
In Silico Analysis of the Antagonist Effect of Enoxaparin on the ApoE4–Amyloid-Beta (Aβ) Complex at Different pH Conditions

 , , , , , , ,
, , , , , , ,  and
and
Abstract
:1. Introduction
2. Computational Details
2.1. System Preparation
2.1.1. ApoE4 and A Structures
2.1.2. Enoxaparin Molecule
2.1.3. Molecular Docking between ApoE4 and Ligands
2.2. Md Simulations
2.3. MM/PBSA Calculations
2.4. Structure and Data Analysis
3. Results and Discussion
3.1. Enoxaparin (Enx) Structure
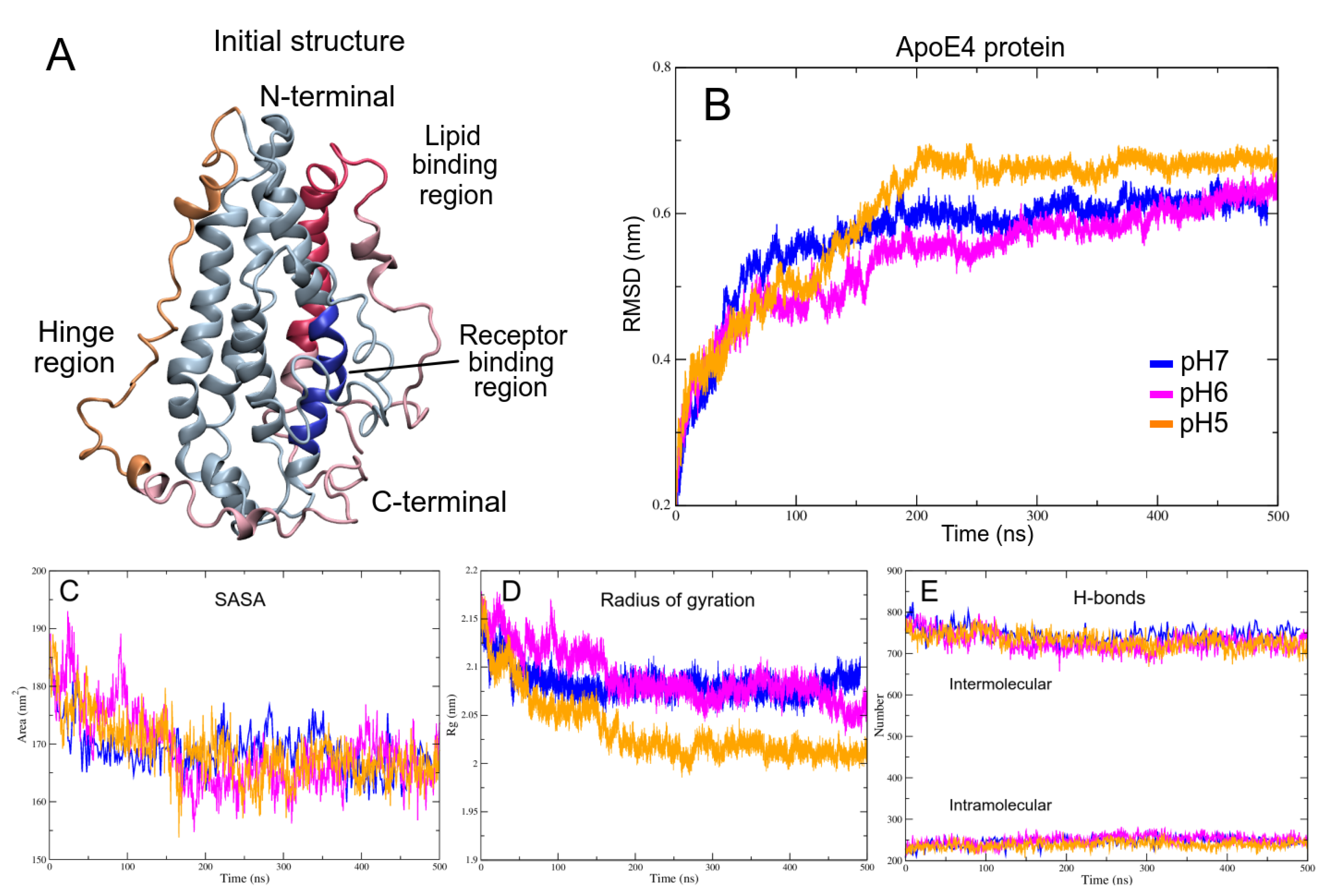
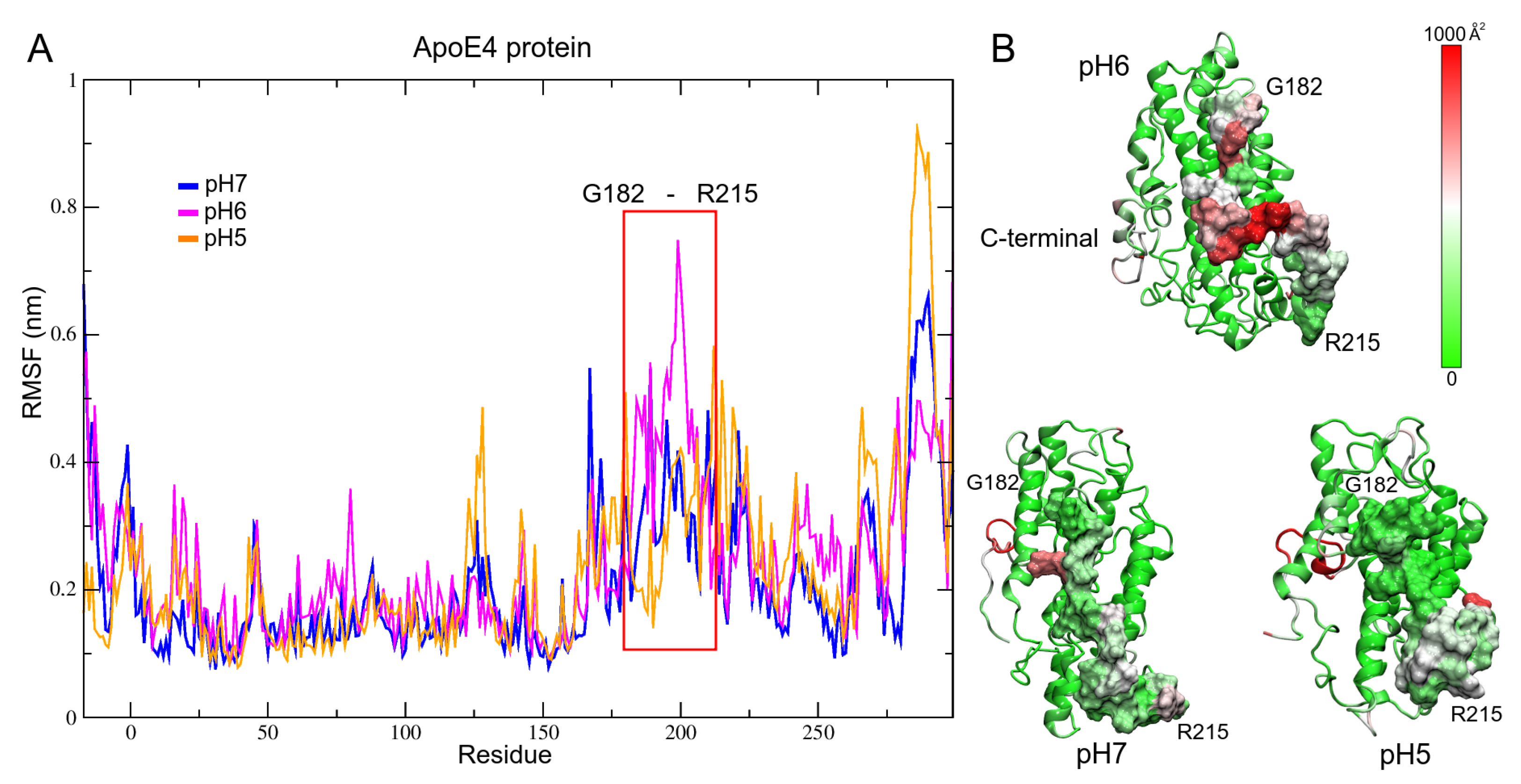
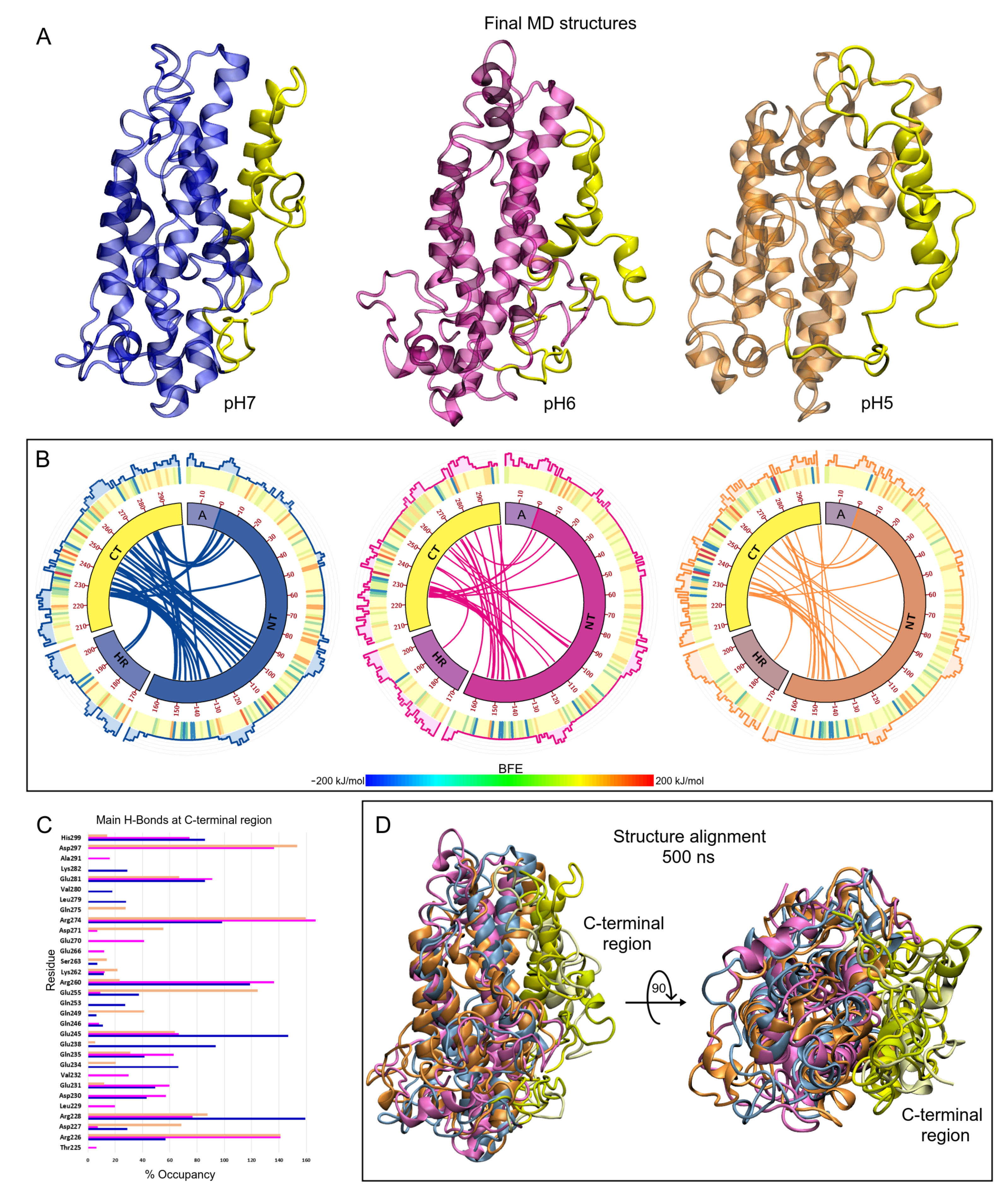
3.2. pH Effect on Isolated ApoE4 Structure
ApoE4 Closed-Conformation Structure
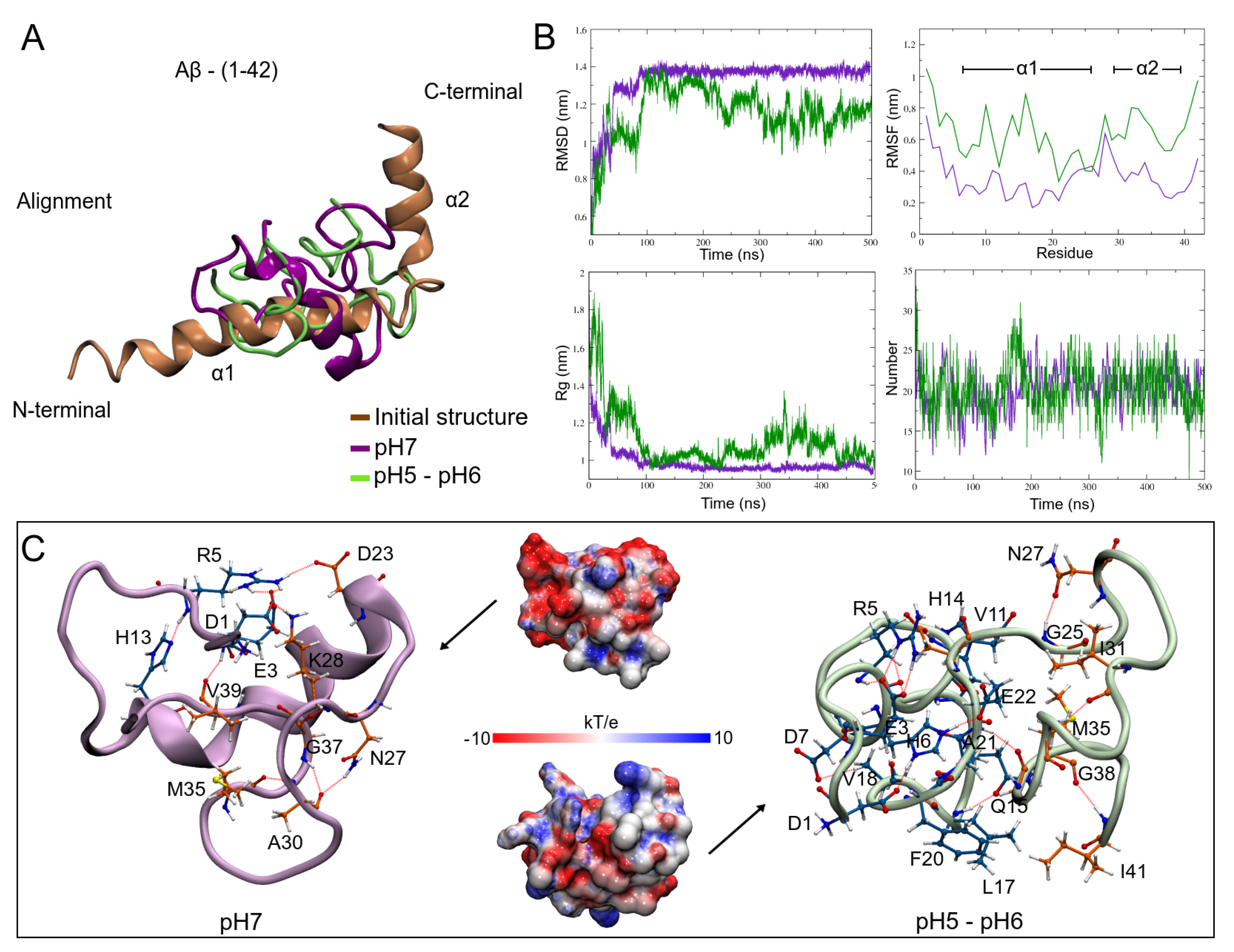
3.3. pH Effect on Isolated A Structure
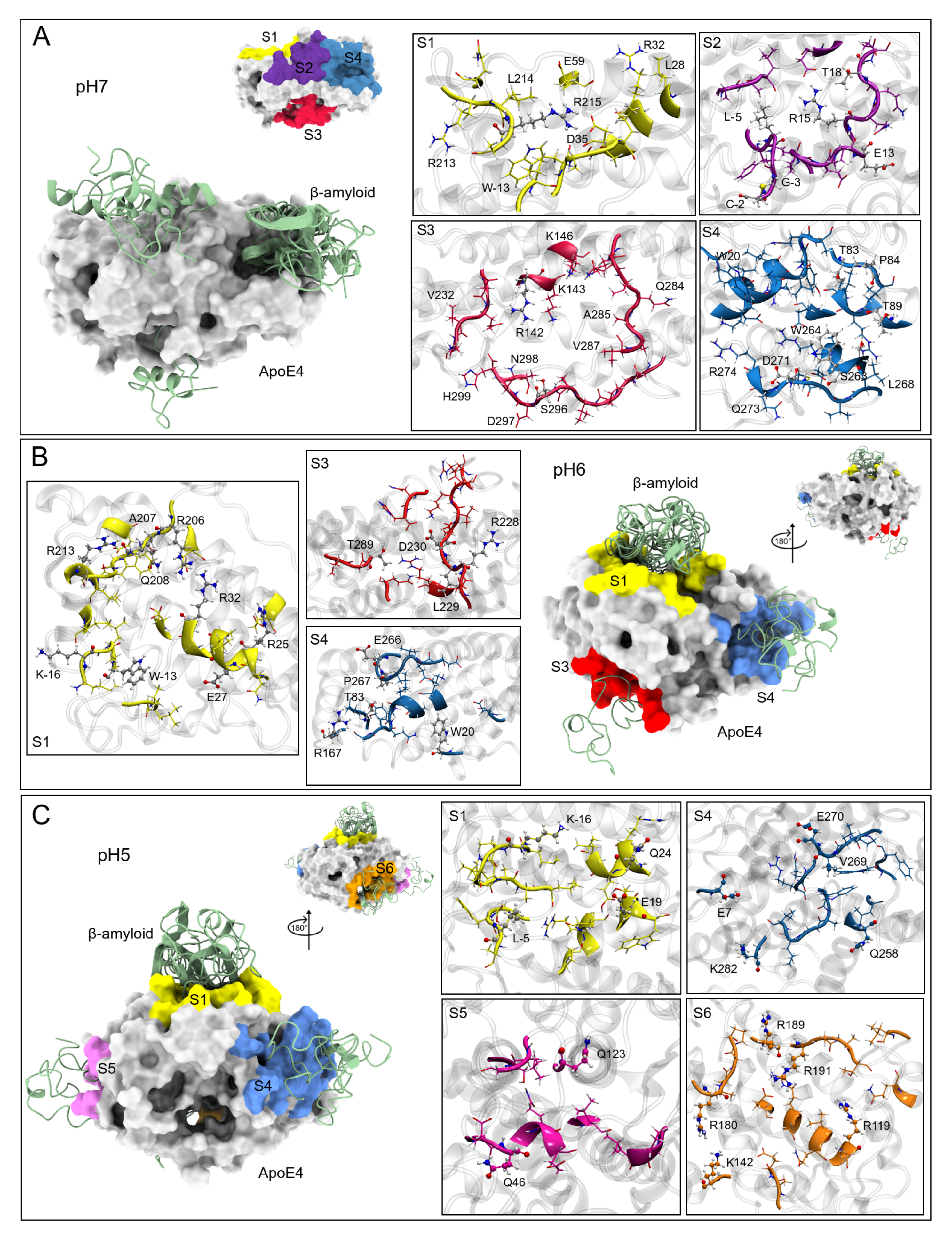
3.4. ApoE4 Complexes
Interaction Sites
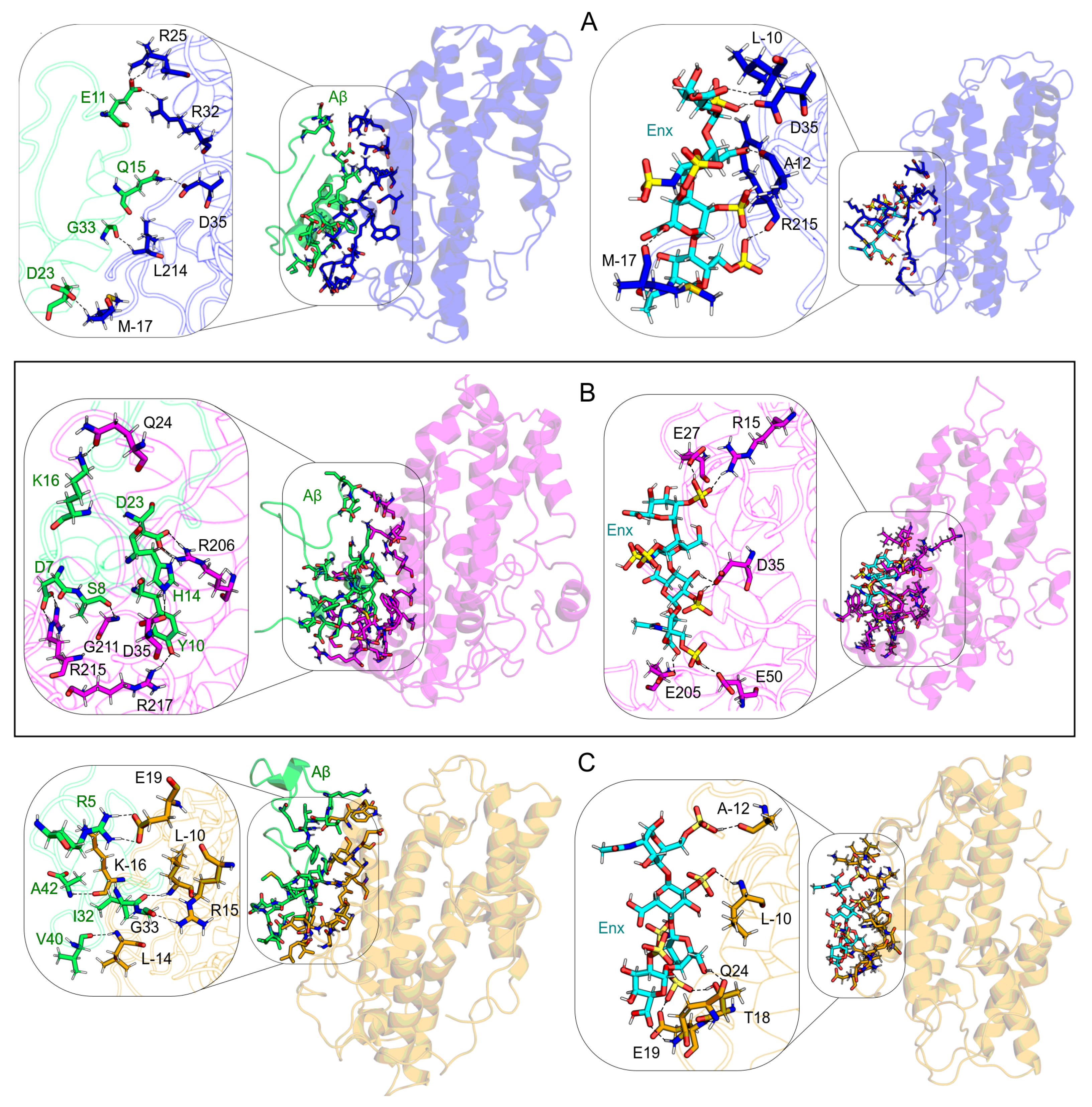
3.5. ApoE4-Ligand Complexes after MD Calculations
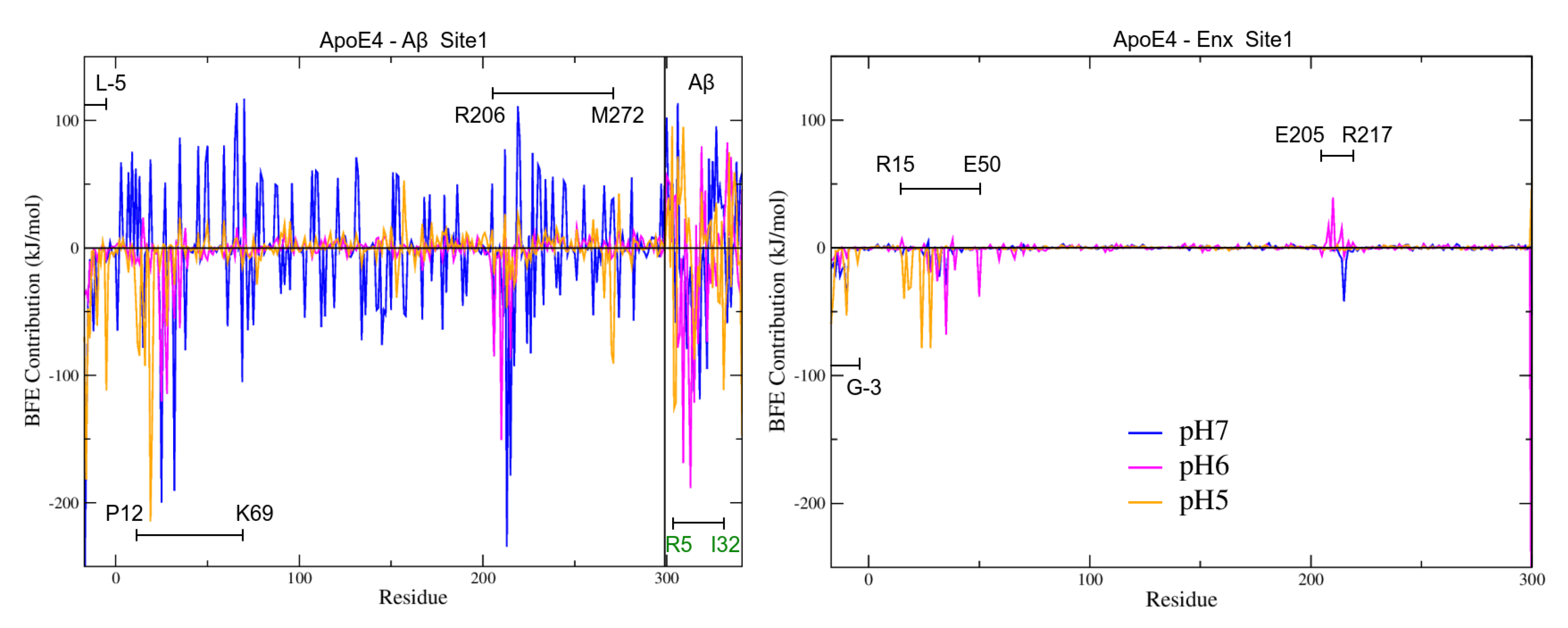
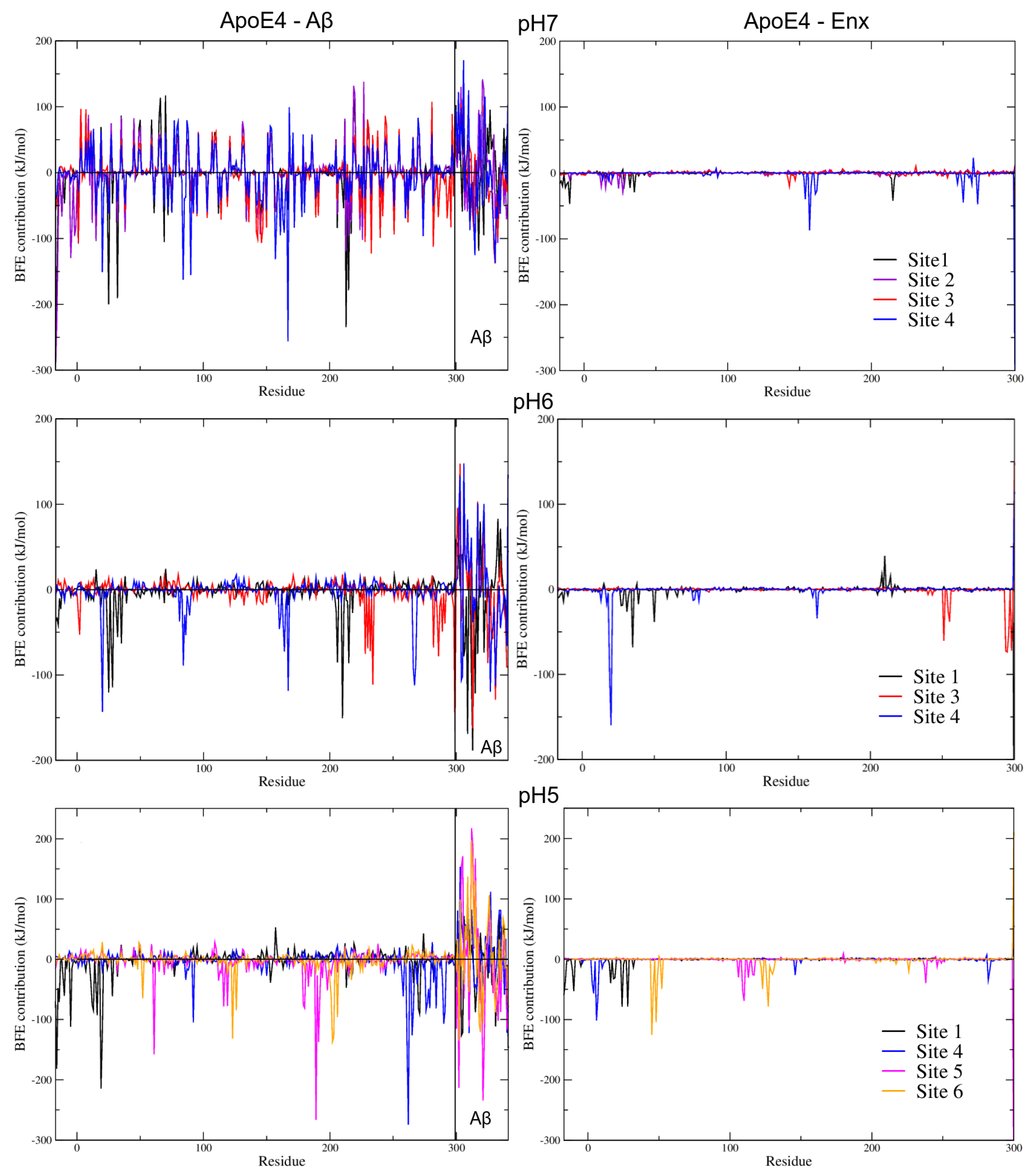
3.6. Binding Free Energies (BFE) Analysis
3.7. Intermolecular Contact and BFE Analyses
3.7.1. S1 Site
3.7.2. BFE Contribution on the Interaction Sites
3.8. Study Limitations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Long, J.M.; Holtzman, D.M. Alzheimer disease: An update on pathobiology and treatment strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef] [PubMed]
- Schindler, S.E.; McConathy, J.; Ances, B.M.; Diamond, M.I. Advances in diagnostic testing for Alzheimer disease. Mo. Med. 2013, 110, 401–405. [Google Scholar] [PubMed]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Kiddle, S.J.; Voyle, N.; Dobson, R.J.B. A Blood Test for Alzheimer’s Disease: Progress, Challenges, and Recommendations. J. Alzheimer’s Dis. 2018, 64, S289–S297. [Google Scholar] [CrossRef]
- Wisniewski, T.; Drummond, E. Future horizons in Alzheimer’s disease research. Prog. Mol. Biol. Transl. Sci. 2019, 168, 223–241. [Google Scholar]
- Rabinovici, G.D. Late-onset Alzheimer disease. Continuum Lifelong Learn. Neurol. 2019, 25, 14–33. [Google Scholar] [CrossRef]
- Wisniewski, T.; Frangione, B. Apolipoprotein E: A pathological chaperone protein in patients with cerebral and systemic am-yloid. Neurosci. Lett. 1992, 135, 235–238. [Google Scholar] [CrossRef]
- Wisniewski, T.; Golabek, A.; Matsubara, E.; Ghiso, J.; Frangione, B. Apolipoprotein E: Binding to soluble Alzheimer’s beta-amyloid. Biochem. Biophys. Res. Commun. 1993, 192, 359–365. [Google Scholar] [CrossRef]
- Wisniewski, T.; Lalowski, M.; Golabek, A.A.; Vogel, T.; Frangione, B. Is Alzheimer’s disease an apolipoprotein E amyloidosis? The Lancet 1995, 345, 956–958. [Google Scholar] [CrossRef]
- Potter, H.; Wisniewski, T. Apolipoprotein E: Essential catalyst of the Alzheimer amyloid cascade. Int. J. Alzheimer’s Dis. 2012, 2012, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roda, A.R.; Montoliu-Gaya, L.; Villegas, S. The role of apolipoprotein E isoforms in Alzheimer’s disease. J. Alzheimer’s Dis. 2019, 68, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.N.; Clarnette, R.; Fisher, C.; Broe, G.A.; Brooks, W.S.; Montgomery, P.; Gandy, S.E. ApoE genotypes in Australia: Roles in early and late onset Alzheimer’s disease and Down’s syndrome. Neuroreport 1995, 6, 1513–1516. [Google Scholar] [CrossRef]
- Martins, R.N.; Villemagne, V.; Sohrabi, H.R.; Chatterjee, P.; Shah, T.M.; Verdile, G.; Fraser, P.; Taddei, K.; Gupta, V.B.; Rainey-Smith, S.R.; et al. Alzheimer’s disease: A journey from amyloid peptides and oxidative stress, to biomarker tech-nologies and disease prevention strategies-gains from AIBL and DIAN cohort studies. J. Alzheimer’s Dis. 2018, 62, 965–992. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.C.; Roe, C.M.; Xiong, C.; Fagan, A.M.; Goate, A.M.; Holtzman, D.M.; Mintun, M.A. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann. Neurol. 2010, 67, 122–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural insights and links to Alzheimer disease pathogenesis. Neuron 2021, 109, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Xu, H.; Bu, G. ApoE and Aβ in Alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Morrow, J.A.; Hatters, D.M.; Lu, B.; Höchtl, P.; Oberg, K.A.; Rupp, B.; Weisgraber, K.H. Apolipoprotein E4 forms a molten globule: A potential basis for its association with disease. J. Biol. Chem. 2002, 277, 50380–50385. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Li, Q.; Wang, J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc. Natl. Acad. Sci. USA 2011, 108, 14813–14818. [Google Scholar] [CrossRef] [Green Version]
- Weisgraber, K.H. Apolipoprotein E: Structure-function relationships. Adv. Protein Chem. 1994, 45, 249–302. [Google Scholar]
- Winkler, K.; Scharnagl, H.; Tisljar, U.; Hoschutzky, H.; Friedrich, I.; Hoffmann, M.M.; Huttinger, M.; Wieland, H.; Marz, W. Competition of Aβ amyloid peptide and apolipoprotein E for receptor-mediated endocy-tosis. J. Lipid Res. 1999, 40, 447–455. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C. Holtzman, D.M. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ associa-tion in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youmans, K.L.; Tai, L.M.; Nwabuisi-Heath, E.; Jungbauer, L.; Kanekiyo, T.; Gan, M.; Kim, J.; Eimer, W.A.; Estus, S.; Rebeck, G.W.; et al. APOE4-specific changes in Aβ accumulation in a new transgenic mouse model of Alz-heimer disease. J. Biol. Chem. 2012, 287, 41774–41786. [Google Scholar] [CrossRef] [Green Version]
- Bales K., R.; Liu, F.; Wu, S.; Lin, S.; Koger, D.; DeLong, C.; Hansen, J.C.; Sullivan, P.M.; Paul, S.M. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J. Neurosci. 2009, 29, 6771–6779. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Zhang, J.; Liu, Q.; Liu, C.C.; Zhang, L.; Bu, G. Heparan Sulphate Proteoglycan and the Low-Density Lipoprotein Receptor-Related Protein 1 Constitute Major Pathways for Neuronal Amyloid-β Uptake. J. Neurosci. 2011, 31, 1644–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, H.; Hung, A.C.; Klaver, D.W.; Suzuki, T.; Freeman, C.; Narkowicz, C.; Jacobson, G.A.; Small, D.H. Effects of heparin and enoxaparin on APP processing and Aβ production in primary corti-cal neurons from Tg2576 mice. PLoS ONE 2011, 6, e23007. [Google Scholar] [CrossRef] [Green Version]
- Patey, S.J.; Yates, E.A.; Turnbull, J.E. Novel heparan sulphate analogues: Inhibition of beta-secretase cleavage of amyloid precursor protein. Biochem. Soc. Trans. 2005, 33, 1116–1118. [Google Scholar] [CrossRef]
- Bergamaschini, L.; Rossi, E.; Vergani, C.; De Simoni, M.G. Alzheimer’s disease: Another target for heparin therapy. Sci. World J. 2009, 9, 891–908. [Google Scholar] [CrossRef] [Green Version]
- Andrade, S.; Ramalho, M.J.; Loureiro, J.A.; Pereira, M.D.C. Natural compounds for Alzheimer’s disease therapy: A systematic review of preclinical and clinical studies. Int. J. Mol. Sci. 2019, 20, 2313. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, K. Direct oral anticoagulants: A new therapy against Alzheimer’s disease? NRR 2021, 16, 1556. [Google Scholar] [CrossRef]
- Schützmann, M.P.; Hasecke, F.; Bachmann, S.; Zielinski, M.; Hänsch, S.; Schröder, G.F.; Zempel, H.; Hoyer, W. Endo-lysosomal Aβ concentration and pH trigger formation of Aβ oligomers that potently induce Tau missorting. Nat. Commun. 2021, 12, 4634. [Google Scholar] [CrossRef] [PubMed]
- Decker, Y.; Németh, E.; Schomburg, R.; Chemla, A.; Fülöp, L.; Menger, M.D.; Liu, Y.; Fassbender, K. Decreased pH in the aging brain and Alzheimer’s disease. Neurobiol. Aging 2021, 101, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Chang, P.T. Acidic pH promotes the formation of toxic fibrils from β-amyloid peptide. Brain Res. 2001, 893, 287–291. [Google Scholar] [CrossRef]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, C.R.; Olsson, M.H.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. Available online: https://www.uniprot.org/uniprot/P02649 (accessed on 18 June 2021). [CrossRef]
- Dong, J.; Peters-Libeu, C.A.; Weisgraber, K.H.; Segelke, B.W.; Rupp, B.; Capila, I.; Hernáiz, M.J.; LeBrun, L.A.; Linhardt, R.J. Interaction of the N-terminal domain of apolipoprotein E4 with heparin. Biochemistry 2001, 40, 2826–2834. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [Green Version]
- Crescenzi, O.; Tomaselli, S.; Guerrini, R.; Salvadori, S.; D’Ursi, A.M.; Temussi, P.A.; Picone, D. Solution structure of the Alzheimer amyloid β-peptide (1–42) in an apolar microenvi-ronment: Similarity with a virus fusion domain. Eur. J. Biochem. 2002, 269, 5642–5648. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, D.V.; Zaretskaia, M.V. Intracellular ion changes induced by the exposure to beta-amyloid can be explained by the for-mation of channels in the lysosomal membranes. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119145. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, D.V.; Zaretskaia, M.V. Flow cytometry method to quantify the formation of beta-amyloid membrane ion channels. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1863, 183506. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Danani, A. Molecular simulations of amyloid beta assemblies. Adv. Phys. X 2020, 5, 1770627. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atom-ic-level energy minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Timmer, N.M.; van Dijk, L.; van der Zee, C.E.; Kiliaan, A.; de Waal, R.M.; Verbeek, M.M. Enoxaparin treatment administered at both early and late stages of amyloid β deposition improves cognition of APPswe/PS1dE9 mice with differential effects on brain Aβ levels. Neurobiol. Dis. 2010, 40, 340–347. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView 6.0. 16; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Ritchie, J.P. Electron density distribution analysis for nitromethane, nitromethide, and nitramide. J. Am. Chem. Soc. 1985, 107, 1829–1837. [Google Scholar] [CrossRef]
- Ritchie, J.P.; Bachrach, S.M. Some methods and applications of electron density distribution analysis. J. Comput. Chem. 1987, 8, 499–509. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodda, L.S.; Vilseck, J.Z.; Tirado-Rives, J.; Jorgensen, W.L. 1.14* CM1A-LBCC: Localized bond-charge corrected CM1A charges for condensed-phase simulations. Phys. Chem. B 2017, 121, 3864–3870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodda, L.S.; Cabeza de Vaca, I.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Duhovny, D.; Nussinov, R.; Wolfson, H.J. Efficient unbound docking of rigid molecules. In International Workshop on Algorithms in Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2002; Volume 40, pp. 185–200. [Google Scholar]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33 (Suppl. S2), W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins Struct. Funct. Bioinf. 2007, 69, 139–159. [Google Scholar] [CrossRef]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36 (Suppl. S2), W229–W232. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Kutzner, C.; Páll, S.; Fechner, M.; Esztermann, A.; de Groot, B.L.; Grubmüller, H. More bang for your buck: Improved use of GPU nodes for GROMACS 2018. J. Comput. Chem. 2019, 40, 2418–2431. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.; Postma, J.V.; van Gunsteren, W.F.; DiNola, A.R.H.J.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Martoňák, R.; Laio, A.; Parrinello, M. Predicting crystal structures: The Parrinello-Rahman method revisited. Phys. Rev. Lett. 2003, 90, 075503. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics Poisson-Boltzmann surface area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.P.; Muchmore, S.W. Large-scale application of high-throughput molecular mechanics with Poisson- Boltzmann sur-face area for routine physics-based scoring of protein-ligand complexes. J. Med. Chem. 2009, 52, 3159–3165. [Google Scholar] [CrossRef]
- Rastelli, G.; Rio, A.D.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, L.; DeLano, W. PyMOL. The PyMOL Molecular Graphics System, Version 2. Available online: https://pymol.org/2/ (accessed on 10 November 2021).
- Turner, P.; Grace, X. Version 5.1. 19. Center for Coastal and Land-Margin Research; Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005. [Google Scholar]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein–ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [Green Version]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calcula-tions. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- WR Inc. Mathematica, Version 12.1; WR Inc.: Champaign, IL, USA, 2020. [Google Scholar]
- Shinohara, M.; Petersen, R.C.; Dickson, D.W.; Bu, G. Brain regional correlation of amyloid-β with synapses and apolipoprotein E in non-demented individuals: Potential mechanisms underlying regional vulnerability to amy-loid-β accumulation. Acta Neuropathol. 2013, 125, 535–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, F.; Li, A.; Xiong, M.; Bien-Ly, N.; Jiang, H.; Zhang, Y.; Finn, M.B.; Hoyle, R.; Keyser, J.; Lefton, K.B. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Investig. 2018, 128, 2144–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enoxaparin. (s.f.). NCBI. Retrieved 12 November 2021. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Heparin (accessed on 10 April 2021).
- Gastegger, M.; Marquetand, P. Molecular dynamics with neural network potentials. arXiv 2020, arXiv:1812.07676. [Google Scholar]
- Riquelme, M.; Lara, A.; Mobley, D.L.; Verstraelen, T.; Matamala, A.R.; Vohringer-Martinez, E. Hydration free energies in the FreeSolv database calculated with polarized iterative Hirshfeld charges. J. Chem. Inf. Model. 2018, 58, 1779–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, J.; Prasana, J.C.; Muthu, S.; Kuruvilla, T.K.; Saji, R.S. Evaluation of vibrational, electronic, reactivity and bioactivity of propafenone—A spectro-scopic, DFT and molecular docking approach. Chem. Data Coll. 2020, 26, 100360. [Google Scholar]
- Corregidor, P.F.; Zígolo, M.A.; Ottavianelli, E.E. Conformational search, structural analysis, vibrational properties, reactivity study and affinity towards DNA of the novel insecticide flonicamid. J. Mol. Struct. 2021, 1241, 130628. [Google Scholar] [CrossRef]
- Raulin, A.C.; Kraft, L.; Al-Hilaly, Y.K.; Xue, W.F.; McGeehan, J.E.; Atack, J.R.; Serpell, L. The molecular basis for apolipoprotein E4 as the major risk factor for late-onset Alzheimer’s disease. J. Mol. Biol. 2019, 431, 2248–2265. [Google Scholar] [CrossRef]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E: Structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid Res. 2009, 50, S183–S188. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Ahalawat, N.; Mondal, J. tomistic insights into structural differences between E3 and E4 isoforms of apolipoprotein E. Biophys. J. 2017, 113, 2682–2694. [Google Scholar] [CrossRef] [Green Version]
- Thu, T.T.M.; Co, N.T.; Tu, L.A.; Li, M.S. Aggregation rate of amyloid beta peptide is controlled by beta-content in monomeric state. J. Chem. Phys. 2019, 150, 225101. [Google Scholar] [CrossRef]
- Amini, Z.; Fatemi, M.H.; Rauk, A. Molecular dynamics studies of a β-sheet blocking peptide with the full-length amyloid beta peptide of Alzheimer’s disease. Can. J. Chem. 2016, 94, 833–841. [Google Scholar] [CrossRef]
- Greenwald, J.; Riek, R. Biology of amyloid: Structure, function, and regulation. Structure 2010, 18, 1244–1260. [Google Scholar] [CrossRef] [PubMed]
- Toyama, B.H.; Weissman, J.S. Amyloid structure: Conformational diversity and consequences. Annu. Rev. Biochem. 2011, 80, 557–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallardo, R.; Ranson, N.A.; Radford, S.E. Amyloid structures: Much more than just a cross-β fold. Curr. Opin. Struct. Biol. 2020, 60, 7–16. [Google Scholar] [CrossRef]
- Wang, L.; Zeng, R.; Pang, X.; Gu, Q.; Tan, W. The mechanisms of flavonoids inhibiting conformational transition of amyloid-β 42 monomer: A comparative molecular dynamics simulation study. RSC Adv. 2015, 5, 66391–66402. [Google Scholar] [CrossRef]
- Brown, A.M.; Bevan, D.R. Molecular dynamics simulations of amyloid β-peptide (1–42): Tetramer formation and membrane interactions. Biophys. J. 2016, 111, 937–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Lee, J.Y. Tautomeric effect of histidine on the monomeric structure of amyloid β-peptide (1–42). ACS Chem. Neurosci. 2017, 8, 669–675. [Google Scholar] [CrossRef]
- Liu, F.; Ma, Z.; Sang, J.; Lu, F. Edaravone inhibits the conformational transition of amyloid-β42: Insights from molecu-lar dynamics simulations. J. Biomol. Struct. Dyn. 2019, 38, 2377–2388. [Google Scholar] [CrossRef]
- Steckmann, T.; Bhandari, Y.R.; Chapagain, P.P.; Gerstman, B.S. Cooperative structural transitions in amyloid-like aggregation. J. Chem. Phys. 2017, 146, 135103. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Golabek, A.A.; Soto, C.; Vogel, T.; Wisniewski, T. The Interaction between Apolipoprotein E and Alzheimer’s Amyloid β-Peptide Is De-pendent on β-Peptide Conformation. J. Biol. Chem. 1996, 271, 10602–10606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, H.; Dhanasekaran, P.; Nguyen, D.; Baldwin, F.; Weisgraber, K.H.; Wehrli, S.; Phillips, M.C.; Lund-Katz, S. Characterization of the heparin binding sites in human apolipoprotein E. J. Biol. Chem. 2003, 278, 14782–14787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Maréchal, J.D.; Wärmländer, S.; Gräslund, A.; Perálvarez-Marın, A. In Silico Analysis of the Apolipoprotein E and the Amyloid b Peptide. PLoS Comput. Biol. 2010, 6, e1000663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillet, J.N. Alzheimer’s disease: Unraveling APOE4 binding to amyloid-beta peptide and lipids with mo-lecular dynamics and quantum mechanics. J. Biomol. Struct. Dyn. 2021, 39, 5026–5032. [Google Scholar] [CrossRef]
- Kuszczyk, M.A.; Sanchez, S.; Pankiewicz, J.; Kim, J.; Duszczyk, M.; Guridi, M.; Asuni, A.A.; Sulli-van, P.M.; Sullivan, D.M.; Sadowski, M.J. Blocking the interaction between apolipoprotein E and Aβ reduces intraneuronal accumu-lation of Aβ and inhibits synaptic degeneration. Am. J. Pathol. 2013, 182, 1750–1768. [Google Scholar] [CrossRef] [Green Version]
- Cerf, E.; Gustot, A.; Goormaghtigh, E.; Ruysschaert, J.M.; Raussens, V. High ability of apolipoprotein E4 to stabilize amyloid-β peptide oligomers, the patholog-ical entities responsible for Alzheimer’s disease. FASEB J. 2011, 25, 1585–1595. [Google Scholar] [CrossRef]
- Hashimoto, T.; Serrano-Pozo, A.; Hori, Y.; Adams, K.W.; Takeda, S.; Banerji, A.O.; Mitani, A.; Joyner, D.; Thyssen, D.H.; Bacskai, B.J.; et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J. Neurosci. 2012, 32, 15181–15192. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | SASA | RMSD | RMSF | RG | H-Bonds | ||
|---|---|---|---|---|---|---|---|
| Total | Axis | Intra | Inter | ||||
| ApoE4 | |||||||
| 1.68 ± 0.15 | |||||||
| pH7 | 169.05 ± 4.02 | 0.61 ± 0.01 | 0.21 ± 0.12 | 2.08 ± 0.01 | 1.72 ± 0.13 | 246 ± 10 | 746 ± 19 |
| 1.68 ± 0.16 | |||||||
| 1.70 ± 0.17 | |||||||
| pH6 | 168.99 ± 6.59 | 0.59 ± 0.03 | 0.24 ± 0.12 | 2.09 ± 0.03 | 1.69 ± 0.16 | 249 ± 11 | 728 ± 22 |
| 1.71 ± 0.14 | |||||||
| 1.64 ± 0.13 | |||||||
| pH5 | 168.78 ± 5.20 | 0.67 ± 0.01 | 0.24 ± 0.15 | 2.03 ± 0.03 | 1.67 ± 0.11 | 238 ± 9 | 732 ± 19 |
| 1.66 ± 0.11 | |||||||
| Amyloid- | |||||||
| 0.81 ± 0.09 | |||||||
| pH7 | 33.17 ± 2.05 | 1.38 ± 0.01 | 0.35 ± 0.12 | 0.99 ± 0.08 | 0.80 ± 0.09 | 20 ± 3 | 105 ± 6 |
| 0.81 ± 0.09 | |||||||
| 0.88 ± 0.13 | |||||||
| pH5–6 rep-1 | 37.23 ± 2.27 | 1.16 ± 0.13 | 0.64 ± 0.16 | 1.10 ± 0.15 | 0.91 ± 0.17 | 20 ± 3 | 105 ± 7 |
| 0.90 ± 0.15 | |||||||
| 0.82 ± 0.09 | |||||||
| pH5–6 rep-2 | 34.21 ± 2.13 | 1.31 ± 0.14 | 0.53 ± 0.13 | 1.02 ± 0.10 | 0.83 ± 0.13 | 18 ± 3 | 106 ± 7 |
| 0.83 ± 0.11 | |||||||
| 0.87 ± 0.11 | |||||||
| pH5–6 rep-3 | 35.98 ± 2.08 | 1.29 ± 0.17 | 0.66 ± 0.18 | 1.06 ± 0.09 | 0.87 ± 0.11 | 20 ± 4 | 106 ± 8 |
| 0.86 ± 0.10 | |||||||
| Site S1 | Site S2 | Site S3 | Site S4 | |
|---|---|---|---|---|
| pH7 | V-15, W-13, A-12, A-11, L-10, L28, R32, D35, E59, Q208, R213, L214, R215, A216 | A-12, A-11, L-10, F-6, L-5, A-4, G-3, C-2, E9, P10, E11, P12, E13, R15, Q16, Q17, T18, E19, E27 | R142, K143, K146, A241, E281, K282, V283, Q284, A285, V287, P295, S296, D297, N298, H299 | W20, T83, P84, V85, T89, R92, L93, L93, D154, K157, R158, A160, V161, Y162, Q163, A164, R260, S263, W264, L268, Q270, D271, M272, R274 |
| Site S1 | Site S3 | Site S4 | ||
| pH6 | M-17, K-16, V-15, L-14, W-13, L-10, L-9, Q24, R25, E27, L28, A29, G31, R32, D35, R38, W39, E50, E59, E70, R206, A207, Q208, A209, W210, G211, R213, L214, M218 | Q4, R142, D227, R228, L229, D230, E231, V232, K233, E234, R240, V287, G288, T289, N298, H299 | E13, W20, T83, P84, K157, A160, V161, Y162, Q163, A164, G165, R167, E266, P267, L268, V269, E270, D271 | |
| Site S1 | Site S4 | Site S5 | Site S6 | |
| pH5 | M-17, K-16, V-15, L-14, W-13, A-12, A-11, L-10, T-7, F-6, L-5, A-4, P12, R15, Q16, T18, E19, W20, Q24, R25, E27, L28 | E109, R112, G113, V116, Q117, R119, G120, R180, L181, G182, P183, R189, R191, A192, A193, T194, Q204, L216, A237, E238, K242 | E45, Q46, Q48, E49, L52, Q123, L126, G127, S129, P202, L203 | E7, E13, Q258, L261, W264, F265, P267, L268, V269, E270, Q273, R274, W276, A277, G278, L279, V280, K282 |
| A Complex (site) | |||||
|---|---|---|---|---|---|
| Sol1 (s4) | |||||
| Sol2 (s1) | |||||
| Sol3 (s4) | |||||
| Sol4 (s4) | |||||
| Sol5 (s4) | |||||
| Sol6 (s2) | |||||
| Sol7 (s4) | |||||
| Sol8 (s4) | |||||
| Sol9 (s2) | |||||
| Sol10 (s3) | |||||
| Enx complex (site) | |||||
| Sol1 (s4) | |||||
| Sol2 (s2) | |||||
| Sol3 (s1) | |||||
| Sol4 (s3) |
| A Complex (site) | |||||
|---|---|---|---|---|---|
| Sol1 (s1) | |||||
| Sol2 (s1) | |||||
| Sol3 (s4) | |||||
| Sol4 (s1) | |||||
| Sol5 (s1) | |||||
| Sol6 (s4) | |||||
| Sol7 (s1) | |||||
| Sol8 (s1) | |||||
| Sol9 (s1) | |||||
| Sol10 (s3) | |||||
| Enx complex (site) | |||||
| Sol1 (s1) | |||||
| Sol2 (s3) | |||||
| Sol3 (s4) |
| A Complex (site) | |||||
|---|---|---|---|---|---|
| Sol1 (s1) | |||||
| Sol2 (s1) | |||||
| Sol3 (s1) | |||||
| Sol4 (s1) | |||||
| Sol5 (s4) | |||||
| Sol6 (s5) | |||||
| Sol7 (s4) | |||||
| Sol8 (s5) | |||||
| Sol9 (s6) | |||||
| Sol10 (s1) | |||||
| Enx complex (site) | |||||
| Sol1 (s1) | |||||
| Sol2 (s4) | |||||
| Sol3 (s5) | |||||
| Sol4 (s6) |
| No. | pH7 | pH6 | pH5 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ApoE4 | A | ApoE4 | ApoE4 | A | ApoE4 | ApoE4 | A | ApoE4 | |
| 1 | M-17(−292) | F19(−119) | L-10(−47) | R25(−126) | H14(−191) | D35(−67) | E19(−215) | A42(−147) | Q24(−78) |
| 2 | R213(−234) | K16(−119) | R215(−42) | W210(−121) | Y10(−160) | E50(−38) | K-16(−182) | R5(−126) | L28(−78) |
| 3 | R25(−200) | D23(−95) | D35(−29) | L28(−110) | K16(−132) | G31(−26) | W20(−128) | H6(−121) | M-17(−59) |
| 4 | R32(−191) | V12(−79) | R32(−23) | R206(−109) | H13(−106) | E27(−19) | L-5(−112) | I32(−111) | L-10(−53) |
| 5 | R215(−178) | Q15(−78) | W-13(−22) | R32(−85) | D23(−87) | L28(−19) | Q16(−92) | K16(−88) | Q16(−40) |
| 6 | K-16(−158) | H6(−59) | V-15(−20) | R215(−80) | V18(−76) | W39(−17) | D271(−91) | L17(−54) | K-16(−38) |
| 7 | K69(−105) | L34(−59) | L28(−18) | E27(−72) | D7(−68) | W-13(−14) | E13(−85) | G33(−48) | T18(−32) |
| 8 | R217(−93) | V36(−47) | A-12(−17) | K69(−52) | S8(−67) | M-17(−10) | E270(−82) | A30(−44) | E19(−31) |
| 9 | R206(−84) | V18(−41) | M-17(−16) | D35(−48) | I41(−57) | E59(−10) | P12(−76) | G29(−42) | A-11(−18) |
| 10 | R226(−83) | E22(−35) | A-11(−14) | V-15(−40) | G25(−41) | R32(−10) | L-14(−71) | S26(−31) | R32(−13) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilar-Pineda, J.A.; Paco-Coralla, S.G.; Febres-Molina, C.; Gamero-Begazo, P.L.; Shrivastava, P.; Vera-López, K.J.; Davila-Del-Carpio, G.; López-C, P.; Gómez, B.; Lino Cardenas, C.L. In Silico Analysis of the Antagonist Effect of Enoxaparin on the ApoE4–Amyloid-Beta (Aβ) Complex at Different pH Conditions. Biomolecules 2022, 12, 499. https://doi.org/10.3390/biom12040499
Aguilar-Pineda JA, Paco-Coralla SG, Febres-Molina C, Gamero-Begazo PL, Shrivastava P, Vera-López KJ, Davila-Del-Carpio G, López-C P, Gómez B, Lino Cardenas CL. In Silico Analysis of the Antagonist Effect of Enoxaparin on the ApoE4–Amyloid-Beta (Aβ) Complex at Different pH Conditions. Biomolecules. 2022; 12(4):499. https://doi.org/10.3390/biom12040499
Chicago/Turabian StyleAguilar-Pineda, Jorge Alberto, Silvana G. Paco-Coralla, Camilo Febres-Molina, Pamela L. Gamero-Begazo, Pallavi Shrivastava, Karin J. Vera-López, Gonzalo Davila-Del-Carpio, Patricia López-C, Badhin Gómez, and Christian L. Lino Cardenas. 2022. "In Silico Analysis of the Antagonist Effect of Enoxaparin on the ApoE4–Amyloid-Beta (Aβ) Complex at Different pH Conditions" Biomolecules 12, no. 4: 499. https://doi.org/10.3390/biom12040499