
Novel In Silico Insights into Rv1417 and Rv2617c as Potential Protein Targets: The Importance of the Medium on the Structural Interactions with Exported Repetitive Protein (Erp) of Mycobacterium tuberculosis

 , , and
, , and 
Abstract
:1. Introduction
2. Computational Details
2.1. System Preparation
Erp, Rv1417 and Rv2617c Proteins
2.2. MD Simulations
2.3. Molecular Docking Calculations
2.4. Structure and Data Analysis
3. Results and Discussion
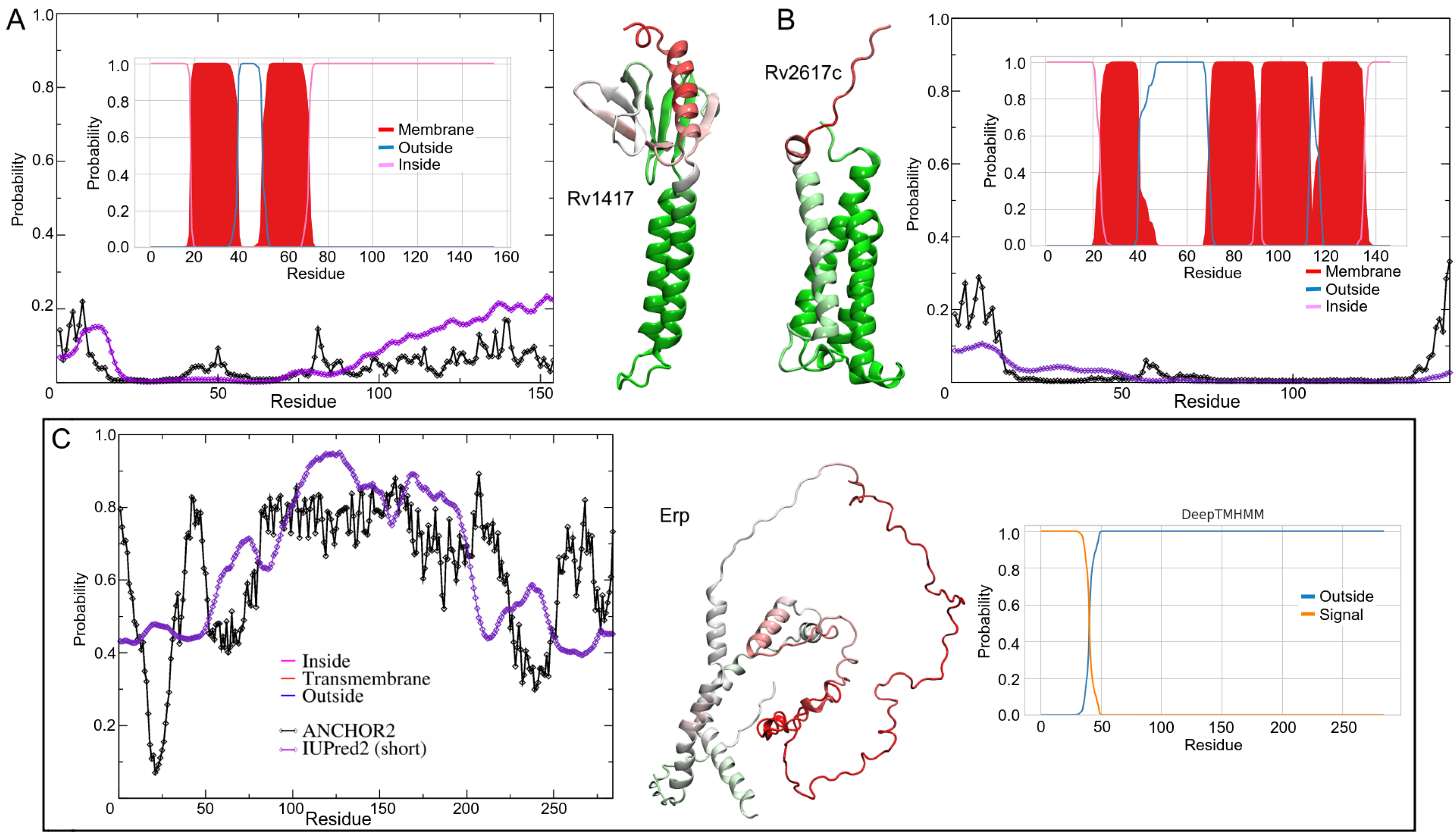
3.1. Structure Characterization
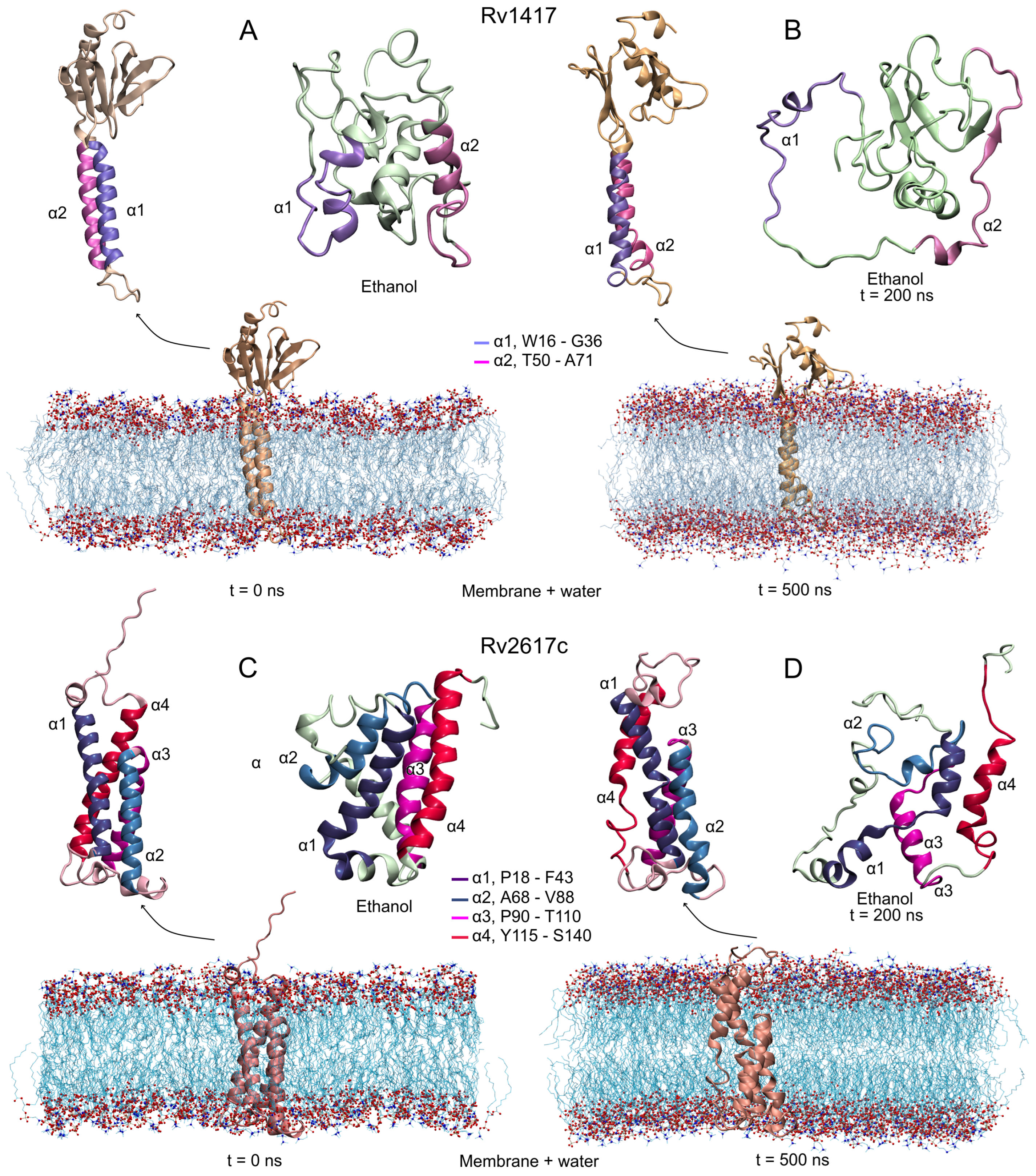
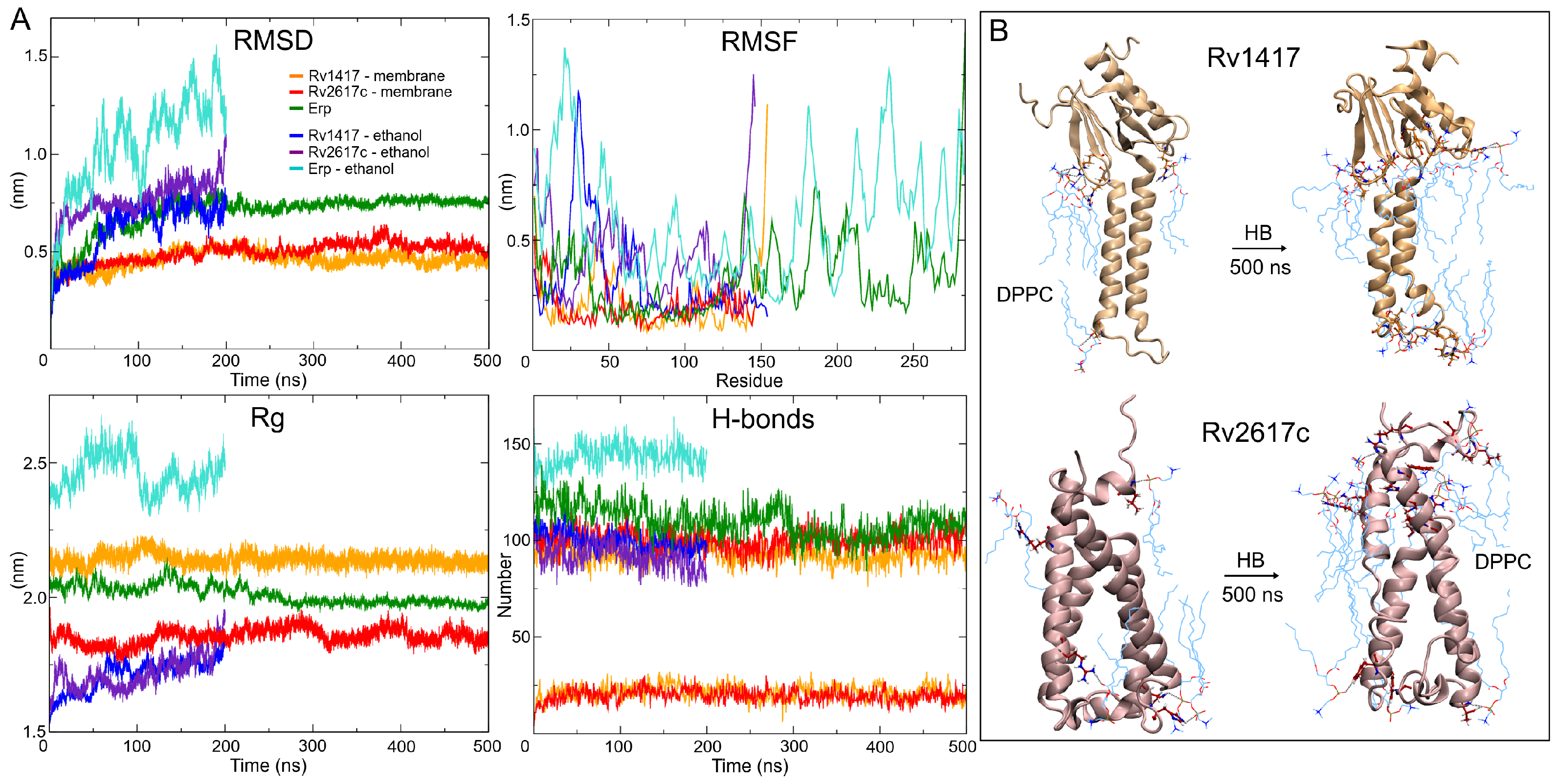
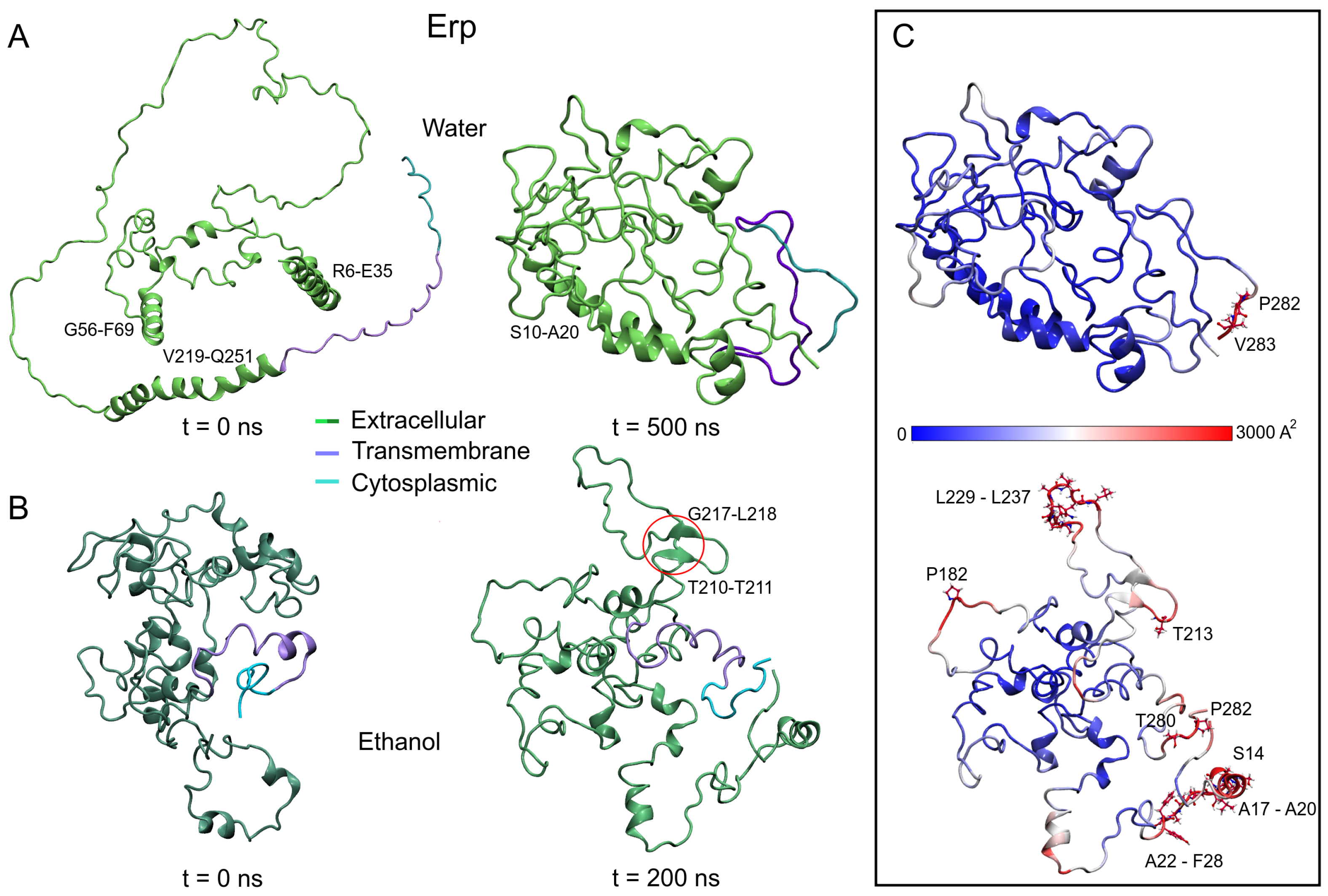
3.2. Protein Structures at Different Medium Conditions
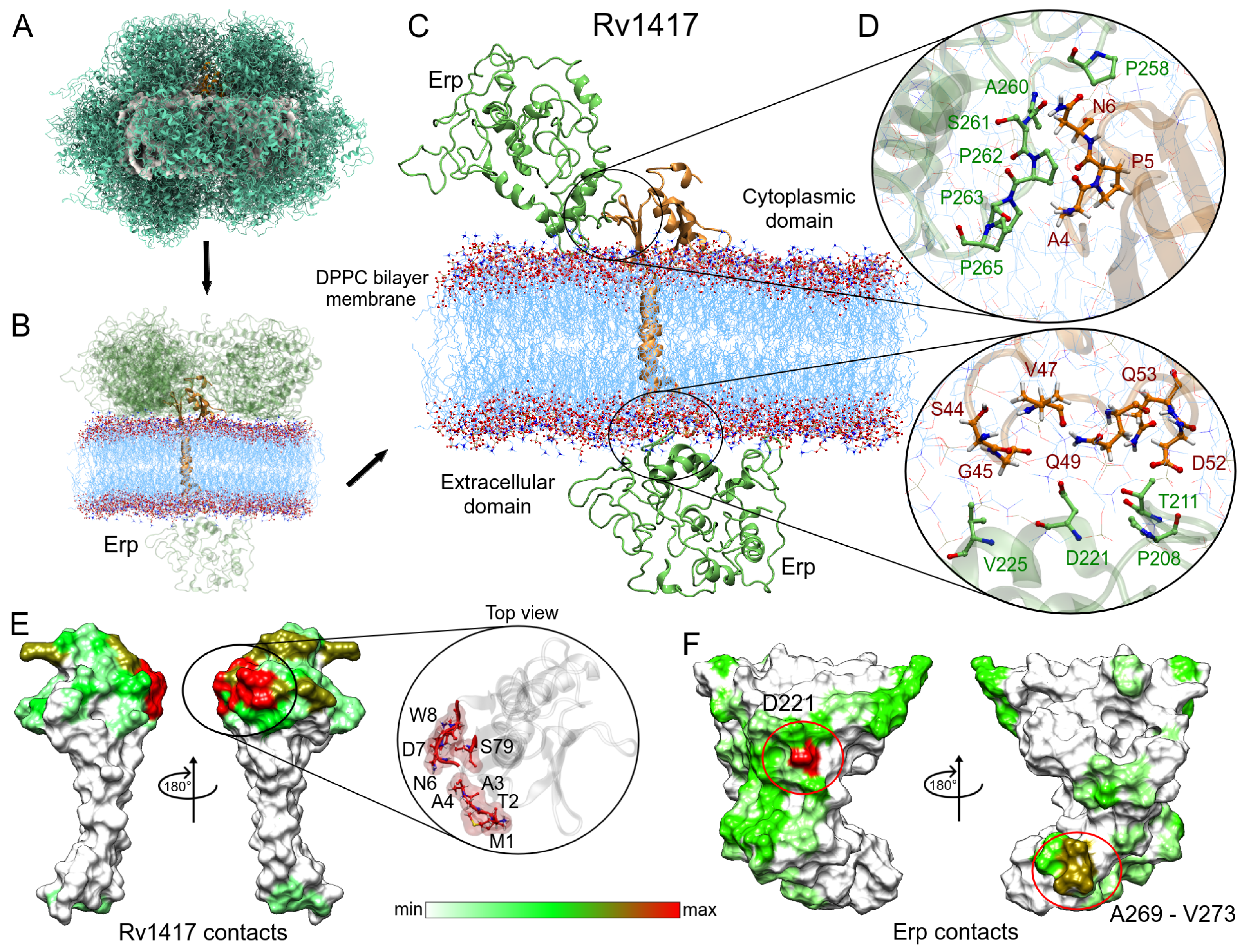
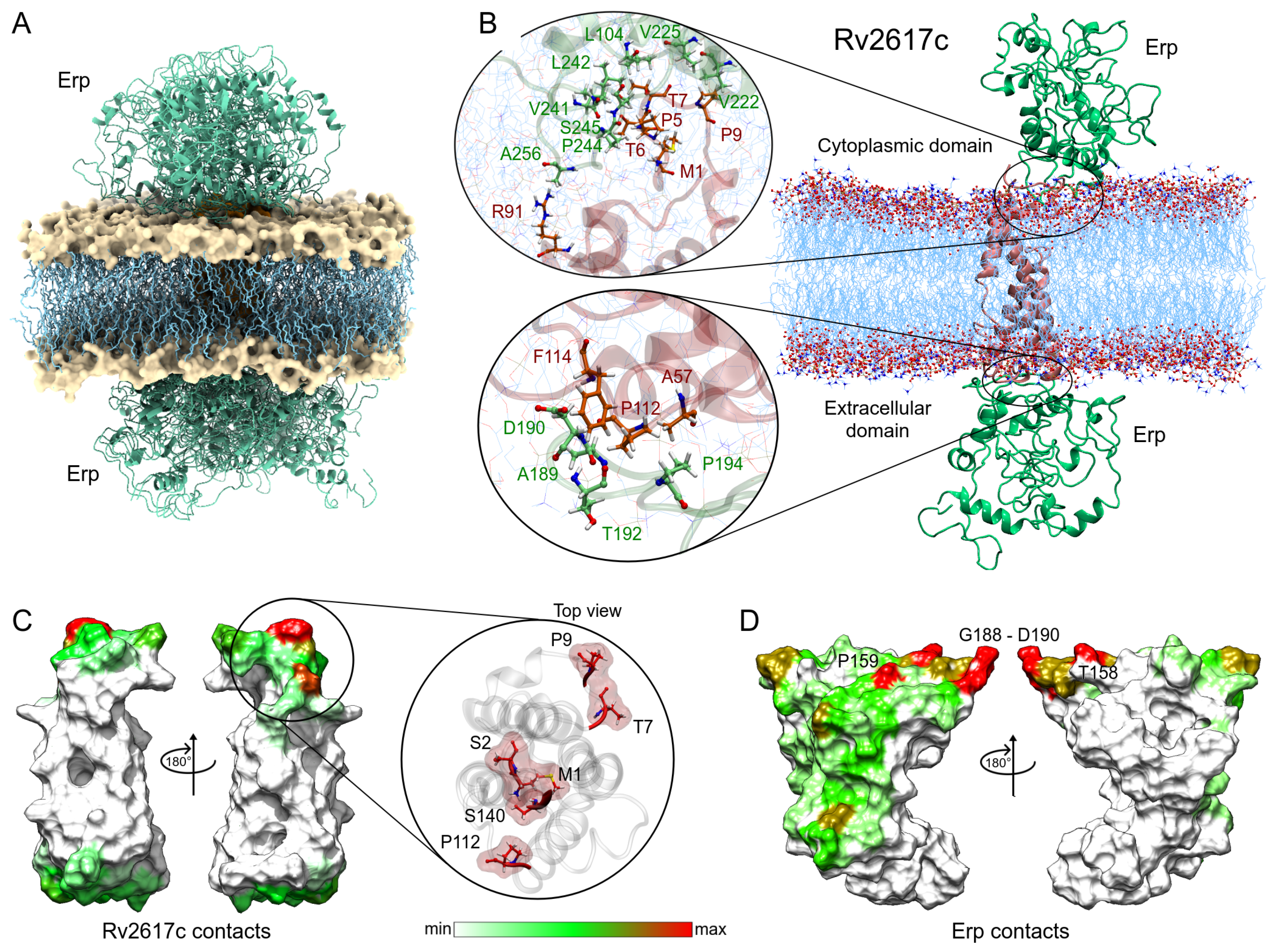
3.2.1. Rv1417 and Rv2617c Proteins
3.2.2. Polar Environment Affects the Compactness of Erp Structure
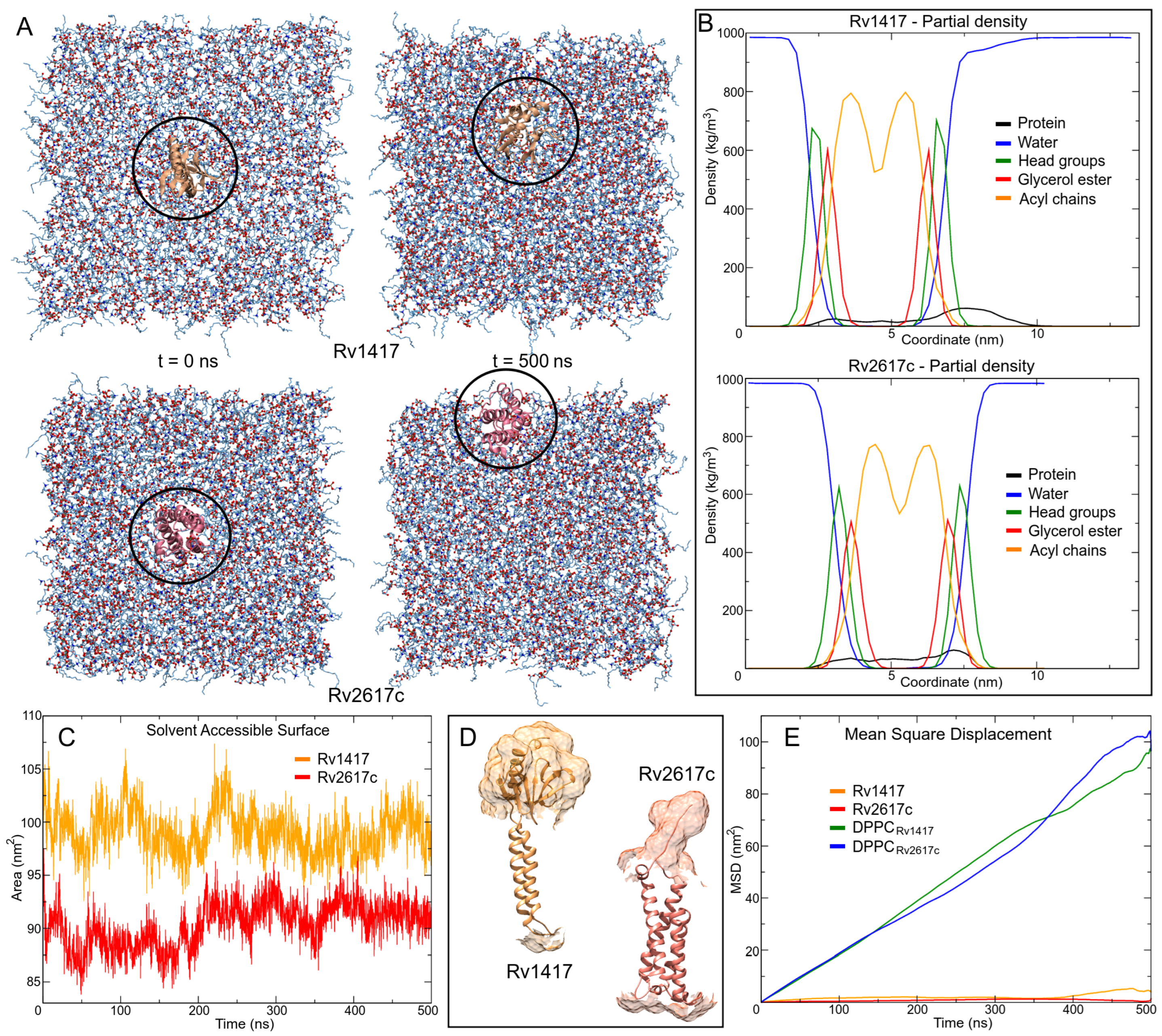
3.3. Protein–Protein Complexes
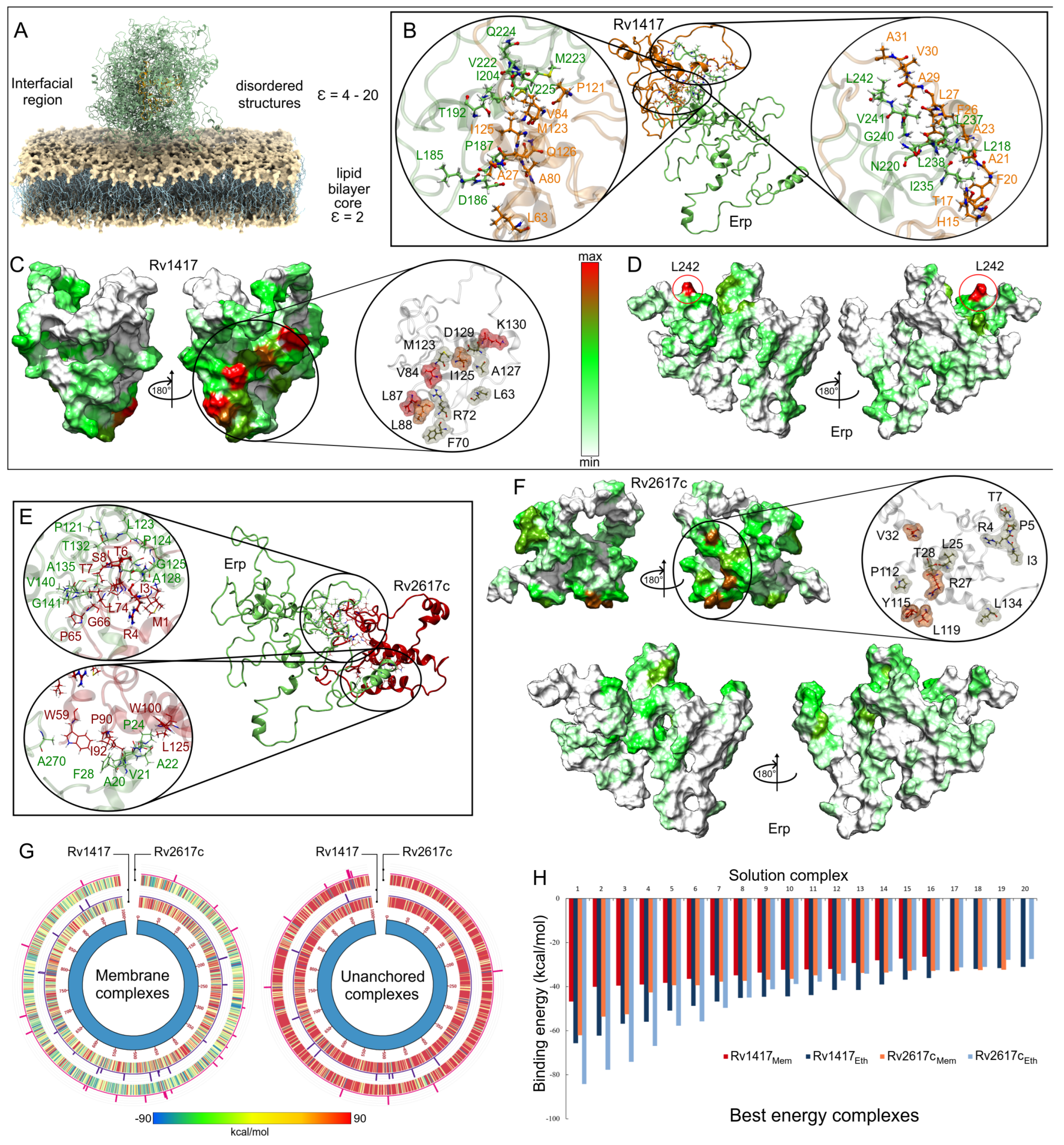
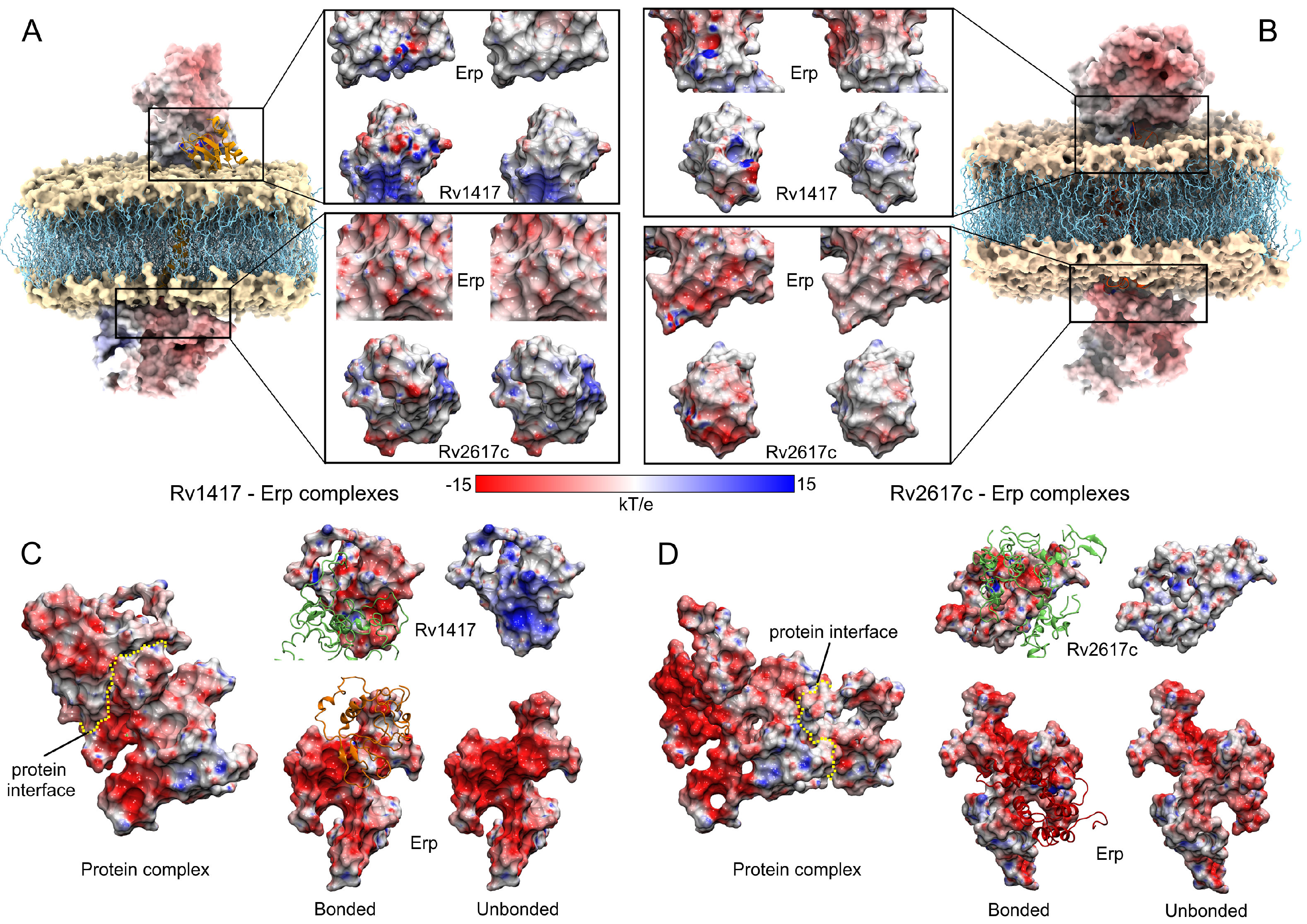
3.3.1. Membrane Systems
3.3.2. Unanchored Complexes
3.3.3. The Erp Protein Is the Main Guideline for Protein Interactions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McQuaid, C.F.; McCreesh, N.; Read, J.M.; Sumner, T.; Houben, R.M.; White, R.G.; Harris, R.C.; CMMID COVID-19 Working Group. The potential impact of COVID-19-related disruption on tuberculosis burden. Eur. Respir. J. 2020, 56, 2001718. [Google Scholar] [CrossRef] [PubMed]
- Antonio-Arques, V.; Franch-Nadal, J.; Caylà, J.A. Diabetes y tuberculosis: Una sindemia complicada por la COVID-19. Med. Clín. 2021, 157, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Hogan, A.B.; Jewell, B.L.; Sherrard-Smith, E.; Vesga, J.F.; Watson, O.J.; Whittaker, C.; Hamlet, A.; Smith, J.A.; Winskill, P.; Verity, R.; et al. Potential impact of the COVID-19 pandemic on HIV, tuberculosis, and malaria in low-income and middle-income countries: A modelling study. Lancet Glob. Health 2020, 8, e1132–e1141. [Google Scholar] [CrossRef]
- Pai, M.; Kasaeva, T.; Swaminathan, S. COVID-19’s Devastating Effect on Tuberculosis Care—A Path to Recovery. N. Engl. J. Med. 2022, 386, 1490–1493. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Tuberculosis Report 2021; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Tuberculosis. Available online: https://www.who.int/es/news-room/fact-sheets/detail/tuberculosis (accessed on 25 March 2022).
- Sandhu, G.K. Tuberculosis: Current situation, challenges and overview of its control programs in India. J. Glob. Infect. Dis. 2011, 3, 143. [Google Scholar] [CrossRef]
- Esaulova, E.; Das, S.; Singh, D.K.; Choreño-Parra, J.A.; Swain, A.; Arthur, L.; Rangel-Moreno, J.; Ahmed, M.; Singh, B.; Gupta, A.; et al. The immune landscape in tuberculosis reveals populations linked to disease and latency. Cell Host Microbe 2021, 29, 165–178. [Google Scholar] [CrossRef]
- Mori, G.; Morrison, M.; Blumenthal, A. Microbiome-immune interactions in tuberculosis. PLoS Pathog. 2021, 17, e1009377. [Google Scholar] [CrossRef]
- Ruswanto, R.; Mardianingrum, R.; Lestari, T.; Nofianti, T.; Rahayuningsih, N. Synthesis and molecular docking of isonicotinohydrazide derivatives as anti-tuberculosis candidates. Malays. J. Fundam. Appl. Sci. 2019, 15, 367–371. [Google Scholar] [CrossRef]
- Shukla, R.; Shukla, H.; Sonkar, A.; Pandey, T.; Tripathi, T. Structure-based screening and molecular dynamics simulations offer novel natural compounds as potential inhibitors of Mycobacterium tuberculosis isocitrate lyase. J. Biomol. Struct. Dyn. 2018, 36, 2045–2057. [Google Scholar] [CrossRef]
- Mondoni, M.; Saderi, L.; Sotgiu, G. Novel treatments in multidrug-resistant tuberculosis. Curr. Opin. Pharmacol. 2021, 59, 103–115. [Google Scholar] [CrossRef]
- Saxena, S.; Abdullah, M.; Sriram, D.; Guruprasad, L. Discovery of novel inhibitors of Mycobacterium tuberculosis MurG: Homology modelling, structure based pharmacophore, molecular docking, and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2018, 36, 3184–3198. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Singh, S.K.; Singh, P.P.; Singh, V.K.; Rai, A.C.; Srivastava, A.K.; Shukla, L.; Kesawat, M.S.; Kumar Jaiswal, A.; Chung, S.M.; et al. Potential Anti-Mycobacterium tuberculosis Activity of Plant Secondary Metabolites: Insight with Molecular Docking Interactions. Antioxidants 2021, 10, 1990. [Google Scholar] [CrossRef] [PubMed]
- Ganaie, A.A.; Trivedi, G.; Kaur, A.; Jha, S.S.; Anand, S.; Rana, V.; Singh, A.; Kumar, S.; Sharma, C. Interaction of Erp protein of Mycobacterium tuberculosis with Rv2212 enhances intracellular survival of Mycobacterium smegmatis. J. Bacteriol. 2016, 198, 2841–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moule, M.G.; Cirillo, J.D. Mycobacterium tuberculosis dissemination plays a critical role in pathogenesis. Front. Cell. Infect. Microbiol. 2020, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, L. Mycobacterium tuberculosis pathogenicity viewed through the lens of molecular Koch’s postulates. Curr. Opin. Microbiol. 2020, 54, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Sharma, A.; Bouzeyen, R.; Deep, A.; Sharma, H.; Mangalaparthi, K.K.; Datta, K.K.; Kidwai, S.; Gowda, H.; Varadarajan, R.; et al. VapBC22 toxin-antitoxin system from Mycobacterium tuberculosis is required for pathogenesis and modulation of host immune response. Sci. Adv. 2020, 6, eaba6944. [Google Scholar] [CrossRef] [PubMed]
- Madacki, J.; Fiol, G.M.; Brosch, R. Update on the virulence factors of the obligate pathogen Mycobacterium tuberculosis and related tuberculosis-causing mycobacteria. Infect. Genet. Evol. 2019, 72, 67–77. [Google Scholar] [CrossRef]
- Kocíncová, D.; Sondén, B.; Bordat, Y.; Pivert, E.; de Mendonça-Lima, L.; Gicquel, B.; Reyrat, J.M. The hydrophobic domain of the mycobacterial Erp protein is not essential for the virulence of Mycobacterium tuberculosis. Infect. Immun. 2004, 72, 2379–2382. [Google Scholar] [CrossRef] [Green Version]
- Cosma, C.L.; Klein, K.; Kim, R.; Beery, D.; Ramakrishnan, L. Mycobacterium marinum Erp is a virulence determinant required for cell wall integrity and intracellular survival. Infect. Immun. 2006, 74, 3125–3133. [Google Scholar] [CrossRef] [Green Version]
- Berthet, F.X.; Lagranderie, M.; Gounon, P.; Laurent-Winter, C.; Ensergueix, D.; Chavarot, P.; Thouron, F.; Maranghi, E.; Pelicic, V.; Portnoï, D.; et al. Attenuation of virulence by disruption of the Mycobacterium tuberculosis erp gene. Science 1998, 282, 759–762. [Google Scholar] [CrossRef]
- Forrellad, M.A.; Vázquez, C.L.; Blanco, F.C.; Klepp, L.I.; García, E.A.; Rocha, R.V.; Luciana, V.; Bigi, M.M.; Gutierrez, M.G.; Bigi, F. Rv2617c and P36 are virulence factors of pathogenic mycobacteria involved in resistance to oxidative stress. Virulence 2019, 10, 1026–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klepp, L.I.; Soria, M.; Blanco, F.C.; Bianco, M.V.; Santangelo, M.P.; Cataldi, A.A.; Bigi, F. Identification of two proteins that interact with the Erp virulence factor from Mycobacterium tuberculosis by using the bacterial two-hybrid system. BMC Mol. Biol. 2009, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthet, F.X.; Rauzier, J.; Lim, E.M.; Philipp, W.; Gicquel, B.; Portnoï, D. Characterization of the Mycobacterium tuberculosis erp gene encoding a potential cell surface protein with repetitive structures. Microbiology 1995, 141, 2123–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exported Repetitive Protein, Accession Code P9WIQ7. Available online: https://www.uniprot.org/uniprot/P9WIQ7 (accessed on 20 September 2021).
- Uncharacterized Protein Rv1417, Accession Code P9WLY1. Available online: https://www.uniprot.org/uniprot/P9WLY1 (accessed on 20 September 2021).
- Probable Transmembrane Protein, Accession Code I6XER9. Available online: https://www.uniprot.org/uniprot/I6XER9 (accessed on 20 September 2021).
- Uniprot. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef] [Green Version]
- Kandt, C.; Ash, W.L.; Tieleman, D.P. Setting up and running molecular dynamics simulations of membrane proteins. Methods 2007, 41, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Lide, D.R. CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA, 2010; Volume 90. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Kutzner, C.; Páll, S.; Fechner, M.; Esztermann, A.; de Groot, B.L.; Grubmüller, H. More bang for your buck: Improved use of GPU nodes for GROMACS 2018. J. Comput. Chem. 2019, 40, 2418–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Duhovny, D.; Nussinov, R.; Wolfson, H.J. Efficient unbound docking of rigid molecules. In Proceedings of the International Workshop on Algorithms in Bioinformatics, Rome, Italy, 17–21 September 2002; pp. 185–200. [Google Scholar]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins Struct. Funct. Bioinform. 2007, 69, 139–159. [Google Scholar] [CrossRef]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef]
- Hurwitz, N.; Wolfson, H.J. Memdock: An α-Helical Membrane Protein Docking Algorithm. In Computational Design of Membrane Proteins; Springer: New York, NY, USA, 2021; pp. 111–117. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Turner, P.J. XMGRACE; Version 5.1.19; Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005.
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Mathematica; Version 12.0; Wolfram Research, Inc.: Champaign, IL, USA, 2019.
- Lee, H.; Kim, H. Membrane topology of transmembrane proteins: Determinants and experimental tools. Biochem. Biophys. Res. Commun. 2014, 453, 268–276. [Google Scholar] [CrossRef]
- Gong, J.; Chen, Y.; Pu, F.; Sun, P.; He, F.; Zhang, L.; Li, Y.; Ma, Z.; Wang, H. Understanding membrane protein drug targets in computational perspective. Curr. Drug Targets 2019, 20, 551–564. [Google Scholar] [CrossRef]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Armenteros, J.J.A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kumar, N.; Shukla, S.; Kumar, S.; Suryawanshi, A.; Chaudhry, U.; Ramachandran, S.; Maiti, S. Intrinsically disordered protein from a pathogenic mesophile Mycobacterium tuberculosis adopts structured conformation at high temperature. Proteins Struct. Funct. Bioinform. 2008, 71, 1123–1133. [Google Scholar] [CrossRef]
- Anurag, M.; Dash, D. Unraveling the potential of intrinsically disordered proteins as drug targets: Application to Mycobacterium tuberculosis. Mol. BioSyst. 2009, 5, 1752–1757. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Kaur, G.; Pandey, B.; Kumar, A.; Garewal, N.; Grover, A.; Kaur, J. Drug targeted virtual screening and molecular dynamics of LipU protein of Mycobacterium tuberculosis and Mycobacterium leprae. J. Biomol. Struct. Dyn. 2019, 37, 1254–1269. [Google Scholar] [CrossRef]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Erdős, G.; Dosztányi, Z. Analyzing protein disorder with IUPred2A. Curr. Protoc. Bioinform. 2020, 70, e99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherepanov, D.A.; Feniouk, B.A.; Junge, W.; Mulkidjanian, A.Y. Low dielectric permittivity of water at the membrane interface: Effect on the energy coupling mechanism in biological membranes. Biophys. J. 2003, 85, 1307–1316. [Google Scholar] [CrossRef] [Green Version]
- Fealey, M.E.; Binder, B.P.; Uversky, V.N.; Hinderliter, A.; Thomas, D.D. Structural impact of phosphorylation and dielectric constant variation on synaptotagmin’s IDR. Biophys. J. 2018, 114, 550–561. [Google Scholar] [CrossRef] [Green Version]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Janek, K.; Behlke, J.; Zipper, J.; Fabian, H.; Georgalis, Y.; Beyermann, M.; Bienert, M.; Krause, E. Water-soluble β-sheet models which self-assemble into fibrillar structures. Biochemistry 1999, 38, 8246–8252. [Google Scholar] [CrossRef]
- Sanderson, J.M. Resolving the kinetics of lipid, protein and peptide diffusion in membranes. Mol. Membr. Biol. 2012, 29, 118–143. [Google Scholar] [CrossRef] [Green Version]
- Leekumjorn, S.; Sum, A.K. Molecular simulation study of structural and dynamic properties of mixed DPPC/DPPE bilayers. Biophys. J. 2006, 90, 3951–3965. [Google Scholar] [CrossRef] [Green Version]
- Regan, D.; Williams, J.; Borri, P.; Langbein, W. Lipid bilayer thickness measured by quantitative DIC reveals phase transitions and effects of substrate hydrophilicity. Langmuir 2019, 35, 13805–13814. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Membrane Systems | Semi-Polar Systems |
|---|---|---|
| NVT equilibrium | ||
| Constraints | all bonds | h-bonds |
| Cutoff | 1.2 nm | 1.1 nm |
| T | V-rescale | V-rescale |
| Temperature | 323.15 K | 309.65 K |
| 0.1 ps | 0.5 ps | |
| NPT equilibrium | ||
| Constraints | all bonds | h-bonds |
| Cutoff | 1.2 nm | 1.1 nm |
| T | Nose–Hoover | V-rescale |
| Temperature | 323.15 K | 309.65 K |
| 0.1 ps | 0.1 ps | |
| P | Parrinello–Rahman | Parrinello–Rahman |
| P type | semi-isotropic | isotropic |
| Pressure | 1.0 bar | 1.0 bar |
| 5.0 ps | 2.0 ps | |
| Production phase | ||
| Constraints | h-bonds | h-bonds |
| Cutoff | 1.2 nm | 1.2 nm |
| T | Nose-Hoover | Nose-Hoover |
| Temperature | 309.65 K | 309.65 K |
| 0.5 ps | 0.1 ps | |
| P | Parrinello–Rahman | Parrinello–Rahman |
| P type | semi-isotropic | isotropic |
| Pressure | 1.0 bar | 1.0 bar |
| 2.0 ps | 1.0 ps | |
| Time trajectory | 500 ns | 200 ns |
| System | RMSD | RMSF | RG | H-Bonds | H-Bonds | |||
|---|---|---|---|---|---|---|---|---|
| Total | x Axis | y Axis | z Axis | + DPPC | ||||
| Membrane and water—500 ns | ||||||||
| Rv1417 | 0.46 ± 0.05 | 0.22 ± 0.14 | 2.14 ± 0.02 | 2.03 ± 0.03 | 2.01 ± 0.03 | 1.00 ± 0.05 | 93 ± 5 | 21 ± 4 |
| Rv2617c | 0.50 ± 0.06 | 0.20 ± 0.07 | 1.85 ± 0.03 | 1.73 ± 0.04 | 1.71 ± 0.03 | 0.98 ± 0.04 | 100 ± 5 | 20 ± 3 |
| Erp | 0.65 ± 0.12 | 0.33 ± 0.17 | 2.04 ± 0.02 | 1.72 ± 0.12 | 1.55 ± 0.15 | 1.70 ± 0.14 | 114 ± 8 | - |
| Ethanol—200 ns | ||||||||
| Rv1417 | 0.63 ± 0.14 | 0.32 ± 0.21 | 1.71 ± 0.06 | 1.41 ± 0.09 | 1.43 ± 0.04 | 1.36 ± 0.05 | 100 ± 5 | - |
| Rv2617c | 0.77 ± 0.11 | 0.44 ± 0.19 | 1.72 ± 0.06 | 1.24 ± 0.06 | 1.51 ± 0.06 | 1.44 ± 0.07 | 92 ± 7 | - |
| Erp | 1.05 ± 0.25 | 0.60 ± 0.26 | 2.46 ± 0.07 | 2.04 ± 0.09 | 1.90 ± 0.06 | 2.07 ± 0.10 | 143 ± 7 | - |
| Rv1417 | Erp | Rv2617c | Erp | |
| Membrane systems | M1, T2, A3, A4, N6, D7, W8, S79, A80, E97, D116, D117, R146, Y147, R148, R154 | D221, A269, A270, A271, A272 V273, P274, P275 | M1, S2, P5, T6, T7, P9, Q50, N53, M54, A57, D62, T67, A68, G111, P112, F114, S140, G141, R145, P146 | H43, E44, T158, P159, G160, T180, G188, A189, D190, G191, T192, Y193, P194, T213, P244, S245 |
| Rv1417 | Erp | Rv2617c | Erp | |
| Unanchored systems | L27, A29, H32, A34, G35, G36, L37, L38, K40, A55, M56, L59, L63, A66, L68, F70, R72, R76, G81, S83, V84, N86, L87, L88, D90, F103, Y119, P121, M123, I125, Q126, A127, V128, D129, K130, D131, R146 | T11, T34, E44, T80, A85, P107, A145, T147, A150, L151, P154, T158, D186, Y193, P194, I195, L196, P208, T211, T213, G216, N227, V241, L242, M243, P244, M247, Q251, P258, A260, P265 | I3, R4, P5, T6, T7, Q14, Y20, Y23, V24, L25, R27, T28, F30, V32, M72, L74, A77, T110, P112, G113, F114, Y115, D116, A118, L119, L124, L134, A135, H139 | A20, V21, S23, P24, C25, F28, L58, F69, L127, A128, A135, P138, V140, G141, D186, G202, S206, S212, N227, M243, Q248, Q251, N252, A255, A256, P262, A269, P274 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paco-Chipana, M.; Febres-Molina, C.; Aguilar-Pineda, J.A.; Gómez, B. Novel In Silico Insights into Rv1417 and Rv2617c as Potential Protein Targets: The Importance of the Medium on the Structural Interactions with Exported Repetitive Protein (Erp) of Mycobacterium tuberculosis. Polymers 2022, 14, 2577. https://doi.org/10.3390/polym14132577
Paco-Chipana M, Febres-Molina C, Aguilar-Pineda JA, Gómez B. Novel In Silico Insights into Rv1417 and Rv2617c as Potential Protein Targets: The Importance of the Medium on the Structural Interactions with Exported Repetitive Protein (Erp) of Mycobacterium tuberculosis. Polymers. 2022; 14(13):2577. https://doi.org/10.3390/polym14132577
Chicago/Turabian StylePaco-Chipana, Margot, Camilo Febres-Molina, Jorge Alberto Aguilar-Pineda, and Badhin Gómez. 2022. "Novel In Silico Insights into Rv1417 and Rv2617c as Potential Protein Targets: The Importance of the Medium on the Structural Interactions with Exported Repetitive Protein (Erp) of Mycobacterium tuberculosis" Polymers 14, no. 13: 2577. https://doi.org/10.3390/polym14132577