
Microglia: The Real Foe in HIV-1-Associated Neurocognitive Disorders?

 , , and
, , and
Abstract
:1. Introduction
2. Microglia Biology
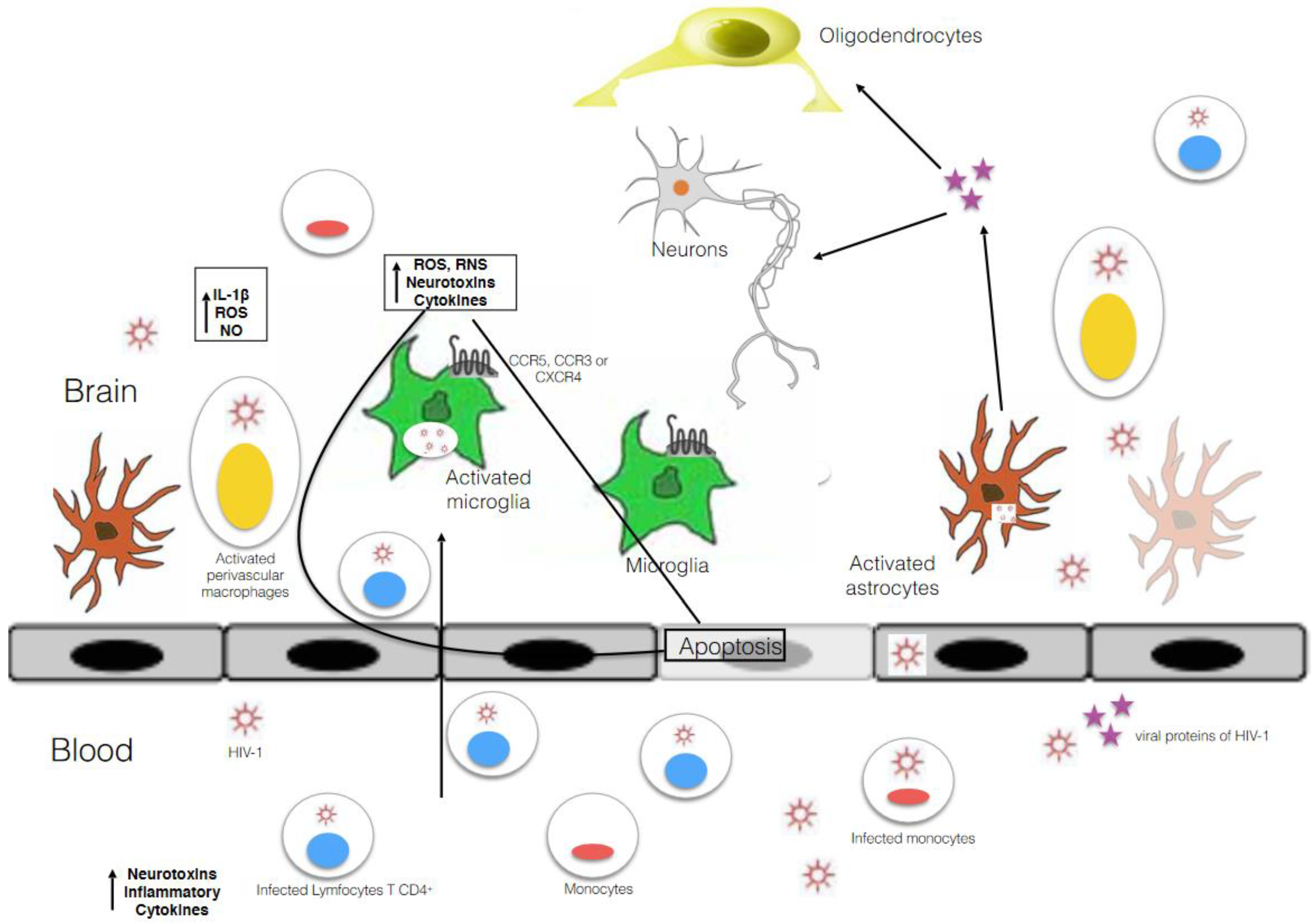
3. Microglia as Main HIV-1 Reservoir in the CNS
4. The Entry of HIV-1 into Microglial Cells and Their Susceptibility to the Virus. Relevant Studies about the Mechanism of Infection and Molecular Mechanisms Involved in Establishing and Maintaining HIV-1 Latency in Microglia, and Procedures to Study HIV Infection in Microglia
5. General and Basic Mechanisms of Latency
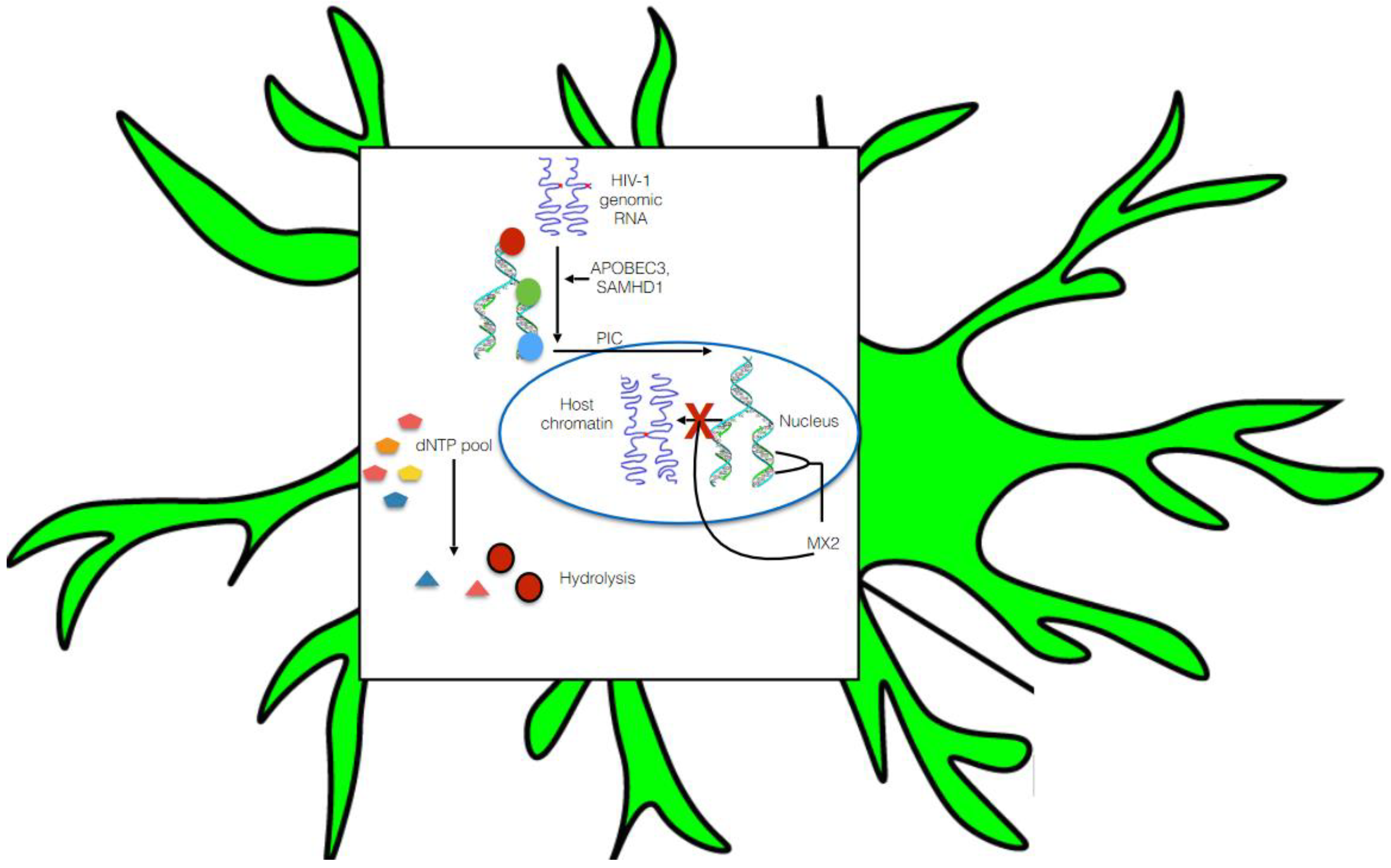
5.1. Pre-Integration Latency
5.2. Post-Integration Latency
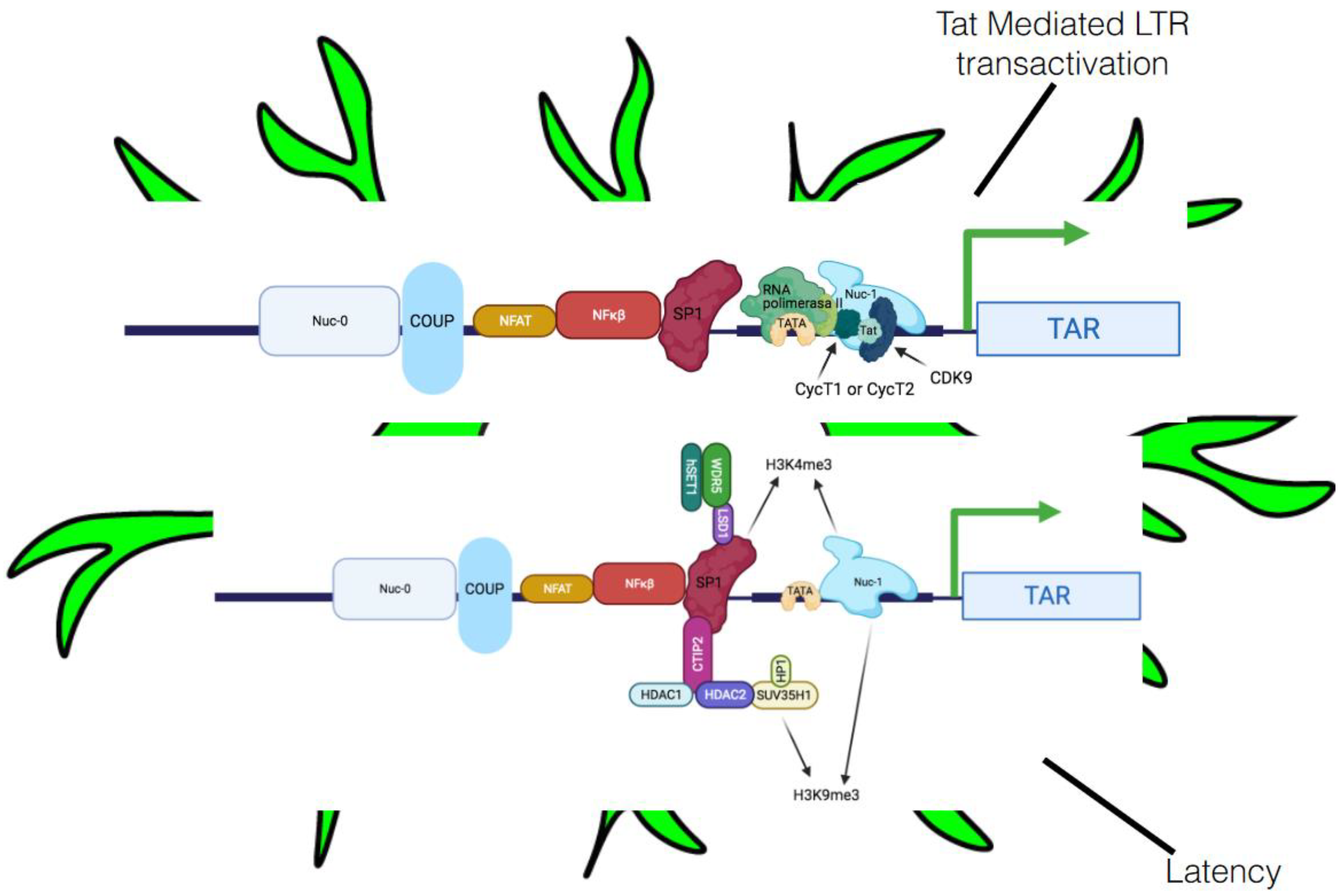
5.2.1. Transcriptional Interference
5.2.2. Host Integration, Heterochromatin, and Epigenetic Alterations
5.2.3. Involvement of Crucial Host Transcription Factors and Viral Proteins in Latency
5.2.4. MicroRNA and HIV Latency in HAND
6. Targeting the HIV-1 Reservoirs as Innovative Approaches against HAND
6.1. Early and Intensified Antiretroviral Therapy
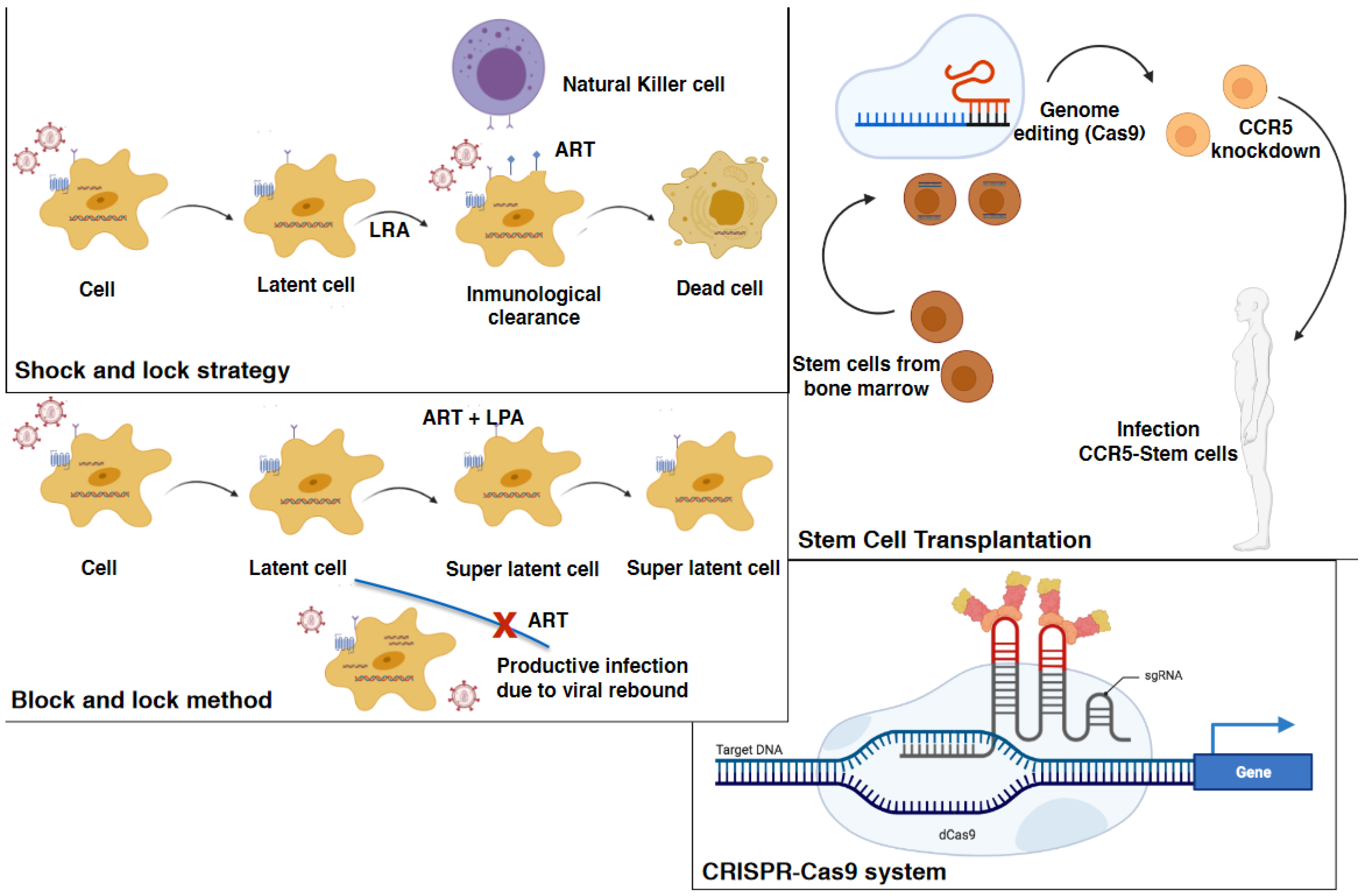
6.2. Immune-Based Strategies
6.3. Stem Cell Transplantation
6.4. Permanent HIV Suppression
6.5. Gene Therapy and the “Kick and Kill” and “Shock and Kill” Therapeutic Strategies
6.6. Progress and Limitations of Treating HAND by Targeting Microglia
7. Combined Antiretroviral Therapy Treatment for HAND
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Law, K.M.; Satija, N.; Esposito, A.M.; Chen, B.K. Cell-to-cell spread of HIV and viral pathogenesis. Adv. Virus Res. 2016, 95, 43–85. [Google Scholar] [PubMed]
- Borrajo, A.; Ranazzi, A.; Pollicita, M.; Bruno, R.; Modesti, A.; Alteri, C.; Perno, C.F.; Svicher, V.; Aquaro, S. Effects of Amprenavir on HIV-1 Maturation, Production and Infectivity Following Drug Withdrawal in Chronically-Infected Monocytes/Macrophages. Viruses 2017, 9, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García, M.; Buzón, M.J.; Benito, J.M.; Rallón, N. Peering into the HIV reservoir. Rev. Med. Virol. 2018, 28, e1981. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.M.; Margolis, D.M. HIV persistence on antiretroviral therapy and barriers to a cure. Adv. Exp. Med. Biol. 2018, 1075, 165–185. [Google Scholar] [CrossRef]
- Borrajo, A.; Ranazzi, A.; Pollicita, M.; Bellocchi, M.C.; Salpini, R.; Mauro, M.V.; Ceccherini-Silberstein, F.; Perno, C.F.; Svicher, V.; Aquaro, S. Different Patterns of HIV-1 Replication in MACROPHAGES is Led by Co-Receptor Usage. Medicina 2019, 55, 297. [Google Scholar] [CrossRef] [Green Version]
- Borrajo, A.; Spuch, C.; Penedo, M.A.; Olivares, J.M.; Agís-Balboa, R.C. Important role of microglia in HIV-1 associated neurocognitive disorders and the molecular pathways implicated in its pathogenesis. Ann. Med. 2020, 53, 43–69. [Google Scholar] [CrossRef] [PubMed]
- Strain, M.C.; Letendre, S.; Pillai, S.K.; Russell, T.; Ignacio, C.C.; Gunthard, H.F.; Good, B.; Smith, D.M.; Wolinsky, S.M.; Furtado, M.; et al. Genetic composition of human immunodeficiency virus type 1 in cerebrospinal fluid and blood without treatment and during failing antiretroviral therapy. J. Virol. 2005, 79, 1772–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell. Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamers, S.L.; Salemi, M.; Galligan, D.C.; Morris, A.; Gray, R.; Fogel, G.; Zhao, L.; McGrath, M.S. Human immunodeficiency virus-1 evolutionary patterns associated with pathogenic processes in the brain. J. Neurovirol. 2010, 16, 230–241. [Google Scholar] [CrossRef] [Green Version]
- Buckley, S.; Byrnes, S.; Cochrane, C.; Roche, M.; Estes, J.D.; Selemidis, S.; Angelovich, T.A.; Churchill, M.J. The role of oxidative stress in HIV—Associated neurocognitive disorders. Brain Behav. Immun. Health 2021, 13, 100235. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Lu, Q.; Farrell, M.; Lappin, J.M.; Shi, J.; Lu, L.; Bao, Y. Global prevalence and burden of HIV—Associated neurocognitive disorder: A meta-analysis. Neurology 2020, 95, e2610–e2621. [Google Scholar] [CrossRef]
- Bougea, A.; Spantideas, N.; Galanis, P.; Gkekas, G.; Thomaides, T. Optimal treatment of HIV—Associated neurocognitive disorders: Myths and reality. A critical review. Ther. Adv. Infect. Dis. 2019, 6, 2049936119838228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Underwood, J.; Robertson, K.R.; Winston, A. Could antiretroviral neurotoxicity play a role in the pathogenesisof cognitive impairment in treated HIV disease? AIDS 2015, 29, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Celis, V.; Valiente-Echeverría, F.; Soto-Rifo, R.; Toro-Ascuy, D. New Challenges of HIV-1 Infection: How HIV-1 Attacks and Resides in the Central Nervous System. Cells 2019, 8, 1245. [Google Scholar] [CrossRef] [Green Version]
- Periyasamy, P.; Thangaraj, A.; Bendi, V.S.; Buch, S. HIV-1 Tat-mediated microglial inflammation involves a novel miRNA-34a-NLRC5-NFκB signaling axis. Brain. Behav. Immun. 2019, 80, 227–237. [Google Scholar] [CrossRef]
- Chivero, E.T.; Guo, M.-L.; Periyasamy, P.; Liao, K.; Callen, S.E.; Buch, S. HIV-1 Tat primes and activates microglial NLRP3 inflammasome-mediated neuroinflammation. J. Neurosci. 2017, 37, 3599–3609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abassi, M.; Morawski, B.M.; Nakigozi, G.; Nakasujja, N.; Kong, X.; Meya, D.B.; Robertson, K.; Gray, R.; Wawer, M.J.; Sacktor, N.; et al. Cerebrospinal fluid biomarkers and HIV-associated neurocognitive disorders in HIV-infected individuals in Rakai, Uganda. J. Neurovirol. 2017, 23, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Aquaro, S.; Borrajo, A.; Pellegrino, M.; Svicher, V. Mechanisms underlying of antiretroviral drugs in different cellular reservoirs with a focus on macrophages. Virulence 2020, 11, 400–413. [Google Scholar] [CrossRef]
- Wong, M.E.; Jaworowski, A.; Hearps, A.C. The HIV reservoir in monocytes and macrophages. Front. Immunol. 2019, 10, 1435. [Google Scholar] [CrossRef] [Green Version]
- Penedo, M.A.; Rivera-Baltanás, T.; Pérez-Rodríguez, D.; Allen, J.; Borrajo, A.; Alonso-Crespo, D.; Fernández-Pereira, C.; Nieto-Araujo, M.; Ramos-García, S.; Barreiro-Villar, C.; et al. The role of dopamine receptors in lymphocytes and their changes in schizophrenia. Brain Behav. Immun. Health 2021, 12, 100199. [Google Scholar] [CrossRef]
- Eisele, E.; Siliciano, R.F. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity 2012, 37, 377–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churchill, M.J.; Wesselingh, S.L.; Cowley, D.; Pardo, C.A.; McArthur, J.C.; Brew, B.J.; Gorry, P.R. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann. Neurol. 2009, 66, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.; Kang, G.; Hattler, J.B.; Galadima, H.I.; Zhang, J.; Li, Q.; Kim, W.K. Macrophages but not astrocytes harbor HIV DNA in the brains of HIV-1-infected aviremic individuals on suppressive antiretroviral therapy. J. Neuroimmune Pharmacol. 2019, 14, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Cenker, J.J.; Stultz, R.D.; McDonald, D. Brain Microglial cells are highly susceptible to HIV-1 infection and spread. AIDS Res. Hum. Retrovir. 2017, 33, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Koppensteiner, H.; Brack-Werner, R.; Schindler, M. Macrophages and their relevance in human immunodeficiency virus type I infection. Retrovirology 2012, 9, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Réu, P.; Khosravi, A.; Bernard, S.; Mold, J.E.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; et al. The lifespan and turnover of microglia in the human brain. Cell Rep. 2017, 20, 779–784. [Google Scholar] [CrossRef] [Green Version]
- Lawson, L.J.; Perry, V.H.; Gordon, S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience 1992, 48, 405–415. [Google Scholar] [CrossRef]
- Jäkel, S.; Dimou, L. Glial cells and their function in the adult brain: A journey through the history of their ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Durafourt, B.A.; Moore, C.S.; Zammit, D.A.; Johnson, T.A.; Zaguia, F.; Guiot, M.C.; Bar-Or, A.; Antel, J.P. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia 2012, 60, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Sivakumar, V.; Zou, Z.; Ling, E.A. Microglia-derived proinflammatory cytokines tumor necrosis factor-alpha and interleukin-1beta induce Purkinje neuronal apoptosis via their receptors in hypoxic neonatal rat brain. Brain Struct. Funct. 2014, 219, 151–170. [Google Scholar] [CrossRef]
- Geffin, R.; Martinez, R.; Perez, R.; Issac, B.; McCarthy, M. Apolipoprotein E-dependent differences in innate immune responses of maturing human neuroepithelial progenitor cells exposed to HIV-1. J. Neuroimmune Pharmacol. 2013, 8, 1010–1026. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; Rathakrishnan, P.; Xiao, H.; Gao, T.; Duong, D.M.; Pennington, M.W.; Lah, J.J.; Seyfried, N.T.; et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 24. [Google Scholar] [CrossRef] [Green Version]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [Green Version]
- Wraith, D.C.; Nicholson, L.B. The adaptive immune system in diseases of the central nervous system. J. Clin. Investig. 2012, 122, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Taipa, R.; das Neves, S.P.; Sousa, A.L.; Fernandes, J.; Pinto, C.; Correia, A.P.; Santos, E.; Pinto, P.S.; Carneiro, P.; Costa, P.; et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol. Aging 2019, 76, 125–132. [Google Scholar] [CrossRef]
- Rodrigues-Amorim, D.; Rivera-Baltanás, T.; Spuch, C.; Caruncho, H.J.; González-Fernandez, A.; Olivares, J.M.; Agis-Balboa, R.C. Cytokines dysregulation in schizophrenia: A systematic review of psychoneuroimmune relationship. Schizophr. Res. 2018, 197, 19–33. [Google Scholar] [CrossRef]
- Omeragic, A.A.; Kayode, O.; Hoque, M.T.; Bendayan, R. Potential pharmacological approaches for the treatment of HIV-1 associated neurocognitive disorders. Fluids Barriers CNS 2020, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, S.D.; Alldred, M.J.; Gunnam, S.M.; Schiroli, C.; Lee, S.H.; Morgello, S.; Fischer, T. Expression profiling suggests microglial impairment in human immunodeficiency virus neuropathogenesis. Ann. Neurol. 2018, 83, 406–417. [Google Scholar] [CrossRef]
- Vera, J.H.; Guo, Q.; Cole, J.H.; Boasso, A.; Greathead, L.; Kelleher, P.; Rabiner, E.A.; Kalk, N.; Bishop, C.; Gunn, R.N.; et al. Neuroinflammation in treated HIV-positive individuals: A TSPO PET study. Neurology 2016, 86, 1425–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinharay, S.; Hammoud, D.A. Brain PET imaging: Value for understanding the pathophysiology of HIV-Associated Neurocognitive Disorder (HAND). Curr. HIV/AIDS Rep. 2019, 16, 66–75. [Google Scholar] [CrossRef]
- Osipova, E.D.; Semyachkina-Glushkovskaya, O.V.; Morgun, A.V.; Pisareva, N.V.; Malinovskaya, N.A.; Boitsova, E.B.; Pozhilenkova, E.A.; Belova, O.A.; Salmin, V.V.; Taranushenko, T.E.; et al. Gliotransmitters and cytokines in the control of blood-brain barrier permeability. Rev. Neurosci. 2018, 29, 567–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumi, N.; Nishioku, T.; Takata, F.; Matsumoto, J.; Watanabe, T.; Shuto, H.; Yamauchi, A.; Dohgu, S.; Kataoka, Y. Lipopolysaccharide-activated microglia induce dysfunction of the blood–brain barrier in rat microvascular endothelial cells co-cultured with microglia. Cell. Mol. Neurobiol. 2010, 30, 247–253. [Google Scholar] [CrossRef]
- Sheng, Z.; Liu, Y.; Li, H.; Zheng, W.; Xia, B.; Zhang, X.; Yong, V.W.; Xue, M. Efficacy of minocycline in acute ischemic stroke: A systematic review and meta-analysis of rodent and clinical studies. Front. Neurol. 2018, 9, 1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.C.; Partridge, A.T.; Sell, C.; Torres, C.; Martín-García, J. Fate of microglia during HIV-1 infection: From activation to senescence? Glia 2017, 65, 431–446. [Google Scholar] [CrossRef]
- Burdo, T.H.; Lackner, A.; Williams, K.C. Monocyte/macrophages and their role in HIV neuropathogenesis. Immunol. Rev. 2013, 254, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Fischer-Smith, T.; Tedaldi, E.M.; Rappaport, J. CD163/CD16 coexpression by circulating monocytes/macrophages in HIV: Potential biomarkers for HIV infection and AIDS progression. AIDS Res. Hum. Retrovir. 2008, 24, 417–421. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.W.; Calderon, T.M.; Lopez, L.; Carvallo-Torres, L.; Gaskill, P.J.; Eugenin, E.A.; Morgello, S.; Berman, J.W. Mechanisms of HIV entry into the CNS: Increased sensitivity of HIV infected CD14+CD16+ monocytes to CCL2 and key roles of CCR2, JAM-A, and ALCAM in diapedesis. PLoS ONE 2013, 8, e69270. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Abbas, W.; Herbein, G. HIV-1 latency in monocytes/macrophages. Viruses 2014, 6, 1837–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sénécal, V.; Barat, C.; Tremblay, M.J. The delicate balance between neurotoxicity and neuroprotection in the context of HIV-1 infection. Glia 2020, 10, 255–280. [Google Scholar] [CrossRef] [PubMed]
- Wesselingh, S.L.; Takahashi, K.; Glass, J.D.; McArthur, J.C.; Griffin, J.W.; Griffin, D.E. Cellular localization of tumor necrosis factor mRNA in neurological tissue from HIV-infected patients by combined reverse transcriptase/polymerase chain reaction in situ hybridization and immunohistochemistry. J. Neuroimmunol. 1997, 74, 1–8. [Google Scholar] [CrossRef]
- Choi, S.S.; Lee, H.J.; Lim, I.; Satoh, J.; Kim, S.U. Human astrocytes: Secretome profiles of cytokines and chemokines. PLoS ONE 2014, 9, e92325. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Milne, S.; Das, B.; Dobrowolski, C.; Rojas, R.; Karn, J. Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells. Retrovirology 2017, 14, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Mesa, Y.; Jay, T.R.; Checkley, M.A.; Luttge, B.; Dobrowolski, C.; Valadkhan, S.; Landreth, G.E.; Karn, J.; Alvarez-Carbonell, D. Immortalization of primary microglia: A new platform to study HIV regulation in the central nervous system. J. Neurovirol. 2017, 23, 47–66. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Carbonell, D.; Ye, F.; Ramanath, N.; Dobrowolski, C.; Karn, J. The glucocorticoid receptor is a critical regulator of HIV latency in human microglial cells. J. Neuroimmune Pharmacol. 2019, 14, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Mlcochova, P.; Sutherland, K.A.; Watters, S.A.; Bertoli, C.; de Bruin, R.A.; Rehwinkel, R.; Neil, S.J.; Lenzi, G.M.; Kim, B.; Khwaja, A.; et al. A G1-like state allows HIV-1 to bypass SAMHD1 restriction in macrophages. EMBO J. 2017, 36, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Cosenza, M.A.; Zhao, M.-L.; Si, Q.; Lee, S.C. Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis. Brain Pathol. 2002, 12, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.J.; Gorry, P.R.; Cowley, D.; Lal, L.; Sonza, S.; Purcell, D.F.; Thompson, K.A.; Gabuzda, D.; McArthur, J.C.; Pardo, C.A.; et al. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J. Neurovirol. 2006, 12, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.A.; Cherry, C.L.; Bell, J.E.; McLean, C.A. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am. J. Pathol. 2011, 179, 1623–1629. [Google Scholar] [CrossRef] [Green Version]
- Edén, A.; Nilsson, S.; Hagberg, L.; Fuchs, D.; Zetterberg, H.; Svennerholm, B.; Gisslén, M. Asymptomatic cerebrospinal fluid HIV-1 viral blips and viral escape during antiretroviral therapy: A longitudinal study. J. Infect. Dis. 2016, 214, 1822–1825. [Google Scholar] [CrossRef] [Green Version]
- Bavaro, D.F.; Calamo, A.; Lepore, L.; Fabrizio, C.; Saracino, A.; Angarano, G.; Monno, L. Cerebrospinal fluid compartmentalization of HIV-1 and correlation with plasma viral load and blood–brain barrier damage. Infection 2019, 47, 441–446. [Google Scholar] [CrossRef]
- Harbison, C.; Zhuang, K.; Gettie, A.; Blanchard, J.; Knight, H.; Didier, P.; Cheng-Mayer, C.; Westmoreland, S. Giant cell encephalitis and microglial infection with mucosally transmitted simian-human immunodeficiency virus SHIVSF162P3N in rhesus macaques. J. Neurovirol. 2014, 20, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Whitney, J.B.; Hill, A.L.; Sanisetty, S.; Penaloza-MacMaster, P.; Liu, J.; Shetty, M.; Parenteau, L.; Cabral, C.; Shields, J.; Blackmore, S.; et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 2014, 512, 74–77. [Google Scholar] [CrossRef]
- Llewellyn, G.N.; Alvarez-Carbonell, D.; Chateau, M.; Karn, J.; Cannon, P.M. HIV-1 infection of microglial cells in a reconstituted humanized mouse model and identification of compounds that selectively reverse HIV latency. J. Neurovirol. 2018, 24, 192–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathews, S.; Branch Woods, A.; Katano, I.; Makarov, E.; Thomas, M.B.; Gendelman, H.E.; Poluektova, L.Y.; Ito, M.; Gorantla, S. Human Interleukin-34 facilitates microglia-like cell differentiation and persistent HIV-1 infection in humanized mice. Mol. Neurodegener. 2019, 14, 12. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Cheng, Y.; Sravanam, S.; Mathews, S.; Gorantla, S.; Poluektova, L.Y.; Dash, P.K.; Gendelman, H.E. Immune activations and viral tissue compartmentalization during progressive HIV-1 infection of humanized mice. Front. Immunol. 2019, 10, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albright, A.V.; Shieh, J.T.; O’Connor, M.J.; González-Scarano, F. Characterization of cultured microglia that can be infected by HIV-1. J. Neurovirol. 2000, 6 (Suppl. 1), S53–S60. [Google Scholar] [PubMed]
- Peudenier, S.; Hery, C.; Montagnier, L.; Tardieu, M. Human microglial cells: Characterization in cerebral tissue and in primary culture, and study of their susceptibility to HIV-1 infection. Ann. Neurol. 1991, 29, 152–161. [Google Scholar] [CrossRef]
- Gougeon, M.L. Alarmins and central nervous system inflammation in HIV-associated neurological disorders. J. Intern. Med. 2017, 281, 433–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcagno, A.; Atzori, C.; Romito, A.; Vai, D.; Audagnotto, S.; Stella, M.L.; Montrucchio, C.; Imperiale, D.; Di Perri, G.; Bonora, S. Blood brain barrier impairment is associated with cerebrospinal fluid markers of neuronal damage in HIV-positive patients. J. Neurovirol. 2016, 22, 88–92. [Google Scholar] [CrossRef]
- Desplats, P.; Dumaop, W.; Smith, D.; Adame, A.; Everall, I.; Letendre, S.; Ellis, R.; Cherner, M.; Grant, I.; Masliah, E. Molecular and pathologic insights from latent HIV-1 infection in the human brain. Neurology 2013, 80, 1415–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawat, P.; Spector, S.A. Development and characterization of a human microglia cell model of HIV-1 infection. J. Neurovirol. 2017, 23, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salemi, M.; Rife, B. Phylogenetics and phyloanatomy of HIV/SIV intra-host compartments and reservoirs: The key role of the central nervous system. Curr. HIV Res. 2016, 14, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Chahroudi, A.; Silvestri, G. Animal models to achieve an HIV cure. Curr. Opin. HIV AIDS 2016, 11, 432–441. [Google Scholar] [CrossRef] [Green Version]
- Gama, L.; Abreu, C.; Shirk, E.N.; Queen, S.E.; Beck, S.E.; Metcalf Pate, K.A.; Bullock, B.T.; Zink, M.C.; Mankowski, J.L.; Clements, J.E. SIV latency in macrophages in the CNS. Curr. Top. Microbiol. Immunol. 2018, 417, 111–130. [Google Scholar] [CrossRef]
- Abreu, C.; Shirk, E.N.; Queen, S.E.; Mankowski, J.L.; Gama, L.; Clements, J.E. A quantitative approach to SIV functional latency in brain macrophages. J. Neuroimmune Pharmacol. 2019, 14, 23–32. [Google Scholar] [CrossRef]
- Marsden, M.D.; Zack, J.A. Humanized mouse models for human immunodeficiency virus infection. Annu. Rev. Virol. 2017, 4, 393–412. [Google Scholar] [CrossRef]
- Avalos, C.R.; Abreu, C.M.; Queen, S.E.; Li, M.; Price, S.; Shirk, E.N.; Engle, E.L.; Forsyth, E.; Bullock, B.T.; Mac Gabhann, F.; et al. Brain macrophages in simian immunodeficiency virus-infected, antiretroviral-suppressed macaques: A functional latent reservoir. mBio 2017, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Gama, L.; Abreu, C.M.; Shirk, E.N.; Price, S.L.; Li, M.; Laird, G.M.; Pate, K.A.; Wietgrefe, S.W.; O’Connor, S.L.; Pianowski, L.; et al. Reactivation of simian immunodeficiency virus reservoirs in the brain of virally suppressed macaques. AIDS 2017, 31, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Barber, S.A.; Gama, L.; Dudaronek, J.M.; Voelker, T.; Tarwater, P.M.; Clements, J.E. Mechanism for the establishment of transcriptional HIV latency in the brain in a simian immunodeficiency virus–macaque model. J. Infect. Dis. 2006, 193, 963–970. [Google Scholar] [CrossRef] [Green Version]
- Asahchop, E.L.; Meziane, O.; Mamik, M.K.; Chan, W.F.; Branton, W.G.; Resch, L.; Gill, M.J.; Haddad, E.; Guimond, J.V.; Wainberg, M.A.; et al. Reduced antiretroviral drug efficacy and concentration in HIV-infected microglia contributes to viral persistence in brain. Retrovirology 2017, 14, 47. [Google Scholar] [CrossRef]
- Al-Harti, L.; Joseph, J.; Nath, A. Astrocytes as an HIV CNS reservoir: Highlights and reflections of an NIMH-sponsored symposium. J. Neurovirol. 2018, 24, 665–669. [Google Scholar] [CrossRef]
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Molecular mechanisms of HIV latency. J. Clin. Investig. 2016, 126, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Jung, A.; Maier, R.; Vartanian, J.P.; Bocharov, G.; Jung, V.; Fischer, U.; Meese, E.; Wain-Hobson, S.; Meyerhans, A. Recombination: Multiply infected spleen cells in HIV patients. Nature 2002, 418, 6894. [Google Scholar] [CrossRef]
- Lenasi, T.; Contreras, X.; Peterlin, B.M. Transcriptional interference antagonizes proviral gene expression to promote HIV latency. Cell Host Microbe 2008, 4, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Mbonye, U.; Karn, J. Transcriptional control of HIV latency: Cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology 2014, 454–455, 328–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadlowsky, J.K.; Wong, J.Y.; Graham, A.C.; Dobrowolski, C.; Devor, R.L.; Adams, M.D.; Fujinaga, K.; Karn, J. Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol. Cell. Biol. 2014, 34, 1911–1928. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.A.; Greene, W.C. Host factors regulating post-integration latency of HIV. Trends Microbiol. 2005, 13, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Lassen, K.G.; Ramyar, K.X.; Bailey, J.R.; Zhou, Y.; Siliciano, R.F. Nuclear retention of multiply spliced HIV-1 RNA in resting CD4+ T cells. PLoS Pathog. 2006, 2, e68. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Zhou, Y.; Kieffer, T.L.; Ruff, C.T.; Buck, C.; Siliciano, R.F. Molecular characterization of preintegration latency in human immunodeficiency virus type 1 infection. J. Virol. 2002, 76, 8518–8531. [Google Scholar] [CrossRef] [Green Version]
- Miyagi, E.; Schwartzkopff, F.; Plishka, R.; Buckler-White, R.; Clouse, K.A.; Strebel, K. APOBEC3G-independent reduction in virion infectivity during long-term HIV-1 replication in terminally differentiated macrophages. Virology 2008, 379, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Goujon, C.; Moncorge, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef]
- Liu, L.; Oliveira, N.M.; Cheney, K.M.; Pade, C.; Dreja, H.; Bergin, A.M.; Borgdorff, V.; Beach, D.H.; Bishop, C.L.; Dittmar, M.T.; et al. A whole genome screen for HIV restriction factors. Retrovirology 2011, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [Green Version]
- Bouchat, S.; Gatot, J.S.; Kabeya, K.; Cardona, C.; Colin, L.; Herbein, G.; De Wit, S.; Clumeck, N.; Lambotte, O.; Rouzioux, C.; et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4+ T cells from HIV-1-infected HAART-treated patients. AIDS 2012, 26, 1473–1482. [Google Scholar] [CrossRef]
- Tripathy, M.K.; Abbas, W.; Herbein, G. Epigenetic regulation of HIV-1 transcription. Epigenomics 2011, 3, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Landry, S.; Halin, M.; Lefort, S.; Audet, B.; Vaquero, C.; Mesnard, J.M.; Barbeau, B. Detection, characterization and regulation of antisense transcripts in HIV-1. Retrovirology 2007, 4, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demeret, C.; Vassetzky, Y.; Mechali, M. Chromatin remodelling and DNA replication: From nucleosomes to loop domains. Oncogene 2001, 20, 3086–3093. [Google Scholar] [CrossRef] [Green Version]
- Brady, T.; Kelly, B.; Male, F.; Roth, S.; Bailey, A.; Malani, N.; Bushman, F.D.; Gijsbers, R.; O’Doherty, U.; Bushman, F.D. Quantitation of HIV DNA integration: Effects of differential integration site distributions on Alu-PCR assays. J. Virol. Methods 2013, 189, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Budhiraja, S.; Rice, A.P. Reactivation of latent HIV: Do all roads go through P-TEFb? Future Virol. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartholomeeusen, K.; Fujinaga, K.; Xiang, Y.; Peterlin, B.M. Histone deacetylase inhibitors (HDACis) that release the positive transcription elongation factor b (P-TEFb) from its inhibitory complex also activate HIV transcription. J. Biol. Chem. 2013, 288, 14400–14407. [Google Scholar] [CrossRef] [Green Version]
- Karn, J. The molecular biology of HIV latency: Breaking and restoring the Tat-dependent transcriptional circuit. Curr. Opin. HIV AIDS 2011, 6, 4–11. [Google Scholar] [CrossRef]
- Breuer, D.; Kotelkin, A.; Ammosova, T.; Kumari, N.; Ivanov, A.; Ilatovskiy, A.V.; Beullens, M.; Roane, P.R.; Bollen, M.; Petukhov, M.G.; et al. CDK2 regulates HIV-1 transcription by phosphorylation of CDK9 on serine 90. Retrovirology 2012, 9, 94. [Google Scholar] [CrossRef] [Green Version]
- Heidemann, M.; Hintermair, C.; Voss, K.; Eick, D. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim. Biophys. Acta 2013, 1829, 55–62. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Shibata, H.; Handa, H. Transcription elongation factors DSIF and NELF: Promoter-proximal pausing and beyond. Biochim. Biophys. Acta 2013, 1829, 98–104. [Google Scholar] [CrossRef]
- Rice, A.P. P-TEFb as a target to reactivate latent HIV: Two Brds are now in hand. Cell Cycle 2013, 12, 392–393. [Google Scholar] [CrossRef] [Green Version]
- Chiang, K.; Rice, A.P. MicroRNA-mediated restriction of HIV-1 in resting CD4+ T cells and monocytes. Viruses 2012, 4, 1390–1409. [Google Scholar] [CrossRef] [Green Version]
- Peterlin, B.M.; Brogie, J.E.; Price, D.H. 7SK snRNA: A noncoding RNA that plays a major role in regulating eukaryotic transcription. Wiley Interdiscip. Rev. RNA 2012, 3, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Muniz, L.; Egloff, S.; Ughy, B.; Jady, B.E. Kiss, Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog. 2010, 6, e1001152. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Xiang, Y.; Fujinaga, K.; Bartholomeeusen, K.; Nilson, K.A.; Price, D.H.; Peterlin, B.M. Release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein (snRNP) activates hexamethylene bisacetamide-inducible protein (HEXIM1) transcription. J. Biol. Chem. 2014, 289, 9918–9925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Douce, V.; Cherrier, T.; Riclet, R.; Rohr, O.; Schwartz, C. The many lives of CTIP2: From aids to cancer and cardiac hypertrophy. J. Cell. Physiol. 2014, 229, 533–537. [Google Scholar] [CrossRef] [Green Version]
- Le Douce, V.; Colin, L.; Redel, L.; Cherrier, T.; Herbein, G.; Aunis, D.; Rohr, O.; Van Lint, C.; Schwartz, C. LSD1 cooperates with CTIP2 to promote HIV-1 transcriptional silencing. Nucleic Acids Res. 2012, 40, 1904–1915. [Google Scholar] [CrossRef]
- Ravimohan, S.; Gama, L.; Barber, S.A.; Clements, J.E. Regulation of SIV mac 239 basal long terminal repeat activity and viral replication in macrophages: Functional roles of two CCAAT/enhancer-binding protein beta sites in activation and interferon beta-mediated suppression. J. Biol. Chem. 2010, 285, 2258–2273. [Google Scholar] [CrossRef] [Green Version]
- Henderson, L.J.; Narasipura, S.D.; Adarichev, V.; Kashanchi, F.; Al-Harthi, L. Identification of novel T cell factor 4 (TCF-4) binding sites on the HIV long terminal repeat which associate with TCF-4, beta-catenin, and SMAR1 to repress HIV transcription. J. Virol. 2012, 86, 9495–9503. [Google Scholar] [CrossRef] [Green Version]
- Keshavarz, M.; Mirzaei, H.; Salemi, M.; Momeni, F.; Mousavi, M.J.; Sadeghalvad, M.; Arjeini, Y.; Solaymani-Mohammadi, F.; Sadri Nahand, J.; Namdari, H.; et al. Influenza vaccine: Where are we and where do we go? Rev. Med. Virol. 2019, 29, e2014. [Google Scholar] [CrossRef]
- Triboulet, R.; Mari, B.; Lin, Y.L.; Chable-Bessia, C.; Bennasser, Y.; Lebrigand, K.; Lebrigand, K.; Cardinaud, B.; Maurin, T.; Barbry, P.; et al. Suppression of miRNA-silencing pathway by HIV-1 during virus replication. Science 2007, 315, 1579–1582. [Google Scholar] [CrossRef]
- Wang, X.; Ye, L.; Hou, W.; Zhou, Y.; Wang, Y.J.; Metzger, D.S. Cellular miRNA expression correlates with susceptibility of monocytes/macrophages to HIV-1 infection. Blood 2009, 113, 671–674. [Google Scholar] [CrossRef]
- Xu, Z.; Asahchop, E.L.; Branton, W.G.; Gelman, B.B.; Power, C.; Hobman, T.C. MicroRNAs upregulated during HIV infection target peroxisome biogenesis factors: Implications for virus biology, disease mechanisms and neuropathology. PLoS Pathog. 2017, 13, e1006360. [Google Scholar] [CrossRef]
- Yelamanchili, S.V.; Chaudhuri, A.D.; Chen, L.N.; Xiong, H.; Fox, H.S. MicroRNA-21 dysregulates the expression of MEF2C in neurons in monkey and human SIV/HIV neurological disease. Cell Death Dis. 2010, 1, e77. [Google Scholar] [CrossRef]
- Noorbakhsh, F.; Ramachandran, R.; Barsby, N.; Ellestad, K.K.; LeBlanc, A.; Dickie, P.; Baker, G.; Hollenberg, M.D.; Cohen, E.A.; Power, C. MicroRNA profiling reveals new aspects of HIV neurodegeneration: Caspase-6 regulates astrocyte survival. FASEB J. 2010, 24, 1799–1812. [Google Scholar] [CrossRef]
- Sáez-Cirión, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013, 9, e1003211. [Google Scholar] [CrossRef]
- Schuetz, A.; Deleage, C.; Sereti, I.; Rerknimitr, R.; Phanuphak, N.; Phuang-Ngern, Y.; Ngern, Y.; Estes, J.D.; Sandler, N.G.; Sukhumvittaya, S.; et al. Initiation of ART during early acute HIV infection preserves mucosal Th17 function and reverses HIV-related immune activation. PLoS Pathog. 2014, 10, e1004543. [Google Scholar] [CrossRef] [Green Version]
- Sacha, J.B.; Ndhlovu, L.C. Strategies to target non-T-cell HIV reservoirs. Curr. Opin. HIV AIDS 2016, 11, 376–382. [Google Scholar] [CrossRef] [Green Version]
- Ndhlovu, L.C.; Umaki, T.; Chew, G.M.; Chow, D.C.; Agsalda, M.; Kallianpur, K.J.; Paul, R.; Zhang, G.; Ho, E.; Hanks, N.; et al. Treatment intensification with maraviroc (CCR5 antagonist) leads to declines in CD16-expressing monocytes in cART-suppressed chronic HIV-infected subjects and is associated with improvements in neurocognitive test performance: Implications for HIV-associated neurocognitive disease (HAND). J. Neurovirol. 2014, 20, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Lafeuillade, A.; Assi, A.; Poggi, C.; Bresson-Cuquemelle, C.; Jullian, E.; Tamalet, C. Failure of combined antiretroviral therapy intensification with maraviroc and raltegravir in chronically HIV-1 infected patients to reduce the viral reservoir: The IntensHIV randomized trial. AIDS Res. Ther. 2014, 11, 33. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Darcis, G.; Van Lint, C.; Herbein, G. Epigenetic control of HIV-1 post integration latency: Implications for therapy. Clin. Epigenet. 2015, 7, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jilg, N.; Li, J.Z. On the Road to a HIV Cure: Moving Beyond Berlin and London. Infect. Dis. Clin. N. Am. 2019, 33, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Deng, K.; Pertea, M.; Rongvaux, A.; Wang, L.; Durand, C.M.; Ghiaur, G.; Lai, J.; McHugh, H.L.; Hao, H.; Zhang, H.; et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature 2015, 517, 381–385. [Google Scholar] [CrossRef] [Green Version]
- García, F.; León, A.; Gatell, J.M.; Plana, M.; Gallart, T. Therapeutic vaccines against HIV infection. Hum. Vaccin. Immunother. 2012, 8, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Persaud, D.; Luzuriaga, K.; Ziemniak, C.; Muresan, P.; Greenough, T.; Fenton, T.; Blackford, A.; Ferguson, K.; Neu, N.; Cunningham, C.K. Effect of therapeutic HIV recombinant poxvirus vaccines on the size of the resting CD4+ T-cell latent HIV reservoir. AIDS 2011, 25, 2227–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novis, C.L.; Archin, N.M.; Buzon, M.J.; Verdin, E.; Round, J.L.; Lichterfeld, M.; Margolis, D.M.; Planelles, V.; Bosque, A. Reactivation of latent HIV-1 in central memory CD4+ T cells through TLR-1/2 stimulation. Retrovirology 2013, 10, 119. [Google Scholar] [CrossRef] [Green Version]
- Ahlenstiel, C.L.; Symonds, G.; Kent, S.J.; Kelleher, A.D. Block and Lock HIV Cure Strategies to Control the Latent Reservoir. Front. Cell. Infect. Microbiol. 2020, 10, 424. [Google Scholar] [CrossRef]
- Pang, K.M.; Castanotto, D.; Li, H.; Scherer, L.; Rossi, J.J. Incorporation of aptamers in the terminal loop of shRNAs yields aneffective and novel combinatorial targeting strategy. Nucleic Acids Res. 2018, 46, e6. [Google Scholar] [CrossRef] [Green Version]
- Battistini, A.; Sgarbanti, M. HIV-1 latency: An update of molecular mechanisms and therapeutic strategies. Viruses 2014, 6, 1715–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatano, H.; Jain, V.; Hunt, P.W.; Lee, T.H.; Sinclair, E.; Do, T.D.; Hoh, R.; Martin, J.N.; McCune, J.M.; Hecht, F.; et al. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. J. Infect. Dis. 2013, 208, 50–56. [Google Scholar] [CrossRef]
- Kulpa, D.A.; Lawani, M.; Cooper, A.; Peretz, Y.; Ahlers, J.; Sékaly, R.P. PD-1 coinhibitory signals: The link between pathogenesis and protection. Semin. Immunol. 2013, 25, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Titanji, K.; Velu, V.; Chennareddi, L.; Vijay-Kumar, M.; Gewirtz, A.T.; Freeman, G.J.; Amara, R.R. Acute depletion of activated memory B cells involves the PD-1 pathway in rapidly progressing SIV-infected macaques. J. Clin. Investig. 2010, 120, 3878–3890. [Google Scholar] [CrossRef]
- Dyavar Shetty, R.; Velu, V.; Titanji, K.; Bosinger, S.E.; Freeman, G.J.; Silvestri, G.; Amara, R.R. PD-1 blockade during chronic SIV infection reduces hyperimmune activation and microbial translocation in rhesus macaques. J. Clin. Investig. 2012, 122, 1712–1716. [Google Scholar] [CrossRef] [PubMed]
- Mencarelli, A.; Francisci, D.; Renga, B.; D’Amore, C.; Cipriani, S.; Basile, F.; Freeman, G.J.; Amara, R.R. Ritonavir-induced lipoatrophy and dyslipidaemia is reversed by the anti-inflammatory drug leflunomide in a PPAR-γ-dependent manner. Antivir. Ther. 2012, 17, 669–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allers, K.; Hütter, G.; Hofmann, J.; Loddenkemper, C.; Rieger, K.; Thiel, E.; Schneider, T. Evidence for the cure of HIV infection by CCR5Δ32/Δ32 stem cell transplantation. Blood 2011, 117, 2791–2799. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Boritz, E.; Busch, M.; Bentsen, C.; Chun, T.W.; Douek, D.; Eisele, E.; Haase, A.; Ho, Y.C.; Hütter, G.; et al. Challenges in detecting HIV persistence during potentially curative interventions: A study of the Berlin patient. PLoS Pathog. 2013, 9, e1003347. [Google Scholar] [CrossRef]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.; Lee, H.; et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef]
- Jamaluddin, M.S.; Hu, P.W.; Jan, Y.; Siwak, E.B.; Rice, A.P. Short Communication: The Broad-Spectrum Histone Deacetylase Inhibitors Vorinostat and Panobinostat Activate Latent HIV in CD4(+) T Cells In Part Through Phosphorylation of the T-Loop of the CDK9 Subunit of P-TEFb. AIDS Res. Hum. Retrovir. 2016, 32, 169–173. [Google Scholar] [CrossRef] [Green Version]
- Anderson, I.; Low, J.S.; Weston, S.; Weinberger, M.; Zhyvoloup, A.; Labokha, A.A.; Corazza, G.; Kitson, R.A.; Moody, C.J.; Marcello, A.; et al. Heat shock protein 90 controls HIV-1 reactivation from latency. Proc. Natl. Acad. Sci. USA 2014, 111, E1528–E1537. [Google Scholar] [CrossRef] [Green Version]
- Duverger, A.; Wolschendorf, F.; Anderson, J.C.; Wagner, F.; Bosque, A.; Shishido, T.; Jones, J.; Planelles, V.; Willey, C.; Cron, R.Q.; et al. Kinase control of latent HIV-1 infection: PIM-1 kinase as a major contributor to HIV-1 reactivation. J. Virol. 2014, 88, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Panfil, A.R.; London, J.A.; Green, P.; Yoder, K.E. CRISPR/Cas9 genome editing to disable the latent HIV-1 provirus. Front. Microbiol. 2018, 9, 3107. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yin, C.; Zhang, T.; Li, F.; Yang, W.; Kaminski, R.; Fagan, P.R.; Putatunda, R.; Young, W.B.; Khalili, K.; et al. CRISPR/gRNA-directed synergistic activation mediator (SAM) induces specific, persistent and robust reactivation of the HIV-1 latent reservoirs. Sci. Rep. 2015, 5, 16277. [Google Scholar] [CrossRef]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 11461–11466. [Google Scholar] [CrossRef] [Green Version]
- Cary, D.C.; Peterlin, B.M. Targeting the latent reservoir to achieve functional HIV cure. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Limsirichai, P.; Gaj, T.; Schaffer, D.V. CRISPR-mediated activation of latent HIV-1 expression. Mol. Ther. 2016, 24, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Qu, D.; Sun, W.W.; Li, L.; Ma, L.; Sun, L.; Jin, X.; Li, T.; Hou, W.; Wang, J.H. Long noncoding RNA MALAT1 releases epigenetic silencing of HIV-1 replication by displacing the polycomb repressive complex 2 from binding to the LTR promoter. Nucleic Acids Res. 2019, 47, 3013–3027. [Google Scholar] [CrossRef] [Green Version]
- Cheray, M.; Joseph, B. Epigenetics Control Microglia Plasticity. Front. Cell. Neurosci. 2018, 12, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, C.; Bouchat, S.; Marban, C.; Gautier, V.; Van Lint, C.; Rohr, O.; Le Douce, V. On the way to find a cure: Purging latent HIV-1 reservoirs. Biochem. Pharmacol. 2017, 146, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, A.; Yndart, A.; Atluri, V.; Tiwari, S.; Tomitaka, A.; Gupta, P.; Jayant, R.D.; Alvarez-Carbonell, D.; Khalili, K.; Nair, M. Magnetically guided non-invasive CRISPR-Cas9/gRNA delivery across blood-brain barrier to eradicate latent HIV-1 infection. Sci. Rep. 2019, 9, 3928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, A.; Brew, B. HIV-associated neurocognitive disorders: Recent advances in pathogenesis, biomarkers, and treatment. F1000Research 2017, 6, 312. [Google Scholar] [CrossRef]
- Martin-Blondel, G.; Brassat, D.; Bauer, J.; Lassmann, H.; Liblau, R.S. CCR5 blockade for neuroinflammatory diseases--beyond control of HIV. Nat. Rev. Neurol. 2016, 12, 95–105. [Google Scholar] [CrossRef]
- Johnson, T.P.; Patel, K.; Johnson, K.R.; Maric, D.; Calabresi, P.A.; Hasbun, R.; Nath, A. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc. Natl. Acad. Sci. USA 2013, 110, 13588–13593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gates, T.M.; Cysique, L.A.; Siefried, K.J.; Chaganti, J.; Moffat, K.J.; Brew, B.J. Maraviroc-intensified combined antiretroviral therapy improves cognition in virally suppressed HIV-associated neurocognitive disorder. AIDS 2016, 30, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Pacifici, M.; Delbue, S.; Ferrante, P.; Jeansonne, D.; Kadri, F.; Nelson, S.; Velasco-Gonzalez, C.; Zabaleta, J.; Peruzzi, F. Cerebrospinal fluid miRNA profile in HIV-encephalitis. J. Cell. Physiol. 2013, 228, 1070–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, A.; Thangaraj, A.; Chivero, E.T.; Periyasamy, P.; Burkovetskaya, M.E.; Niu, F.; Guo, M.L.; Buch, S. N-Acetylcysteine Reverses Antiretroviral-Mediated Microglial Activation by Attenuating Autophagy-Lysosomal Dysfunction. Front. Neurol. 2020, 11, 840. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Shi, X.; Puthiyakunnon, S.; Zhang, L.; Zeng, Q.; Li, Y.; Boddu, S.; Qiu, J.; Lai, Z.; Ma, C.; et al. CD44-mediated monocyte transmigration across Cryptococcus neoformans-infected brain microvascular endothelial cells is enhanced by HIV-1 gp41-I90 ectodomain. J. Biomed. Sci. 2016, 23, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churchill, M.J.; Deeks, S.G.; Margolis, D.M.; Siliciano, R.F.; Swanstrom, R. HIV reservoirs: What, where and how to target them. Nat. Rev. Microbiol. 2016, 14, 55–60. [Google Scholar] [CrossRef]
- Clark, U.S.; Cohen, R.A. Brain dysfunction in the era of combination antiretroviral therapy: Implications for the treatment of the aging population of HIV-infected individuals. Curr. Opin. Investig. Drugs 2010, 11, 884–900. [Google Scholar]
- Ambrosius, B.; Faissner, S.; Guse, K.; von Lehe, M.; Grunwald, T.; Gold, R.; Grewe, B.; Chan, A. Teriflunomide and monomethylfumarate target HIV-induced neuroinflammation and neurotoxicity. J. Neuroinflamm. 2017, 14, 51. [Google Scholar] [CrossRef] [Green Version]
- Mamik, M.K.; Asahchop, E.L.; Chan, W.F.; Zhu, Y.; Branton, W.G.; McKenzie, B.A.; Cohen, E.A.; Power, C. Insulin treatment prevents neuroinflammation and neuronal injury with restored neurobehavioral function in models of HIV/AIDS neurodegeneration. J. Neurosci. 2016, 36, 10683–10695. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyzed Mechanisms | Study | Reference |
|---|---|---|
| Function of HIV-Tat in microglia | E.T. Chivero, M.-L. Guo, P. Periyasamy, K. Liao, S.E. Callen, S. Buch, HIV-1 Tat primes and activates microglial NLRP3 inflammasome-mediated neuroinflammation (2017) | [16] |
| Detection of infected microglia in patients whose viral level is suppressed but died from an HIV-1 unrelated outcome | A. Ko, G. Kang, J.B. Hattler, H.I. Galadima, J. Zhang, Q. Li, W.K. Kim, Macrophages but not astrocytes harbor HIV DNA in the brains of HIV-1-infected aviremic individuals on suppressive antiretroviral therapy (2019) | [23] |
| SAMHD1 restriction take place in in vitro differentiated macrophages and in freshly isolated macrophages from the lungs, abdomen, and brain | J.J. Cenker, R.D. Stultz, D. McDonald, Brain Microglial Cells Are Highly Susceptible to HIV-1 Infection and Spread (2017) | [24] |
| Over stimulation of microglia contributes to HAND | R. Geffin, R. Martinez, R. Perez, B. Issac, M. McCarthy, Apolipoprotein E-dependent differences in innate immune responses of maturing human neuroepithelial progenitor cells exposed to HIV-1 (2013) | [36] |
| Increased release of proinflammatory chemokines by microglia | S.L. Wesselingh, K. Takahashi, J.D.Glass, J.C. McArthur, J.W. Griffin, D.E. Griffin, Cellular localization of tumor necrosis factor mRNA in neurological tissue from HIV-infected patients by combined reverse transcriptase/polymerase chain reaction in situ hybridization and immunohisto-chemistry (1997) D. Alvarez-Carbonell, Y. Garcia-Mesa, S. Milne, B. Das, C. Dobrowolski, R. Rojas, J. Karn J, Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells (2017) | [56,58] |
| HIV-1 replication in microglial cells and their activation are dependent on autophagy activation | S.S. Choi, H.J. Lee, I. Lim, J. Satoh, S.U. Kim, Human astrocytes: secretome profiles of cytokines and chemokines (2014) | [57] |
| HIV-1 replication in microglia using primary cultures obtained from fetal brains or post-mortem assessment of AIDS patients | A.V. Albright, J.T. Shieh, M.J. O’Connor, F. Gonzalez-Scarano, Characterization of cultured microglia that can be infected by HIV-1 (2000) S. Peudenier, C. Hery, L. Montagnier, M. Tardieu, Human microglial cells: characterization in cerebral tissue and in primary culture, and study of their susceptibility to HIV-1 infection (1991) | [58,59] |
| Immortalized human microglial cell line is useful to produce stable cell lines latently infected with HIV-1 proviruses | Y. Garcia-Mesa, T.R. Jay, M.A. Checkley, B. Luttge, C. Dobrowolski, S. Valadkhan, G.E. Landreth, J. Karn, D. Alvarez-Carbonell, Immortalization of primary microglia: A new platform to study HIV regulation in the central nervous system (2017) D. Alvarez-Carbonell, F. Ye, N. Ramanath, C. Dobrowolski, J. Karn, The glucocorticoid receptor is a critical regulator of HIV latency in human microglial cells (2019) | [59,60] |
| Absence of restriction by SAMDH1 is due to its phosphorylation by the cyclin kinase 1 (CDK1) which is induced in microglial cells | P. Mlcochova, K.A. Sutherland, S.A. Watters, C. Bertoli, R.A. de Bruin, J. Rehwinkel, S.J. Neil, G.M. Lenzi, B. Kim, A. Khwaja, M.C. Gage, C. Georgiou, A. Chittka, S. Yona, M. Noursadeghi, G.J. Towers, R.K. Gupta, A G1-like state allows HIV-1 to bypass SAMHD1 restriction in macrophages (2017) | [61] |
| Identification of HIV-1 DNA, RNA and protein in microglial cells of brain autopsies from patients that died from severe form of HAND | M.A. Cosenza, M.-L. Zhao, Q. Si, S.C. Lee, Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis (2002) M.J. Churchill, P.R. Gorry, D. Cowley, L. Lal, S. Sonza, D.F. Purcell, K.A. Thompson, D. Gabuzda, J.C. McArthur, C.A. Pardo, S.L. Wesselingh, Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues (2006) | [62,63] |
| Detection of HIV-1 DNAs in microglial cells and macrophages in brain autopsies from patients whose infection was controlled | K.A. Thompson, C.L. Cherry, J.E. Bell, C.A. McLean, Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals (2011) | [64] |
| Detection of the virus in the CSF in persons under effective cART, who had otherwise undetectable plasma HIV-1 | A. Edén A, S. Nilsson, L. Hagberg, D. Fuchs, H. Zetterberg, B. Svennerholm, M. Gisslén, Asymptomatic cerebrospinal fluid HIV-1 viral blips and viral escape during antiretroviral therapy: a longitudinal study (2016) | [65] |
| Compartmentalization of HIV-1 in the CSF | D.F. Bavaro, A. Calamo A, L. Lepore, C. Fabrizio, A. Saracino, G. Angarano, L. Monno, Cerebrospinal fluid compartmentalization of HIV-1 and correlation with plasma viral load and blood–brain barrier damage (2019) | [66] |
| Macaques model with microglial cells infection is useful to conduct preclinical evaluation of therapeutic interventions aimed at eradicating HIV-1 from the CNS | C. Harbison, K. Zhuang, A. Gettie, J. Blanchard, H. Knight, P. Didier, C. Cheng-Mayer, S. Westmoreland, Giant cell encephalitis and microglial infection with mucosally transmitted simian-human immunodeficiency virus SHIVSF162P3N in rhesus macaques (2014) | [67] |
| Demonstration of the establishment of viral reservoir in animal models | J.B. Whitney, A.L. Hill, S. Sanisetty, P. Penaloza-MacMaster, J. Liu, M. Shetty, L. Parenteau, C. Cabral, J. Shields, S. Blackmore, J.Y. Smith, A.L. Brinkman, L.E. Peter, S.I. Mathew, K.M. Smith, E.N. Borducchi, D.I. Rosenbloom, M.G. Lewis, J. Hattersley, B. Li, J. Hesselgesser, R. Geleziunas, M.L. Robb, J.H. Kim, N.L. Michael, D.H. Barouch, Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys (2014) | [68] |
| Infected microglia by HIV-1 in vivo in humanized mouse models | G.N. Llewellyn, D. Alvarez-Carbonell, M. Chateau, J. Karn, P.M. Cannon, HIV-1 infection of microglial cells in a reconstituted humanized mouse model and identification of compounds that selectively reverse HIV latency (2018) S. Mathews, A. Branch Woods, I. Katano, E. Makarov, M.B. Thomas, H.E. Gendelman, L.Y. Poluektova, M. Ito, S. Gorantla, Human Interleukin-34 facilitates microglia-like cell differentiation and persistent HIV-1 infection in humanized mice (2019) H. Su, Y. Cheng, S. Sravanam, S. Mathews, S. Gorantla, L.Y. Poluektova, P.K. Dash, H.E. Gendelman, Immune activations and viral tissue compartmentalization during progressive HIV-1 infection of humanized mice (2019) | [69,70,71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borrajo López, A.; Penedo, M.A.; Rivera-Baltanas, T.; Pérez-Rodríguez, D.; Alonso-Crespo, D.; Fernández-Pereira, C.; Olivares, J.M.; Agís-Balboa, R.C. Microglia: The Real Foe in HIV-1-Associated Neurocognitive Disorders? Biomedicines 2021, 9, 925. https://doi.org/10.3390/biomedicines9080925
Borrajo López A, Penedo MA, Rivera-Baltanas T, Pérez-Rodríguez D, Alonso-Crespo D, Fernández-Pereira C, Olivares JM, Agís-Balboa RC. Microglia: The Real Foe in HIV-1-Associated Neurocognitive Disorders? Biomedicines. 2021; 9(8):925. https://doi.org/10.3390/biomedicines9080925
Chicago/Turabian StyleBorrajo López, Ana, Maria Aránzazu Penedo, Tania Rivera-Baltanas, Daniel Pérez-Rodríguez, David Alonso-Crespo, Carlos Fernández-Pereira, José Manuel Olivares, and Roberto Carlos Agís-Balboa. 2021. "Microglia: The Real Foe in HIV-1-Associated Neurocognitive Disorders?" Biomedicines 9, no. 8: 925. https://doi.org/10.3390/biomedicines9080925