
Therapeutic Approaches in Lysosomal Storage Diseases


Abstract
:1. Introduction
1.1. General Considerations concerning Rare Diseases
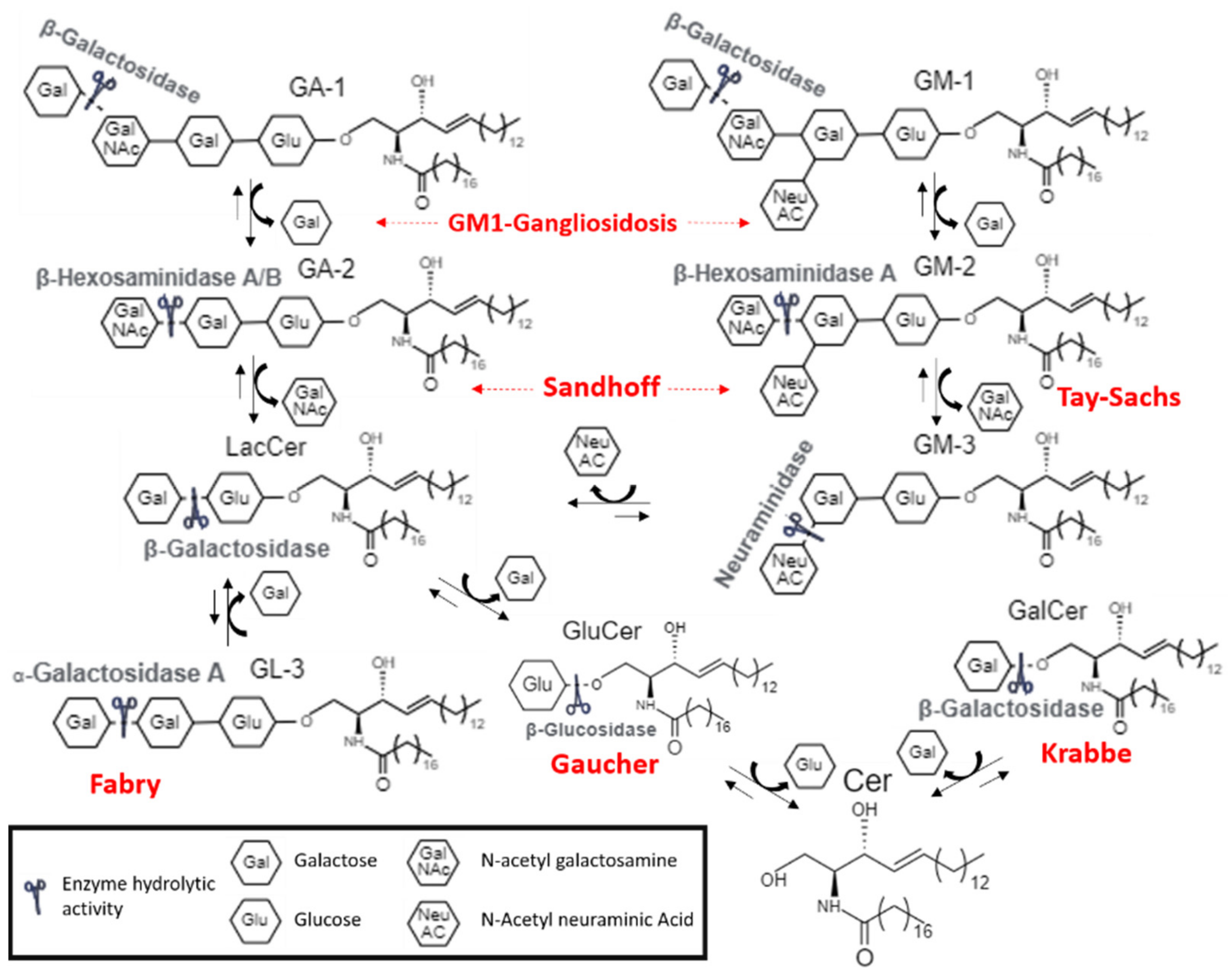
1.2. Molecular Basis of LSDs and Possible Therapeutical Strategies
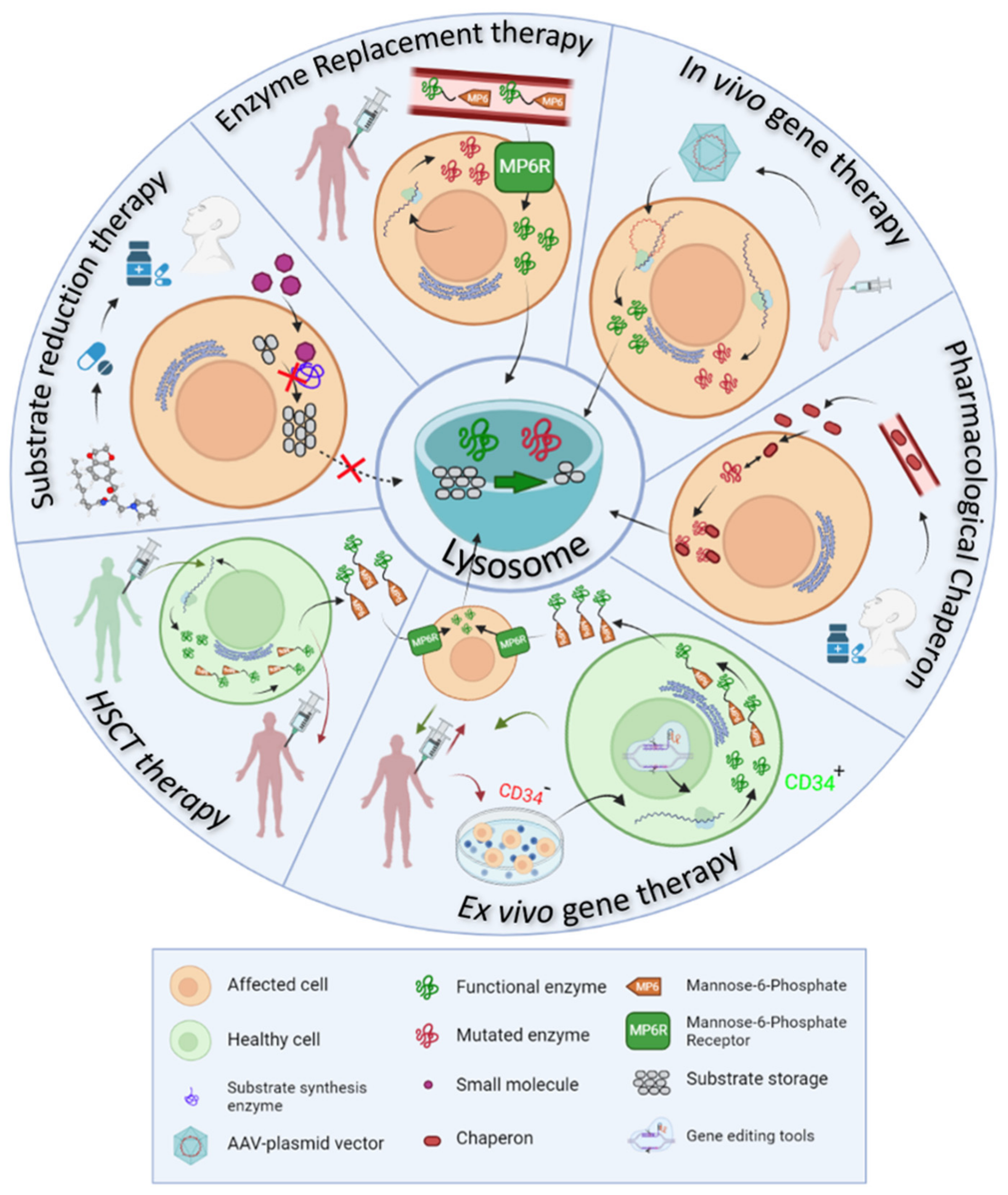
- Enzyme replacement therapy (ERT) consists of the intravenous administration of a properly glycosylated and functional form of the enzyme impaired in the disease, and takes advantage of the MP6R and the endogenous endocytic routes to reach the lysosomes within the target cells.
- Ex vivo gene therapy aims to administer a functional enzyme through the autologous transplant of hematopoietic stem cells, which are genetically modified in vitro.
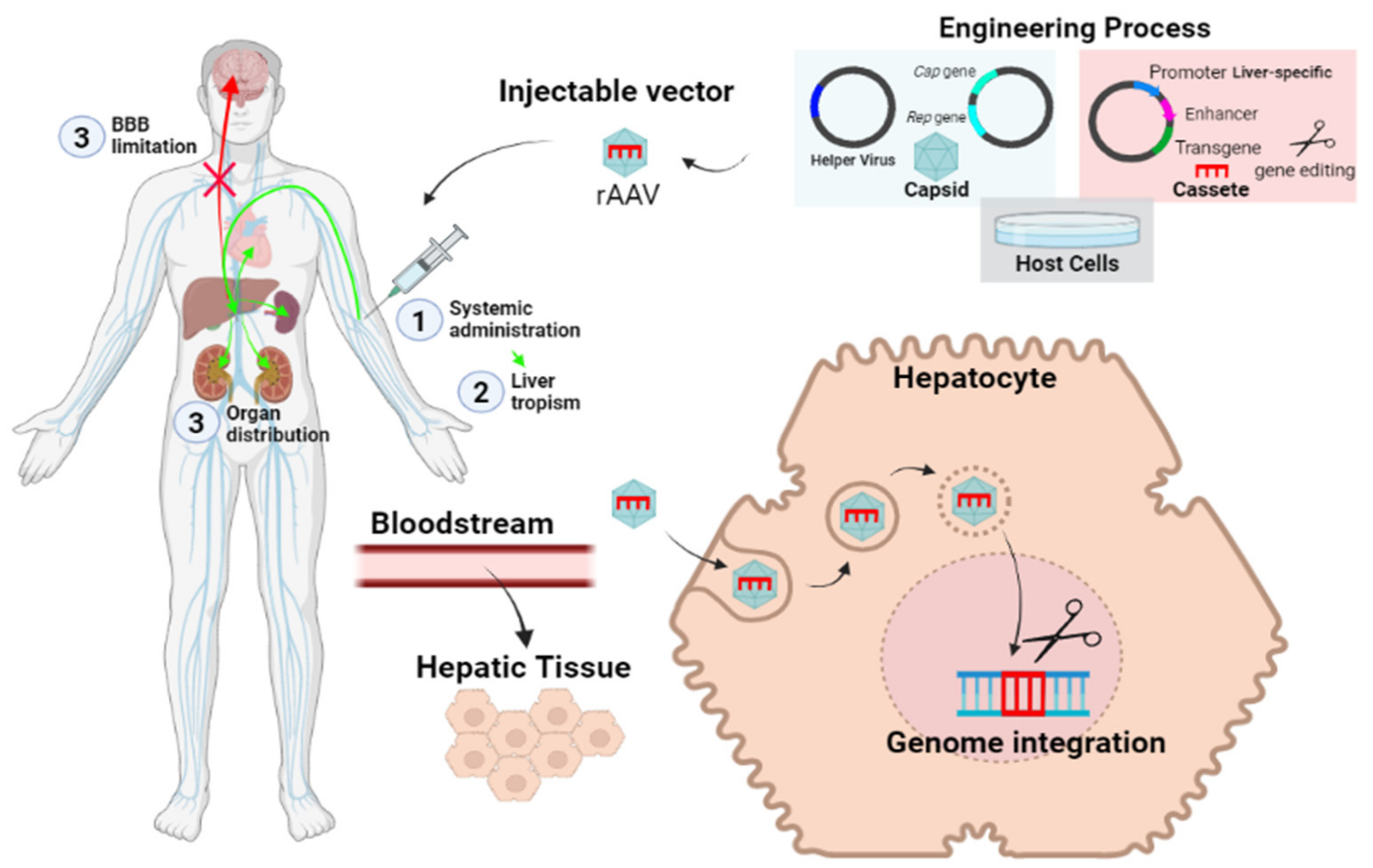
- In vivo gene therapy consists of the direct injection of non-replicating non-hazardous viral vectors, which encode for the functional enzymes of interest in the transduced cells.
- Pharmacological chaperones (PC) stabilize the mutated enzymes to avoid their degradation in the endoplasmic reticulum and therefore, facilitate their translocation to the lysosome and the degradation of accumulated substrates.
- Substrate reduction therapy (SRT) and autophagy regulating drugs aim to target the molecular pathways in which the mutated protein participates, trying to restore the correct balance between synthesis and degradation of the substrates by slowing down the synthesis, accelerating the degradation or regulating vesicle trafficking of the substrates.
2. Enzyme Replacement Therapy
2.1. Available Drugs and ERT Mechanism of Action
2.2. Limitations of Enzymatic Replacement Therapy
2.3. New Generation Enzyme Replacement Therapy
3. Hematopoietic Stem Cell Transplantation
4. Gene Therapy in LSDs
4.1. Ex Vivo Genetic Therapy
4.2. In Vivo Genetic Therapy
5. Small Molecules of Oral Administration
- pharmacological chaperones
- substrate reduction therapies
- autophagy and proteostasis regulators
5.1. Pharmacological Chaperones
5.2. Substrate Reduction Therapy
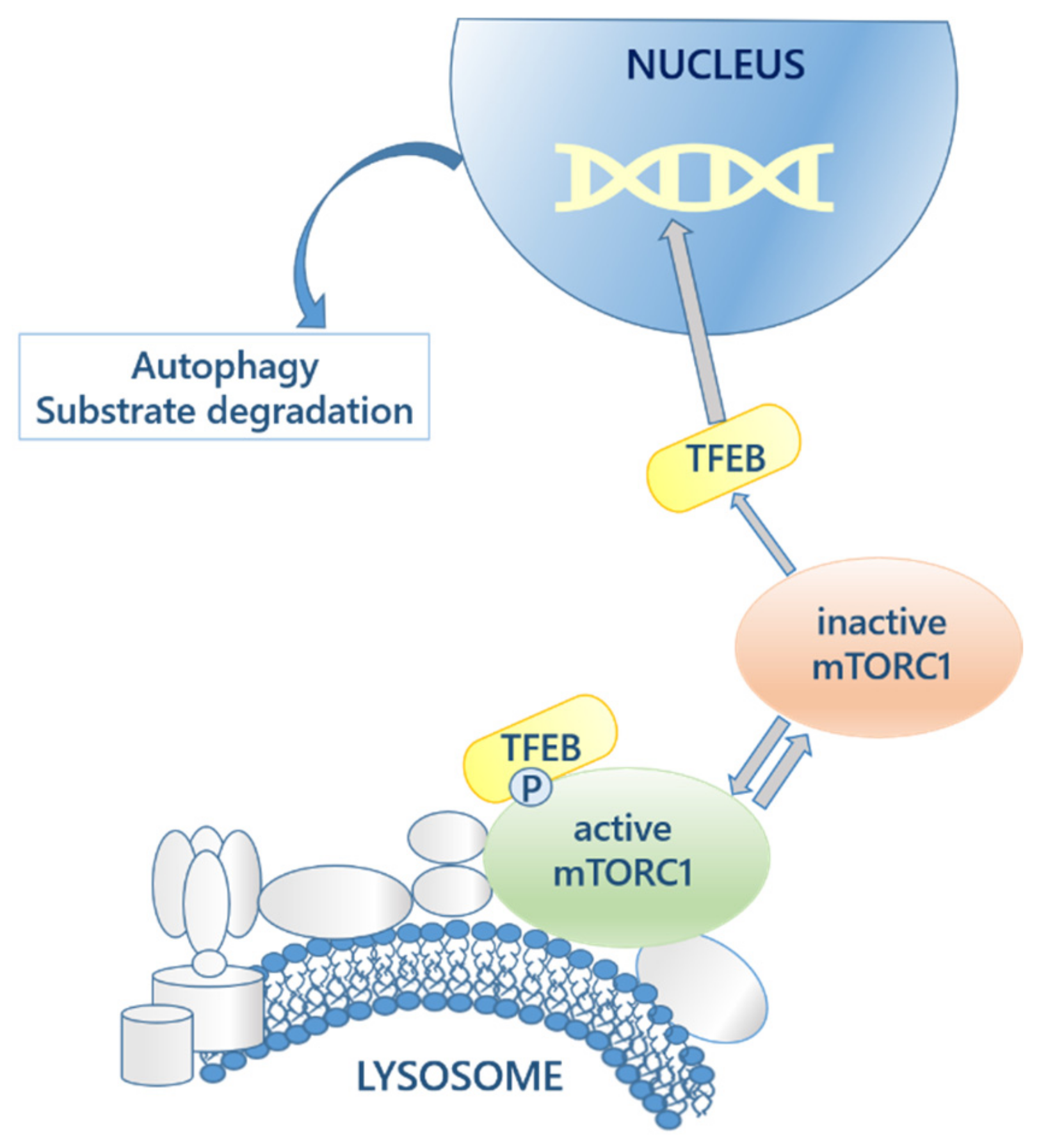
5.3. Autophagy and Proteostasis Regulatory Molecules
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parkinson-Lawrence, E.J.; Shandala, T.; Prodoehl, M.; Plew, R.; Borlace, G.N.; Brooks, D.A. Lysosomal storage Disease: Revealing lysosomal function and physiology. Physiology 2010, 25, 102–115. [Google Scholar] [CrossRef] [Green Version]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Braulke, T.; Bonifacino, J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta-Mol. Cell Res. 2009, 1793, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Reczek, D.; Schwake, M.; Schröder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 Is a Receptor for Lysosomal Mannose-6-Phosphate-Independent Targeting of β-Glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Shea, L.; Raben, N. Autophagy in skeletal muscle: Implications for Pompe disease. Int. J. Clin. Pharmacol. Ther. 2009, 47, S42. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.L.; De Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2007, 17, 119–129. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; A Smith, D.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauhin, W.; Lidove, O.; Masat, E.; Mingozzi, F.; Mariampillai, K.; Ziza, J.-M.; Benveniste, O. Innate and adaptive immune response in fabry disease. JIMD Rep. 2015, 22, 1–10. [Google Scholar] [PubMed] [Green Version]
- Satoh, K. Globotriaosylceramide Induces Endothelial Dysfunction in Fabry Disease. Arter. Thromb. Vasc. Biol. 2014, 34, 2–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lachmann, R.H. Enzyme replacement therapy for lysosomal storage diseases. Curr. Opin. Pediatr. 2011, 23, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.W.; Brady, R.O.; Dambrosia, J.M.; Di Bisceglie, A.M.; Doppelt, S.H.; Hill, S.C.; Mankin, H.J.; Murray, G.J.; Parker, R.I.; Argoff, C.E.; et al. Replacement Therapy for Inherited Enzyme Deficiency—Macrophage-Targeted Glucocerebrosidase for Gaucher’s Disease. N. Engl. J. Med. 1991, 324, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Dinur, T.; Grittner, U.; Revel-Vilk, S.; Becker-Cohen, M.; Istaiti, M.; Cozma, C.; Rolfs, A.; Zimran, A. Impact of Long-Term Enzyme Replacement Therapy on Glucosylsphingosine (Lyso-Gb1) Values in Patients with Type 1 Gaucher Disease: Statistical Models for Comparing Three Enzymatic Formulations. Int. J. Mol. Sci. 2021, 22, 7699. [Google Scholar] [CrossRef] [PubMed]
- Arends, M.; Biegstraaten, M.; Wanner, C.; Sirrs, S.; Mehta, A.; Elliott, P.; Oder, D.; Watkinson, O.T.; Bichet, D.G.; Khan, A.; et al. Agalsidase alfa versus agalsidase beta for the treatment of Fabry disease: An international cohort study. J. Med Genet. 2018, 55, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Schoser, B.; Stewart, A.; Kanters, S.; Hamed, A.; Jansen, J.; Chan, K.; Karamouzian, M.; Toscano, A. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: A systematic review and meta-analysis. J. Neurol. 2017, 264, 621–630. [Google Scholar] [CrossRef]
- Pena, L.D.M.; Barohn, R.J.; Byrne, B.J.; Desnuelle, C.; Goker-Alpan, O.; Ladha, S.; Laforêt, P.; Mengel, K.E.; Pestronk, A.; Pouget, J.; et al. Safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy of the novel enzyme replacement therapy avalglucosidase alfa (neoGAA) in treatment-naïve and alglucosidase alfa-treated patients with late-onset Pompe disease: A phase 1, open-label, multicenter, multinational, ascending dose study. Neuromuscul. Disord. 2019, 29, 167–186. [Google Scholar] [PubMed] [Green Version]
- Jameson, E.; Jones, S.; Remmington, T. Enzyme replacement therapy with laronidase (Aldurazyme®) for treating mucopolysaccharidosis type I. Cochrane Database Syst. Rev. 2019, 6, 1–24. [Google Scholar]
- Whiteman, D.A.H.; Kimura, A. Development of idursulfase therapy for mucopolysaccharidosis type II(Hunter syndrome): The past, the present and the future. Drug Des. Dev. Ther. 2017, 11, 2467–2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuyama, T.; Eto, Y.; Sakai, N.; Minami, K.; Yamamoto, T.; Sonoda, H.; Yamaoka, M.; Tachibana, K.; Hirato, T.; Sato, Y. Iduronate-2-Sulfatase with Anti-human Transferrin Receptor Antibody for Neuropathic Mucopolysaccharidosis II: A Phase 1/2 Trial. Mol. Ther. 2019, 27, 456–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- í Dali, C.; Sevin, C.; Krägeloh-Mann, I.; Giugliani, R.; Sakai, N.; Wu, J.; Wasilewski, M. Safety of intrathecal delivery of recombinant human arylsulfatase A in children with metachromatic leukodystrophy: Results from a phase 1/2 clinical trial. Mol. Genet. Metab. 2020, 131, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Barton, R.W.; Neufeld, E.F. The Hurler corrective factor. Purification and some properties. J. Biol. Chem. 1971, 246, 7773–7779. [Google Scholar] [CrossRef]
- Tian, W.; Ye, Z.; Wang, S.; Schulz, M.A.; Van Coillie, J.; Sun, L.; Chen, Y.-H.; Narimatsu, Y.; Hansen, L.; Kristensen, C.; et al. The glycosylation design space for recombinant lysosomal replacement enzymes produced in CHO cells. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Galpin, J.D.; Tropak, M.B.; Mahuran, D.; Haselhorst, T.; Von Itzstein, M.; Kolarich, D.; Packer, N.; Miao, Y.; Jiang, L.; et al. Production of active human glucocerebrosidase in seeds of Arabidopsis thaliana complex-glycan-deficient (cgl) plants. Glycobiology 2011, 22, 492–503. [Google Scholar] [CrossRef] [Green Version]
- Chiba, Y.; Akeboshi, H. Glycan Engineering and Production of ‘Humanized’ Glycoprotein in Yeast Cells. Biol. Pharm. Bull. 2009, 32, 786–795. [Google Scholar] [CrossRef] [Green Version]
- Oder, D.; Nordbeck, P.; Wanner, C. Long Term Treatment with Enzyme Replacement Therapy in Patients with Fabry Disease. Nephron 2016, 134, 30–36. [Google Scholar] [CrossRef]
- Concolino, D.; Deodato, F.; Parini, R. Enzyme replacement therapy: Efficacy and limitations. Ital. J. Pediatr. 2018, 44, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Sly, W.S.; Vogler, C.; Grubb, J.H.; Levy, B.; Galvin, N.; Tan, Y.; Nishioka, T.; Tomatsu, S. Enzyme therapy in mannose receptor-null mucopolysaccharidosis VII mice defines roles for the mannose 6-phosphate and mannose receptors. Proc. Natl. Acad. Sci. USA 2006, 103, 15172–15177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroobants, S.; Gerlach, D.; Matthes, F.; Hartmann, D.; Fogh, J.; Gieselmann, V.; D’Hooge, R.; Matzner, U. Intracerebroventricular enzyme infusion corrects central nervous system pathology and dysfunction in a mouse model of metachromatic leukodystrophy. Hum. Mol. Genet. 2011, 20, 2760–2769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.C.; Luu, A.R.; Wise, N.; De Angelis, R.; Agrawal, V.; Mangini, L.; Vincelette, J.; Handyside, B.; Sterling, H.; Lo, M.J.; et al. Intracerebroventricular enzyme replacement therapy with β-galactosidase reverses brain pathologies due to GM1 gangliosidosis in mice. J. Biol. Chem. 2020, 295, 13532–13555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, M.; Muro, S. Lysosomal enzyme replacement therapies: Historical development, clinical outcomes, and future perspectives. Adv. Drug Deliv. Rev. 2017, 118, 109–134. Available online: http://europepmc.org/articles/pmc5828774?pdf=render (accessed on 15 November 2021). [CrossRef]
- Rombach, S.M.; Aerts, J.; Poorthuis, B.J.H.M.; Groener, J.E.M.; Donker-Koopman, W.; Hendriks, E.; Mirzaian, M.; Kuiper, S.; Wijburg, F.A.; Hollak, C.E.M.; et al. Long-Term Effect of Antibodies against Infused Alpha-Galactosidase A in Fabry Disease on Plasma and Urinary (lyso)Gb3 Reduction and Treatment Outcome. PLOS ONE 2012, 7, e47805. [Google Scholar] [CrossRef] [PubMed]
- Banugaria, S.G.; Prater, S.N.; Ng, Y.-K.; Kobori, J.A.; Finkel, R.S.; Ladda, R.L.; Chen, Y.-T.; Rosenberg, A.S.; Kishnani, P.S. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: Lessons learned from infantile Pompe disease. Genet. Med. 2011, 13, 729–736. [Google Scholar] [CrossRef] [Green Version]
- Banugaria, S.G.; Prater, S.N.; Patel, T.T.; DeArmey, S.M.; Milleson, C.; Sheets, K.B.; Bali, D.S.; Rehder, C.W.; Raiman, J.A.J.; Wang, R.A.; et al. Algorithm for the Early Diagnosis and Treatment of Patients with Cross Reactive Immunologic Material-Negative Classic Infantile Pompe Disease: A Step towards Improving the Efficacy of ERT. PLOS ONE 2013, 8, e67052. [Google Scholar]
- Lenders, M.; Stappers, F.; Brand, E. In Vitro and In Vivo Amenability to Migalastat in Fabry Disease. Mol. Ther.-Methods Clin. Dev. 2020, 19, 24–34. [Google Scholar] [CrossRef]
- Abasolo, I.; Seras-Franzoso, J.; Moltó-Abad, M.; Díaz-Riascos, V.; Corchero, J.L.; Pintos-Morell, G.; Schwartz, S., Jr. Nanotechnology-based approaches for treating lysosomal storage disorders, a focus on Fabry disease. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 13, e1684. [Google Scholar] [CrossRef]
- Kizhner, T.; Azulay, Y.; Hainrichson, M.; Tekoah, Y.; Arvatz, G.; Shulman, A.; Ruderfer, I.; Aviezer, D.; Shaaltiel, Y. Characterization of a chemically modified plant cell culture expressed human α-Galactosidase-A enzyme for treatment of Fabry disease. Mol. Genet. Metab. 2015, 114, 259–267. [Google Scholar] [CrossRef]
- Schiffmann, R.; Goker-Alpan, O.; Holida, M.; Giraldo, P.; Barisoni, L.; Colvin, R.B.; Jennette, C.J.; Maegawa, G.; Boyadjiev, S.A.; Gonzalez, D.; et al. Pegunigalsidase alfa, a novel PEGylated enzyme replacement therapy for Fabry disease, provides sustained plasma concentrations and favorable pharmacodynamics: A 1-year Phase 1/2 clinical trial. J. Inherit. Metab. Dis. 2019, 42, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Hui, E.K.-W.; Lu, J.Z.; Zhou, Q.-H.; Pardridge, W.M. Reversal of Lysosomal Storage in Brain of Adult MPS-I Mice with Intravenous Trojan Horse-Iduronidase Fusion Protein. Mol. Pharm. 2011, 8, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Pignata, C.; Vajro, P.; Salerno, M. New strategies for the treatment of lysosomal storage diseases (Review). Int. J. Mol. Med. 2012, 31, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Valayannopoulos, V.; Wijburg, F.A. Therapy for the mucopolysaccharidoses. Rheumatology 2011, 50 (Suppl. 5), v49–v59. [Google Scholar] [CrossRef] [Green Version]
- Yoon, I.C.; Bascou, N.A.; Poe, M.D.; Szabolcs, P.; Escolar, M.L. Long-Term Neurodevelopmental Outcomes of Hematopoietic Stem Cell Transplantation for Late-Infantile Krabbe Disease. Blood 2020, 137, 1719–1730. [Google Scholar] [CrossRef]
- Orchard, P.J.; Tolar, J. Transplant Outcomes in Leukodystrophies. Semin. Hematol. 2010, 47, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Ries, M.; Clarke, J.T.; Whybra, C.; Timmons, M.; Robinson, C.; Schlaggar, B.L.; Pastores, G.; Lien, Y.H.; Kampmann, C.; Brady, R.O.; et al. Enzyme-Replacement Therapy With Agalsidase Alfa in Children With Fabry Disease. Pediatrics 2006, 118, 924–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.K.; Shukla, P. Gene editing for cell engineering: Trends and applications. Crit. Rev. Biotechnol. 2016, 37, 672–684. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Benefits Metachromatic Leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medin, J.A.; Khan, A.; Huang, J.; Barber, D.; Rupar, C.A.; Auray-Blais, C.; Fraser, G.; Fowler, D.H.; Keating, A.; West, M.L.; et al. FACTs Fabry gene therapy clinical trial: Two-year data. Mol. Genet. Metab. 2019, 126, S99. [Google Scholar] [CrossRef]
- Fumagalli, F.; Calbi, V.; Sessa, M.; Zambon, A.; Baldoli, C.; Rancoita, P.M.; Acquati, S.; De Mattia, F.; Tucci, F.; Gallo, V.; et al. Lentiviral hematopoietic stem and progenitor cell gene therapy (HSPC-GT) for metachromatic leukodystrophy (MLD): Clinical outcomes from 33 patients. Mol. Genet. Metab. 2020, 129, S59. [Google Scholar] [CrossRef]
- NCT04283227. Available online: https://clinicaltrials.gov/ct2/show/study/NCT04283227?cond=OTL-200&draw=2&rank=1 (accessed on 15 November 2021).
- Khan, A.; Barber, D.L.; Huang, J.; Rupar, C.A.; Rip, J.W.; Auray-Blais, C.; Boutin, M.; O’Hoski, P.; Gargulak, K.; McKillop, W.M.; et al. Lentivirus-mediated gene therapy for Fabry disease. Nat. Commun. 2021, 12, 1–9. [Google Scholar] [CrossRef]
- NCT04145037. Available online: https://clinicaltrials.gov/ct2/show/NCT04145037 (accessed on 15 November 2021).
- NCT03488394. Available online: https://clinicaltrials.gov/ct2/show/NCT03488394?term=NCT03488394&draw=2&rank=1 (accessed on 15 November 2021).
- NCT03566043. Available online: https://clinicaltrials.gov/ct2/show/NCT03566043?cond=RGX-121&draw=2&rank=2 (accessed on 15 November 2021).
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Ortolano, S.; Spuch, C.; Navarro, C. Present and future of adeno associated virus based gene therapy approaches. Recent Pat. Endocr. Metab. Immune Drug Discov. 2012, 6, 47–66. [Google Scholar] [CrossRef] [PubMed]
- Manfredsson, F.P.; Rising, A.C.; Mandel, R.J. AAV9: A potential blood-brain barrier buster. Mol. Ther. 2009, 17, 403–405. [Google Scholar] [CrossRef]
- Biferi, M.G.; Cohen-Tannoudji, M.; García-Silva, A.; Souto-Rodríguez, O.; Viéitez-González, I.; San-Millán-Tejado, B.; Fernández-Carrera, A.; Pérez-Márquez, T.; Teijeira-Bautista, S.; Barrera, S.; et al. Systemic Treatment of Fabry Disease Using a Novel AAV9 Vector Expressing α-Galactosidase A. Mol. Ther.-Methods Clin. Dev. 2020, 20, 1–17. [Google Scholar] [CrossRef]
- Aravalli, R.N.; Belcher, J.D.; Steer, C.J. Liver-targeted gene therapy: Approaches and challenges. Liver Transplant. 2015, 21, 718–737. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Anguela, X.M.; Doyon, Y.; Wechsler, T.; DeKelver, R.C.; Sproul, S.; Paschon, D.E.; Miller, J.C.; Davidson, R.J.; Shivak, D.; et al. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood 2015, 126, 1777–1784. [Google Scholar] [CrossRef] [Green Version]
- Puzzo, F.; Colella, P.; Biferi, M.G.; Bali, D.; Paulk, N.K.; Vidal, P.; Collaud, F.; Simon-Sola, M.; Charles, S.; Hardet, R.; et al. Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid α-glucosidase. Sci. Transl. Med. 2017, 9, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, M.; Huston, M.W.; Pagant, S.; Gan, L.; Martin, S.S.; Sproul, S.; Richards, D.; Ballaron, S.; Hettini, K.; Ledeboer, A.; et al. AAV2/6 Gene Therapy in a Murine Model of Fabry Disease Results in Supraphysiological Enzyme Activity and Effective Substrate Reduction. Mol. Ther.-Methods Clin. Dev. 2020, 18, 607–619. [Google Scholar] [CrossRef] [PubMed]
- NCT04093349. Available online: https://clinicaltrials.gov/ct2/show/NCT04093349?term=SPK-3006&draw=2&rank=1 (accessed on 15 November 2021).
- Hughes, D.A.; Patel, N.; Kinch, R.; Dronfield, L.; Short, G.; Sheridan, R.; Kia, A.; Jeyakumar, J.; Corbau, R.; Nathwani, A. First-in-human study of a liver-directed AAV gene therapy (FLT190) in Fabry disease. Mol. Genet. Metab. 2020, 129, S77–S78. [Google Scholar] [CrossRef]
- NCT04040049. Available online: https://clinicaltrials.gov/ct2/show/NCT04040049?cond=FLT190&draw=2 (accessed on 15 November 2021).
- NTC03580083. Available online: https://clinicaltrials.gov/ct2/show/NCT03580083?cond=RGX-111&draw=2 (accessed on 15 November 2021).
- Bradbury, A.M.; Rafi, M.A.; Bagel, J.H.; Brisson, B.K.; Marshall, M.S.; Salvador, J.P.; Jiang, X.; Swain, G.P.; Prociuk, M.L.; Odonnell, P.A.; et al. AAVrh10 Gene Therapy Ameliorates Central and Peripheral Nervous System Disease in Canine Globoid Cell Leukodystrophy (Krabbe Disease). Hum. Gene Ther. 2018, 29, 785–801. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.M.; Bagel, J.H.; Nguyen, D.; Lykken, E.A.; Salvador, J.P.; Jiang, X.; Swain, G.P.; Assenmacher, C.A.; Hendricks, I.J.; Miyadera, K.; et al. Krabbe disease successfully treated via monotherapy of intrathecal gene therapy. J. Clin. Investig. 2020, 130, 4906–4920. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Nicholls, K.; Giugliani, R.; Bichet, D.G.; Hughes, D.A.; Barisoni, L.M.; Colvin, R.B.; Jennette, J.C.; Skuban, N.; Castelli, J.P.; et al. Efficacy of the pharmacologic chaperone migalastat in a subset of male patients with the classic phenotype of Fabry disease and migalastat-amenable variants: Data from the phase 3 randomized, multicenter, double-blind clinical trial and extension study. Genet. Med. 2019, 21, 1987–1997. [Google Scholar] [CrossRef] [Green Version]
- Pastores, G.M. Miglustat: Substrate reduction therapy for lysosomal storage disorders associated with primary central nervous system involvement. Recent Pat. CNS Drug Discov. 2006, 1, 77–82. [Google Scholar] [CrossRef]
- Cox, T.M.; Drelichman, G.I.; Cravo, R.; Balwani, M.; Burrow, T.A.; Martins, A.M.; Lukina, E.; Rosenbloom, B.E.; Ross, L.H.; Angell, J.; et al. ENCORE, a randomized, controlled, open-label non-inferiority study comparing eliglustat to imiglucerase in Gaucher disease type 1 patients stabilized on enzyme replacement therapy: 24-month results. Mol. Genet. Metab. 2015, 114, S33–S34. [Google Scholar] [CrossRef]
- Patterson, M.C.; Vecchio, D.; Jacklin, E.; Abel, L.; Chadha-Boreham, H.; Luzy, C.; Giorgino, R.; Wraith, J.E. Long-Term Miglustat Therapy in Children With Niemann-Pick Disease Type C. J. Child Neurol. 2010, 25, 300–305. [Google Scholar] [CrossRef]
- Ortolano, S. Small molecules: Substrate inhibitors, chaperones, stop-codon read through, and beyond. J. Inborn. Errors Metab. Screen. 2016, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological chaperone therapy: Preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol. Ther. 2015, 23, 1138–1148. Available online: http://europepmc.org/search?query=(DOI:10.1038/mt.2015.62)%0Ahttp://search.ebscohost.com/login.aspx?direct=true&scope=site&site=ehost-live&db=mdc&AN=25881001%0Ahttps://gateway.proquest.com/openurl?ctx_ver=Z39.88-2004&res_id=xri:pqm&req_dat=xri:pqil:pq_cln (accessed on 15 November 2021). [CrossRef] [PubMed] [Green Version]
- Fan, J.Q.; Ishii, S. Active-site-specific chaperone therapy for Fabry disease: Yin and Yang of enzyme inhibitors. FEBS J. 2007, 274, 4962–4971. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Nicholls, K.; Sunder-Plassmann, G.; Jovanovic, A.; Feldt-Rasmussen, U.; Schiffmann, R.; Giugliani, R.; Jain, V.; Viereck, C.; Castelli, J.P.; et al. Safety of switching to Migalastat from enzyme replacement therapy in Fabry disease: Experience from the Phase 3 ATTRACT study. Am. J. Med. Genet. Part A 2019, 179, 1069–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffmann, R.; Bichet, D.G.; Benjamin, E.; Wu, X.; Giugliani, R. The migalastat GLP-HEK assay is the gold standard for determining amenability in patients with Fabry disease. Mol. Genet. Metab. Rep. 2019, 20, 100494. [Google Scholar] [CrossRef]
- Flanagan, J.J.; Rossi, B.; Tang, K.; Wu, X.; Mascioli, K.; Donaudy, F.; Tuzzi, M.R.; Fontana, F.; Cubellis, M.V.; Porto, C.; et al. The pharmacological chaperone 1-deoxynojirimycin increases the activity and lysosomal trafficking of multiple mutant forms of acid alpha-glucosidase. Hum. Mutat. 2009, 30, 1683–1692. [Google Scholar] [CrossRef]
- Khanna, R.; Powe, A.C.; Lun, Y.; Soska, R.; Feng, J.; Dhulipala, R.; Frascella, M.; Garcia, A.; Pellegrino, L.J.; Xu, S.; et al. The Pharmacological Chaperone AT2220 Increases the Specific Activity and Lysosomal Delivery of Mutant Acid Alpha-Glucosidase, and Promotes Glycogen Reduction in a Transgenic Mouse Model of Pompe Disease. PLOS ONE 2014, 9, e102092. [Google Scholar]
- Guce, A.I.; Clark, N.E.; Salgado, E.N.; Ivanen, D.R.; Kulminskaya, A.A.; Brumer, H.; Garman, S.C. Catalytic Mechanism of Human α-Galactosidase. J. Biol. Chem. 2010, 285, 3625–3632. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Padia, J.; Urban, D.J.; Jadhav, A.; Goker-Alpan, O.; Simeonov, A.; Goldin, E.; Auld, D.; LaMarca, M.E.; Inglese, J.; et al. Three classes of glucocerebrosidase inhibitors identified by quantitative high-throughput screening are chaperone leads for Gaucher disease. Proc. Natl. Acad. Sci. USA 2007, 104, 13192–13197. [Google Scholar] [CrossRef] [Green Version]
- Marugan, J.J.; Zheng, W.; Ferrer, M.; Motabar, O.; Southall, N.; Goldin, E.; Westbroek, W.; Sidransky, E. Discovery, SAR, and Biological Evaluation of a Non-Inhibitory Chaperone for Acid Alpha Glucosidase. Probe Reports from the NIH Molecular Libraries Program; 2010. Available online: http://www.ncbi.nlm.nih.gov/pubmed/23905202 (accessed on 15 November 2021).
- Warnock, D.G.; Bichet, D.-G.; Holida, M.; Goker-Alpan, O.; Nicholls, K.; Thomas, M.; Eyskens, F.; Shankar, S.; Adera, M.; Sitaraman, S.; et al. Oral Migalastat HCl Leads to Greater Systemic Exposure and Tissue Levels of Active α-Galactosidase A in Fabry Patients when Co-Administered with Infused Agalsidase. PLOS ONE 2015, 10, e0134341. [Google Scholar] [CrossRef]
- Shen, J.-S.; Edwards, N.J.; Bin Hong, Y.; Murray, G.J. Isofagomine increases lysosomal delivery of exogenous glucocerebrosidase. Biochem. Biophys. Res. Commun. 2008, 369, 1071–1075. [Google Scholar] [CrossRef] [Green Version]
- Voss, A.A.; Díez-Sampedro, A.; Hirayama, B.A.; Loo, D.D.F.; Wright, E.M. Imino Sugars Are Potent Agonists of the Human Glucose Sensor SGLT3. Mol. Pharmacol. 2006, 71, 628–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiri, M.; Naim, H.Y. Miglustat-induced intestinal carbohydrate malabsorption is due to the inhibition of α-glucosidases, but not β-galactosidases. J. Inherit. Metab. Dis. 2012, 35, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Shayman, J.A. Eliglustat tartrate. Glucosylceramide synthase inhibitor treatment of type 1 gaucher disease. Drugs Future 2010, 35, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Guérard, N.; Oder, D.; Nordbeck, P.; Zwingelstein, C.; Morand, O.; Welford, R.W.; Dingemanse, J.; Wanner, C. Lucerastat, an Iminosugar for Substrate Reduction Therapy: Tolerability, Pharmacodynamics, and Pharmacokinetics in Patients With Fabry Disease on Enzyme Replacement. Clin. Pharmacol. Ther. 2017, 103, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Guérard, N.; Morand, O.; Dingemanse, J. Lucerastat, an iminosugar with potential as substrate reduction therapy for glycolipid storage disorders: Safety, tolerability, and pharmacokinetics in healthy subjects. Orphanet J. Rare Dis. 2017, 12, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Peterschmitt, M.J.; Crawford, N.P.S.; Gaemers, S.J.M.; Ji, A.J.; Sharma, J.; Pham, T.T. Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability of Oral Venglustat in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2020, 10, 86–98. [Google Scholar] [CrossRef]
- Mu, T.; Ong, D.S.T.; Wang, Y.-J.; Balch, W.E.; Yates, J.R.; Segatori, L.; Kelly, J.W. Chemical and Biological Approaches Synergize to Ameliorate Protein-Folding Diseases. Cell 2008, 134, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.; Sardiello, M.; et al. Transcriptional Activation of Lysosomal Exocytosis Promotes Cellular Clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar] [CrossRef]
- Song, W.; Wang, F.; Savini, M.; Ake, A.; Di Ronza, A.; Sardiello, M.; Segatori, L. TFEB regulates lysosomal proteostasis. Hum. Mol. Genet. 2013, 22, 1994–2009. [Google Scholar] [CrossRef] [Green Version]
- Moskot, M.; Montefusco, S.; Jakóbkiewicz-Banecka, J.; Mozolewski, P.; Wegrzyn, A.; di Bernardo, D.; Wegrzyn, G.; Medina, D.L.; Ballabio, A.; Gabig-Cimińska, M. The Phytoestrogen Genistein Modulates Lysosomal Metabolism and Transcription Factor EB (TFEB) Activation. J. Biol. Chem. 2014, 289, 17054–17069. [Google Scholar] [CrossRef] [Green Version]
- Petersen, N.H.; Kirkegaard, T. HSP70 and lysosomal storage disorders: Novel therapeutic opportunities. Biochem. Soc. Trans. 2010, 38, 1479–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LSDs | Affected Enzyme | Available ERT | Development Status | Reference |
|---|---|---|---|---|
| GD | Glucocerebrosidase | Imglucerase | Approved | [18] |
| Velaglucerase | Approved | |||
| Taliglucerase alpha | Approved | |||
| FD | α-Galactosidase A | Agalsidase alpa | Approved | [19] |
| Agalsidase beta | Approved | NCT03018730 | ||
| PRX-102 | Phase III | NCT02795676 | ||
| NCT03180840 | ||||
| PD | α-Glycosidase | Alglucosidase alpha | Approved | [20] |
| Avalglucosidase alfa | Approved | [21] | ||
| VAL-1221 | Phase I-II | NCT02898753 | ||
| ATB200 | Phase III | NCT03729362 | ||
| MPS I | α-L-iduronidase | Laronidase | Approved | [22] |
| MPS II | Iduronate-2-sulfatase | Idursulfasa | Approved | [23] |
| AGT-182 | Phase I | NCT02262338 | ||
| JR-141 | Approved | [24] | ||
| MPS III A B | Heparan N-sulphatase | rhHNS | Phase I-II | NCT01299727 |
| N-acetyl-glucosaminidase | BMN250 | Phase I-II | NCT02754076 | |
| MPS VI | N-acetylgalactosamine-4-sulfatase | Galsufase | Approved | [24] |
| MLD | Arilsulphatase A | HGT1110 | Phase I-II | NCT01887938 |
| TAK611 | Phase II | NCT015128 [25] | ||
| NPA and NPB | Acid Sphingomyelinase | rh-ASM | Phase I | NCT00410566 |
| LSD | Drug Name and Trial ID | Action Mechanism | Trial Phase |
|---|---|---|---|
| GD | AVR-RD-02 (NCT04145037) | CD34+ cells from patients treated with a Lentiviral vector to correct mutations in GBA | I/II |
| FD | AVR-RD-01 (NCT03454893) | CD34+ cells from patients treated with a Lentiviral vector to correct mutations in GLA | I/II |
| MLD | OTL-200 [53] (NCT04283227 III) (NCT03392987) (NCT01560182) | CD34+ cells from patients treated with a Lentiviral vector to correct mutations in ARSA | I/II |
| MPSI | IDUA LV (NCT03488394) | Lentivirus-based vector to correct defects in IDUA gene in CD34+ cells | I/II |
| MPS II | L2SN-transduced lymphocytes | Mononuclear cells from blood are extracted from patients and transduced with a retroviral vector which express iduronate-2-sulphatase. Cells are stimulated in order to enrich the lymphocyte T population which are re-implanted in the same patient | I/II |
| LSD | Drug Name and Trial ID | Action Mechanism | Trial Phase |
|---|---|---|---|
| FD | FLT190 (NCT04040049) | AAV vector (AAV8) which drive the GLA functional gene in the liver | I/II |
| 4D-310 (NCT04519749) | AAV based vector, which express GLA gene under CAG promoter action | I/II | |
| ST920 (NCT04046224) | vector based on AAV (AAV2/6) which drive the GLA functional gene in the liver | I/II | |
| PD | SPK-3006 (NCT04093349) | AAV based vector to express GAA in liver | I/II |
| Raav9-DES-Hgaa (NCT02240407) | Intramuscular AAV9, to express GAA | I/II | |
| AT845 (NCT04174105) | Intravenous AAV8 to express GAA | I/II | |
| MLD | TYF-ARSA (NCT03725670) | Self-inactivating lentiviral vector injected intracerebrally. The vector transports a correct version of ARSA gene. | I/II |
| MPSI | RGX-111 (NCT03580083) | Intracisternal, intracerebroventricular or lumbar puncture of AAV9 which express IDUA | I/II |
| SB913 (NCT03041324) (NCT04628871) | Permanent expression of iduronidase in hepatocytes, obtained by in vivo genetic editing in albumin locus driven by AAV | I/II | |
| MPSII | RGX-121 (NCT0457190) | AAV9-based vector which directs iduronate-2-sulfatase expression | I/II |
| SBFIX | Permanent expression of iduronate-2-sulphatase in hepatocytes, obtained by in vivo genetic editing from albumin locus directed by AAV-based vectors | I/II | |
| MPSIII (A, B) | LYS-SAF302 | AAVrh10 intracerebral injection which express SGSH gene | I/II |
| SAF-301 (NCT02053064) | AAV10 intracerebral injection that express SGSH and SUMF1 genes | I/II | |
| ABO101 (NCT04655911) | AAV9 which express NAGLU gene by intracerebral injection | I/II | |
| MPS VI | AAV2/8.TBG.hARSB | AAV8 to express ARSB gene in liver | I/II |
| Krabbe Disease | AAVrh10 | AAV vector expressing galactosylceramidase is combined with HSCT and delivered intravenously | I/II |
| AAV Hu68 | Intracisternal AAV vector expressing galactosylceramidase | I/II |
| LSD | Compound | Strategy | Reference | Trial Phase |
|---|---|---|---|---|
| FD | Migalastat | PC | Approved [73] | |
| Lucerastat | SRT | NCT03425539 | III | |
| Venglustat | SRT | NCT02489344 | II | |
| GD | Venglustat | PC | NCT02843035 | II |
| Afegostat | PC | NCT00813865 NCT00446550 NCT00433147 | II | |
| Ambroxol | PC | NCT03950050 NCT04388969 | II | |
| Miglustat | SRT | Approved [74] | ||
| Eliglustat | SRT | Approved [75] | ||
| PD | Miglustat | PC | NCT02185651 NCT04327973 NCT04808505 | III |
| Duvoglustat | PC | NCT01380743 NCT00688597 NCT04327973 | II | |
| Gangliosidosis GM-2 | Venglustat | SRT | NCT04221451 | III |
| Pyrimethamina | PC | NCT01102686 | I/II | |
| MPS IV | Odiparcil | SRT | NCT03370653 | II |
| NPC | Miglustat | SRT | Approved [76] | |
| Arimoclomol | Induces HSP70 chaperone synthesis | NCT02612129 | III | |
| Ostat | Histone deacetylase inhibitor that increase mutant NPC1 protein levels | NCT02124083 | I/II |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Pereira, C.; San Millán-Tejado, B.; Gallardo-Gómez, M.; Pérez-Márquez, T.; Alves-Villar, M.; Melcón-Crespo, C.; Fernández-Martín, J.; Ortolano, S. Therapeutic Approaches in Lysosomal Storage Diseases. Biomolecules 2021, 11, 1775. https://doi.org/10.3390/biom11121775
Fernández-Pereira C, San Millán-Tejado B, Gallardo-Gómez M, Pérez-Márquez T, Alves-Villar M, Melcón-Crespo C, Fernández-Martín J, Ortolano S. Therapeutic Approaches in Lysosomal Storage Diseases. Biomolecules. 2021; 11(12):1775. https://doi.org/10.3390/biom11121775
Chicago/Turabian StyleFernández-Pereira, Carlos, Beatriz San Millán-Tejado, María Gallardo-Gómez, Tania Pérez-Márquez, Marta Alves-Villar, Cristina Melcón-Crespo, Julián Fernández-Martín, and Saida Ortolano. 2021. "Therapeutic Approaches in Lysosomal Storage Diseases" Biomolecules 11, no. 12: 1775. https://doi.org/10.3390/biom11121775