low presence of a beta-glucosidase (the FPR_25480), which is present in the starch and sucrose metabolism as well as the cyanoamino acid metabolism.
The strains in the cluster 3 (blue colored) showed low scores for FPR_25170 and high scores for FPR_12120, both glycosides associated with the starch and sucrose metabolism. In turn, the strains of the cluster 4 (light green colored) and the cluster 5 (yellow colored) display overall high frequency profiles. These strains show high homology for enzymes associated to the pyrimidine metabolism. Differences lay in the high score of a 4-alpha-glucanotransferase (FPR_24180) in cluster 4, which participates in the starch and sucrose metabolism, whereas the key glycoside hydrolase in cluster 5 is a purine-nucleoside phosphorylase (FPR_06940), which is part of the metabolisms of purine and the metabolism of nicotinate and nicotinamide.
The clustering model supported by the homology data of F. prausnitzii L2-6 (Figure 6) is somewhat different from the above described for F. prausnitzii SL3-3. The two models present some similar grouping of strains, i.e., the clusters 0 (dark green colored), 3 (blue colored), and 5 (yellow colored). Moerover, the strains F. prausnitzii CNCM I 4540, F. prausnitzii CNCM I 4542, F. prausnitzii CNCM I 4544, F. prausnitzii KLEI1255, F. prausnitzii UBA4703, and F. prausnitzii UBA1082 are grouped together in both models, namely in cluster 2 (pink colored). However, in the first model, this cluster also contains the strain F. prausnitzii CNCM I 4541 whereas in the second model this cluster contains the strain F. prausnitzii 60_20.
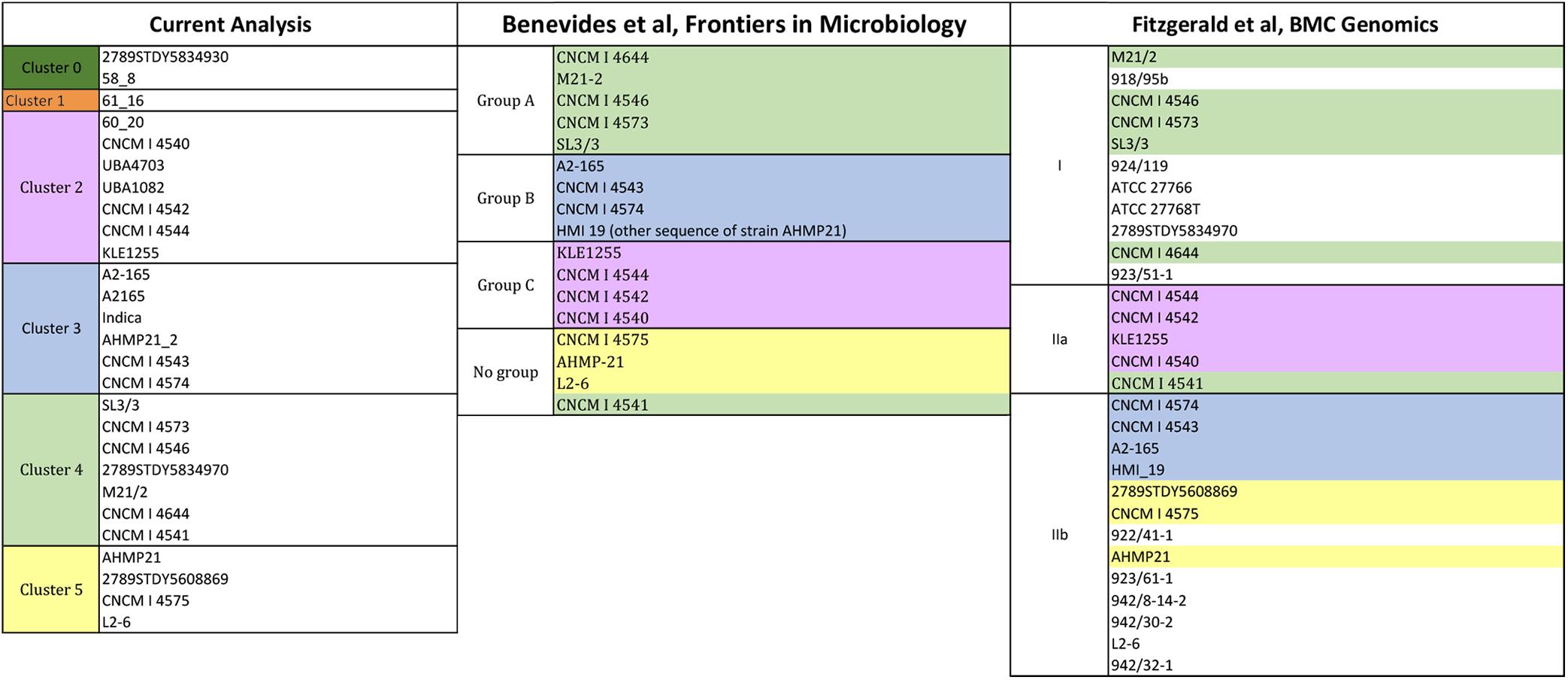
Figure 6. Clustering of strains having F. prausnitzii L2-6 (fpr) as reference. The reference strains is located in cluster 5. Strains per cluster: cluster 0 (58_8 and 2789STDY5834930), in cluster 1 (61_16), in cluster 2 (UBA1082, UBA4703, 60_20, CNCM | 4540, CNCM | 4542, CNCM | 4544, and KLE1255), in cluster 3 (A2165, A2-165, CNCM | 4543, Indica, AHMP-21_2, CNCM | 4574), in cluster 4 (CNCM | 4541, CNCM | 4546, CNCM | 4573, CNCM | 4644, SL3/3, M21/2, and 2789STDY5834970), and in cluster 5 (L2-6, 2789STDY5608869, CNCM | 4575, and AHMP-21).
The strains showing the most different/unique traits (i.e., those more distant from the rest of strains) are the same, i.e., the strains F. prausnitzii 60_20, F. prausnitzii 58_8, F. prausnitzii 61_16, and F. prausnitzii 2789STDY5834930. Also interesting, the strains F. prausnitzii 61_16 and F. prausnitzii 60_20 are together when the F. prausnitzii SL3-3 is used as reference (cluster 1, colored in orange), whereas, for the F. prausnitzii L2-6, the strain F. prausnitzii 61_16 is alone (cluster 1, colored in orange), and the strain F. prausnitzii 60_20 belongs to cluster 2. Looking into the F. prausnitzii SL3-3 homology data, the strains F. prausnitzii 61_16 and F. prausnitzii 60_20 show similar high homology (>93%) with a predicted unsaturated glucuronyl hydrolase (FPR_02050), an adenine/guanine phosphoribosyltransferase (FPR_06700) and a thymidine phosphorylase (FPR_21760), and similar low homology (<55%) for a 4-O-beta-D-mannosyl-D-glucose phosphorylase (FPR_17220) and a beta-1,4-mannooligosaccharide/beta-1,4-mannosyl-N-acetylglucosamine phosphorylase (FPR_17240). In turn, in the second analysis, the two strains present quite different glycolytic profiles. For example, the homology scores obtained for a glycogen/starch/alpha-glucan phosphorylase (FP2_13480) and an alpha-glucosidase (FP2_19180) differ in 40%.
See Supplementary Data Sheet S5 for detailed information on this analysis.
Gene Neighboring
Genomic neighborhoods can provide an important level of information about the cooperative role of genes in metabolic routes. Notably, functionally related genes are often organized into co-expression functional networks. Therefore, the study of the gene neighborhoods related to certain glycoside hydrolases, either very frequent or very specific in the present collection of genomes, was interesting to gain a better understanding about the genes that participate in the regulation or are functionally related to those genes encoding these activities. In this regard, the SEED genome annotation viewer is a useful tool to investigate how glycoside hydrolase genes are co-localized with other key genes in the F. prausnitzii genomes. As an example, a graphical representation of a potential cellobiose utilization cluster is included in Figure 7, showing that genes coding for an ABC transporter, a LacI family transcriptional regulator and a cellobiose phosphorylase (responsible for the phosphorylation of cellobiose and the release of alpha-D-glucose 1-phosphate and D-glucose) seem to be involved in cellobiose uptake. Remarkably, this gene cluster organization is a common feature in other intestinal bacteria (Figure 7). Supplementary Data Sheet S6 presents other examples of gene clusters related to glycoside hydrolase activities in F. prausnitzii.
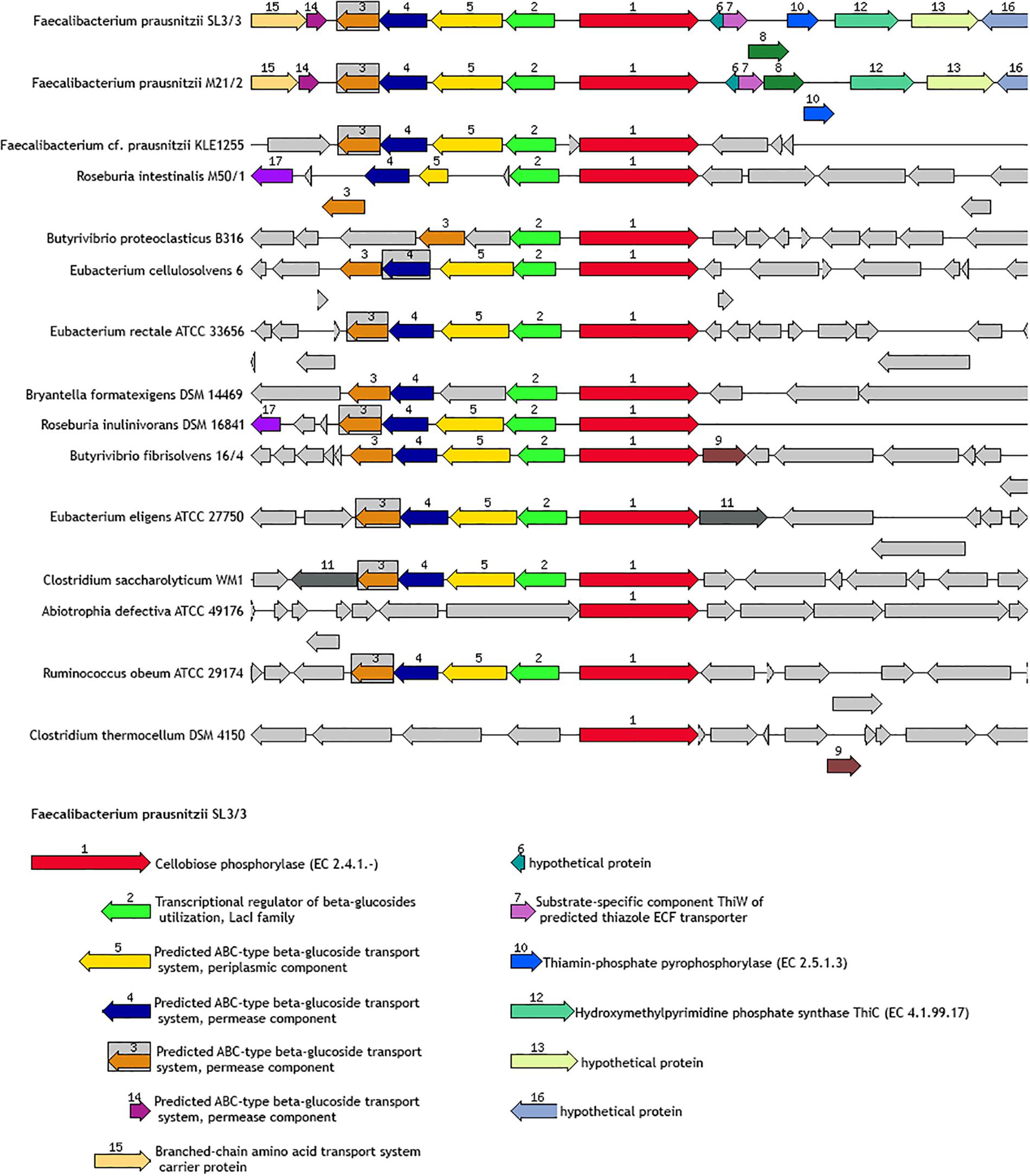
Figure 7. Schematic of the gene neighbor of FPR_05900 (EC.2.4.1.20). This gene encodes a predicted unsatured glucuronyl hydrolase involved in the regulation of bacterial surface properties and related properties.
Discussion
Since F. prausnitzii emerged as one of the most promising next-generation probiotics, the research efforts invested in gaining a deeper understanding about the metabolism of this microorganism have proliferated. Till date, and to the best of our knowledge, the glycolytic activities of F. prausnitzii had not been portrayed.
Therefore, this work presented an in silico approach to the systematic and large-scale study of such metabolic activities in the publicly available genomes. Such approach combined sequence similarity analysis, data clustering and gene neighborhood analysis and took advantage on publicly available functional enzyme and pathway annotations. In total, this study screend the activity of 337 glycoside hydrolases in 27 strains, having two possible strains of reference, i.e., the strains F. prausnitzii SL3/3 and F. prausnitzii L2-6.
Although information about the carbohydrate preferences of F. prausnitzii is scarce, the in silico metabolic predictions inferred here are consistent with what we know about the impact of fermentable sugars on the growth promotion and metabolism of F. prausnitzii. For instance, the broad representation of glycoside hydrolases potentially involved in the metabolism of galactose-containing oligosaccharides points to a theoretical capability to hydrolyse these kind of substrates. Homologs of the beta-D-galactosidase (FPR_27540; EC.3.2.1 23) and the lacto-N-biose phosphorylase (FPR_27530; EC.2.4.1.211) were present in the majority of strains analyzed. Beta-D-galactosidases release terminal non-reducing β-D-galactose residues in galactosides, such as some prebiotic galactooligosaccharides (Garrido et al., 2013), and lactose-N-biose phosphorylase is involved in the metabolism of lacto-N-biose, the major building block of human milk oligosaccharides (Xiao et al., 2010). In this regard, in vitro fermentation experiments and in vivo intervention studies highlighted the ability of galactooligosacharides to increase the population of F. prausnitzii in the human gut microbiota (Azcarate-Peril et al., 2017; Grimaldi et al., 2017). Also, a recent report shows that there is an increase in bacterial taxa belonging to the species Faecalibacterium in the gut microbiota of piglets when fed with key human milk oligosaccharides (Jacobi et al., 2016). This suggests that milk oligosaccharides can contribute to the persistence of F. prausnitzii in the intestine of suckling pigs, and therefore, trigger the health promoting effects attributed to this bacterium. In relation to the lacto-N-biose phosphorylase activity, it is also worth mentioning that enzymes involved in the galacto N-biose/lacto-N-biose pathway play a crucial role in the metabolism of mucin from epithelial cells, an important colonization factor in intestinal bacteria (Turroni et al., 2010; Duranti et al., 2015).
Other commonly used prebiotic substrates are inulin-type fructans, which are linear fructans with beta (2←1) fructosyl-fructose linkages, including oligofructose (normally with a chain length of 2–8) and inulin (with a chain length up to 60 moieties). A starting alfa-D-glucose moiety can be present in the backbone but it is not essential (Roberfroid, 2007). Inulin-type fructans have been found to be metabolized by F. prausnitzii (Dewulf et al., 2013) and to increase the Faecalibacterium population within the human microbiota in intervention studies (Ramirez-Farias et al., 2009; Dewulf et al., 2013; Clarke et al., 2017; Healey et al., 2018). In this regard, the proposed bioinformatics approach identified homologs of alfa and beta glucosidases widely distributed in the 27 genomes of the strains analyzed. These enzymes could be involved in inulin-type fructan degradation (Saulnier et al., 2007; Giraldo et al., 2014). Altogether, the broad presence of the glycoside hydrolase activities discussed above in the F. prausnitzii genomes suggests that this species possesses ecological traits that favor its adaptation to the gut environment.
Moreover, a wide representation of strains within the F. prausnitzii species. Furthermore, the gene neighboring analysis enabled the description of the genetic context surrounding glycoside hydrolase genes. In present analysis detected glycoside hydrolases potentially involved in the metabolism of maltose (FPR_07280; EC.3.2.1.20) and cellobiose (FPR_05900; EC.2.4.1.20) in the genomes of all the strains analyzed. Maltose and cellobiose are added in culture media for F. prausnitzii since these disaccharides specifically promote its growth in laboratory conditions (Martín et al., 2017). Our results support the fact that maltose and cellobiose utilization can be a common metabolic characteristic of F. prausnitzii, rather than a strain-dependent feature, and highlight the importance of including this kind of carbon sources for the isolation particular, genes potentially involved in oligosaccharide metabolism are organized in clusters, which are specific to the uptake and degradation of the substrates. This is in accordance with what has been described for other intestinal bacteria (Pokusaeva et al., 2011; Wei et al., 2014).
In summary, although the suitability of our in silico approach to infer glycoside hydrolase functional maps of F. prausnizii needs to be validated using culturing methods, the results showed in this work are in agreement with the current, limited knowledge on carbohydrate metabolism in this species. After experimental validation, our bioinformatics approach may be reproduced and scaled in order to accommodate the analysis of other strains (or even families and genus), as well as other metabolic activities. This will allow the exploration of novel methodologies to design or obtain targeted prebiotics for F. prausnitzii and other strains of interest.
Author Contributions
BS, FF-R, AM, and AL conceived and designed the study. GB compiled the data and executed all the analyses. All authors drafted the manuscript and read and approved the final version of the manuscript.
Funding
This work was supported by the Spanish “Programa Estatal de Investigación, Desarrollo e Innovación Orientada a los Retos de la Sociedad” (Grant AGL2016-78311-R); and, the Asociación Española Contra el Cáncer (“Obtención de péptidos bioactivos contra el Cáncer Colo-Rectal a partir de secuencias genéticas de microbiomas intestinales,” Grant PS-2016). This work was also supported by the Portuguese Foundation for Science and Technology (FCT) under the scope of the strategic funding of UID/BIO/04469/2013 unit and COMPETE 2020 (POCI-01-0145-FEDER006684). This work was partially supported by the Consellería de Educación, Universidades e Formación Profesional (Xunta de Galicia) under the scope of the strategic funding of ED431C2018/55-GRC Competitive Reference Group.
Conflict of Interest Statement
BS and AM are on the scientific board and are co-founders of Microviable Therapeutics SL.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
SING group thanks CITI (Centro de Investigación, Transferencia e Innovación) from University of Vigo for hosting its IT infrastructure.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00517/full#supplementary-material
TABLE S1 | List of the amino acid sequences of the glycoside hydrolases in two fasta files fpa.fasta (corresponds to F. prausnitzii SL3/3) and fpr.fasta (corresponds to F. prausntizii L2-6).
TABLE S2 | Extended analysis of the homology among F. prausntizii strains.
TABLE S3 | Graphic representation of the clusters and the Davis-Boulding index.
TABLE S4 | List of SEED URLs shown for the gene neighbourhood.
DATA SHEET S1 | Analysis of F. prausnitzii gene orthology.
DATA SHEET S2 | Differents examples of gene neighbourhoods related to glycoside hydrolase activities en F. prausnitzii.
Footnotes
- ^https://www.genome.jp/kegg/mapper.html
- ^http://pubseed.theseed.org/
- ^http://rast.nmpdr.org
- ^http://rast.nmpdr.org/seedviewer.cgi
References
Agarwala, R., Barrett, T., Beck, J., Benson, D. A., Bollin, C., Bolton, E., et al. (2018). Database resources of the national center for biotechnology information. Nucleic Acids Res. 46, D8–D13. doi: 10.1093/nar/gkx1095
Ahmed, S., Macfarlane, G. T., Fite, A., McBain, A. J., Gilbert, P., and Macfarlane, S. (2007). Mucosa-associated bacterial diversity in relation to human terminal ileum and colonic biopsy samples. Appl. Environ. Microbiol. 73, 7435–7442. doi: 10.1128/AEM.01143-07
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Arumugam, M., Raes, J., Pelletier, E., Le Paslier, D., Yamada, T., Mende, D. R., et al. (2011). Enterotypes of the human gut microbiome. Nature 473, 174–180. doi: 10.1038/nature09944
Azcarate-Peril, M. A., Ritter, A. J., Savaiano, D., Monteagudo-Mera, A., Anderson, C., Magness, S. T., et al. (2017). Impact of short-chain galactooligosaccharides on the gut microbiome of lactose-intolerant individuals. Proc. Natl. Acad. Sci. U.S.A. 114, E367–E375. doi: 10.1073/pnas.1606722113
Benevides, L., Burman, S., Martin, R., Robert, V., Thomas, M., Miquel, S., et al. (2017). New insights into the diversity of the genus Faecalibacterium. Front. Microbiol. 8:1790. doi: 10.3389/fmicb.2017.01790
Benson, D. A., Cavanaugh, M., Clark, K., Karsch-Mizrachi, I., Ostell, J., Pruitt, K. D., et al. (2018). GenBank. Nucleic Acids Res. 46, D41–D47. doi: 10.1093/nar/gkx1094
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 37, D233–D238. doi: 10.1093/nar/gkn663
Clarke, S. T., Brooks, S. P. J., Inglis, G. D., Yanke, L. J., Green, J., Petronella, N., et al. (2017). Impact of β2-1 fructan on faecal community change: results from a placebo-controlled, randomised, double-blinded, cross-over study in healthy adults. Br. J. Nutr. 118, 441–453. doi: 10.1017/S0007114517002318
Davies, D. L., and Bouldin, D. W. (1979). A cluster separation measure. IEEE Trans. Pattern Anal. Mach. Intell. 1, 224–227. doi: 10.1109/TPAMI.1979.4766909
Dewulf, E. M., Cani, P. D., Claus, S. P., Fuentes, S., Puylaert, P. G. B., Neyrinck, A. M., et al. (2013). Insight into the prebiotic concept: lessons from an exploratory, double blind intervention study with inulin-type fructans in obese women. Gut 62, 1112–1121. doi: 10.1136/gutjnl-2012-303304
Duranti, S., Milani, C., Lugli, G. A., Turroni, F., Mancabelli, L., Sanchez, B., et al. (2015). Insights from genomes of representatives of the human gut commensal Bifidobacterium bifidum. Environ. Microbiol. 17, 2515–2531. doi: 10.1111/1462-2920.12743
El-Semman, I. E., Karlsson, F. H., Shoaie, S., Nookaew, I., Soliman, T. H., and Nielsen, J. (2014). Genome-scale metabolic reconstructions of Bifidobacterium adolescentis L2-32 and Faecalibacterium prausnitzii A2-165 and their interaction. BMC Syst. Biol. 8:41. doi: 10.1186/1752-0509-8-41
Fitzgerald, C. B., Shkoporov, A. N., Sutton, T. D. S., Chaplin, A. V., Velayudhan, V., Ross, R. P., et al. (2018). Comparative analysis of Faecalibacterium prausnitzii genomes shows a high level of genome plasticity and warrants separation into new species-level taxa. BMC Genomics 19:931. doi: 10.1186/s12864-018-5313-6
Garrido, D., Ruiz-Moyano, S., Jimenez-Espinoza, R., Eom, H.-J., Block, D. E., and Mills, D. A. (2013). Utilization of galactooligosaccharides by Bifidobacterium longum subsp. infantis isolates. Food Microbiol. 33, 262–270. doi: 10.1016/J.FM.2012.10.003
Gevers, D., Kugathasan, S., Denson, L. A., Vázquez-Baeza, Y., Van Treuren, W., Ren, B., et al. (2014). The treatment-naive microbiome in new-onset crohn’s disease. Cell Host. Microbe 15, 382–392. doi: 10.1016/J.CHOM.2014.02.005
Giraldo, M. A., Gonçalves, H. B., Furriel, R., dos, P. M., Jorge, J. A., and Guimarães, L. H. S. (2014). Characterization of the co-purified invertase and β-glucosidase of a multifunctional extract from Aspergillus terreus. World J. Microbiol. Biotechnol. 30, 1501–1510. doi: 10.1007/s11274-013-1570-3
Grimaldi, R., Cela, D., Swann, J. R., Vulevic, J., Gibson, G. R., Tzortzis, G., et al. (2017). In vitro fermentation of B-GOS: impact on faecal bacterial populations and metabolic activity in autistic and non-autistic children. FEMS Microbiol. Ecol. 93:fiw233. doi: 10.1093/femsec/fiw233
Healey, G., Murphy, R., Butts, C., Brough, L., Whelan, K., and Coad, J. (2018). Habitual dietary fibre intake influences gut microbiota response to an inulin-type fructan prebiotic: a randomised, double-blind, placebo-controlled, cross-over, human intervention study. Br. J. Nutr. 119, 176–189. doi: 10.1017/S0007114517003440
Hofmann, M., and Klinkenberg, R. (2013). RapidMiner: Data Mining Use Cases, and Business Analytics Applications. Boca Raton, FL: CRC Press.
Hunter, J. D. (2007). Matplotlib: a 2D Graphics Environment. Comput. Sci. Eng. 9, 90–95. doi: 10.1109/MCSE.2007.55
Jacobi, S. K., Yatsunenko, T., Li, D., Dasgupta, S., Yu, R. K., Berg, B. M., et al. (2016). Dietary isomers of sialyllactose increase ganglioside sialic acid Concentrations in the corpus callosum and cerebellum and modulate the colonic microbiota of formula-fed piglets. J. Nutr. 146, 200–208. doi: 10.3945/jn.115.220152
Joossens, M., Huys, G., Cnockaert, M., De Preter, V., Verbeke, K., Rutgeerts, P., et al. (2011). Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 60, 631–637. doi: 10.1136/gut.2010.223263
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Machiels, K., Joossens, M., Sabino, J., De Preter, V., Arijs, I., Eeckhaut, V., et al. (2014). A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 63, 1275–1283. doi: 10.1136/gutjnl-2013-304833
Martín, R., Miquel, S., Benevides, L., Bridonneau, C., Robert, V., Hudault, S., et al. (2017). Functional characterization of novel Faecalibacterium prausnitzii strains isolated from healthy volunteers: a step forward in the use of F. prausnitzii as a next-generation probiotic. Front. Microbiol. 8:1226. doi: 10.3389/fmicb.2017.01226
Morgan, X. C., Tickle, T. L., Sokol, H., Gevers, D., Devaney, K. L., Ward, D. V., et al. (2012). Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13:R79. doi: 10.1186/gb-2012-13-9-r79
Neyman, J. (1967). “Berkeley symposium on mathematical statistics and probability,” in Proceedings of the Berkeley Symposium on Mathematical Statistics and Probability, eds L. M. Le Cam and J. Neyman (Berkeley, CA: University of California Press).
O’Toole, P. W., Marchesi, J. R., and Hill, C. (2017). Next-generation probiotics: the spectrum from probiotics to live biotherapeutics. Nat. Microbiol. 2:17057. doi: 10.1038/nmicrobiol.2017.57
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Pokusaeva, K., O’Connell-Motherway, M., Zomer, A., Macsharry, J., Fitzgerald, G. F., and van Sinderen, D. (2011). Cellodextrin utilization by bifidobacterium breve UCC2003. Appl. Environ. Microbiol. 77, 1681–1690. doi: 10.1128/AEM.01786-10
Quévrain, E., Maubert, M.-A., Sokol, H., Devreese, B., and Seksik, P. (2016). The presence of the anti-inflammatory protein MAM, from Faecalibacterium prausnitzii, in the intestinal ecosystem. Gut 65:882. doi: 10.1136/gutjnl-2015-311094
Ramirez-Farias, C., Slezak, K., Fuller, Z., Duncan, A., Holtrop, G., and Louis, P. (2009). Effect of inulin on the human gut microbiota: stimulation of Bifidobacterium adolescentis and Faecalibacterium prausnitzii. Br. J. Nutr. 101, 541–550. doi: 10.1017/S0007114508019880
Roberfroid, M. B. (2007). Inulin-type fructans: functional food ingredients. J. Nutr. 137, 2493S–2502S. doi: 10.1093/jn/137.11.2493S
Rossi, O., Khan, M. T., Schwarzer, M., Hudcovic, T., Srutkova, D., Duncan, S. H., et al. (2015). Faecalibacterium prausnitzii strain HTF-F and its extracellular polymeric matrix attenuate clinical parameters in DSS-Induced colitis. PLoS One 10:e0123013. doi: 10.1371/journal.pone.0123013
Rossi, O., van Berkel, L. A., Chain, F., Tanweer Khan, M., Taverne, N., Sokol, H., et al. (2016). Faecalibacterium prausnitzii A2-165 has a high capacity to induce IL-10 in human and murine dendritic cells and modulates T cell responses. Sci. Rep. 6:18507. doi: 10.1038/srep18507
Rowland, I., Gibson, G., Heinken, A., Scott, K., Swann, J., Thiele, I., et al. (2018). Gut microbiota functions: metabolism of nutrients and other food components. Eur. J. Nutr. 57, 1–24. doi: 10.1007/s00394-017-1445-8
Saulnier, D. M. A., Molenaar, D., de Vos, W. M., Gibson, G. R., and Kolida, S. (2007). Identification of prebiotic fructooligosaccharide metabolism in Lactobacillus plantarum WCFS1 through microarrays. Appl. Environ. Microbiol. 73, 1753–1765. doi: 10.1128/AEM.01151-06
Sokol, H., Pigneur, B., Watterlot, L., Lakhdari, O., Bermúdez-Humarán, L. G., Gratadoux, J.-J., et al. (2008). Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. U.S.A. 105, 16731–16736. doi: 10.1073/pnas.0804812105
Stewart, C. S., Hold, G. L., Duncan, S. H., Flint, H. J., and Harmsen, H. J. M. (2002). Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 52, 2141–2146. doi: 10.1099/00207713-52-6-2141
Tilg, H., and Moschen, A. R. (2014). Microbiota and diabetes: an evolving relationship. Gut 63, 1513–1521. doi: 10.1136/gutjnl-2014-306928
Tipton, K. F. (1994). Nomenclature committee of the international union of biochemistry and molecular biology (NC-IUBMB). Enzyme nomenclature. Recommendations 1992. Supplement: corrections and additions. Eur. J. Biochem. 223, 1–5. doi: 10.1111/j.1432-1033.1994.tb18960.x
Turroni, F., Bottacini, F., Foroni, E., Mulder, I., Kim, J.-H., Zomer, A., et al. (2010). Genome analysis of Bifidobacterium bifidum PRL2010 reveals metabolic pathways for host-derived glycan foraging. Proc. Natl. Acad. Sci. U.S.A. 107, 19514–19519. doi: 10.1073/pnas.1011100107
van der Walt, S., Colbert, S. C., and Varoquaux, G. (2011). The NumPy array: a Structure for Efficient Numerical Computation. Comput. Sci. Eng. 13, 22–30. doi: 10.1109/MCSE.2011.37
Wei, H., Fu, Y., Magnusson, L., Baker, J. O., Maness, P.-C., Xu, Q., et al. (2014). Comparison of transcriptional profiles of Clostridium thermocellum grown on cellobiose and pretreated yellow poplar using RNA-Seq. Front. Microbiol. 5:142. doi: 10.3389/fmicb.2014.00142
Xiao, J. Z., Takahashi, S., Nishimoto, M., Odamaki, T., Yaeshima, T., Iwatsuki, K., et al. (2010). Distribution of In Vitro fermentation ability of lacto-TV-Biose I, a major building block of human milk oligosaccharides, in bifidobacteria! strains. Appl. Environ. Microbiol. 76, 54–59. doi: 10.1128/AEM.01683-09
Keywords: fermentable sugars, glycoside hydrolases, bioactivity, Faecalibacterium prausnitzii, computational screening
Citation: Blanco G, Sánchez B, Fdez-Riverola F, Margolles A and Lourenço A (2019) In silico Approach for Unveiling the Glycoside Hydrolase Activities in Faecalibacterium prausnitzii Through a Systematic and Integrative Large-Scale Analysis. Front. Microbiol. 10:517. doi: 10.3389/fmicb.2019.00517
Received: 06 November 2018; Accepted: 28 February 2019;
Published: 04 April 2019.
Edited by:
F. Javier Moreno, Instituto de Investigación en Ciencias de la Alimentación (CIAL), SpainReviewed by:
Rebeca Martín, INRA Centre Jouy-en-Josas, FranceFrancesca Bottacini, University College Cork, Ireland
Copyright © 2019 Blanco, Sánchez, Fdez-Riverola, Margolles and Lourenço. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anália Lourenço, analia@uvigo.es