
OmniSARS2: A Highly Sensitive and Specific RT-qPCR-Based COVID-19 Diagnostic Method Designed to Withstand SARS-CoV-2 Lineage Evolution

 , , , , , , and
, , , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primers and Probe Design
2.2. Patients Samples
2.3. Nucleic Acid Extraction and RT-PCR Reaction Protocol
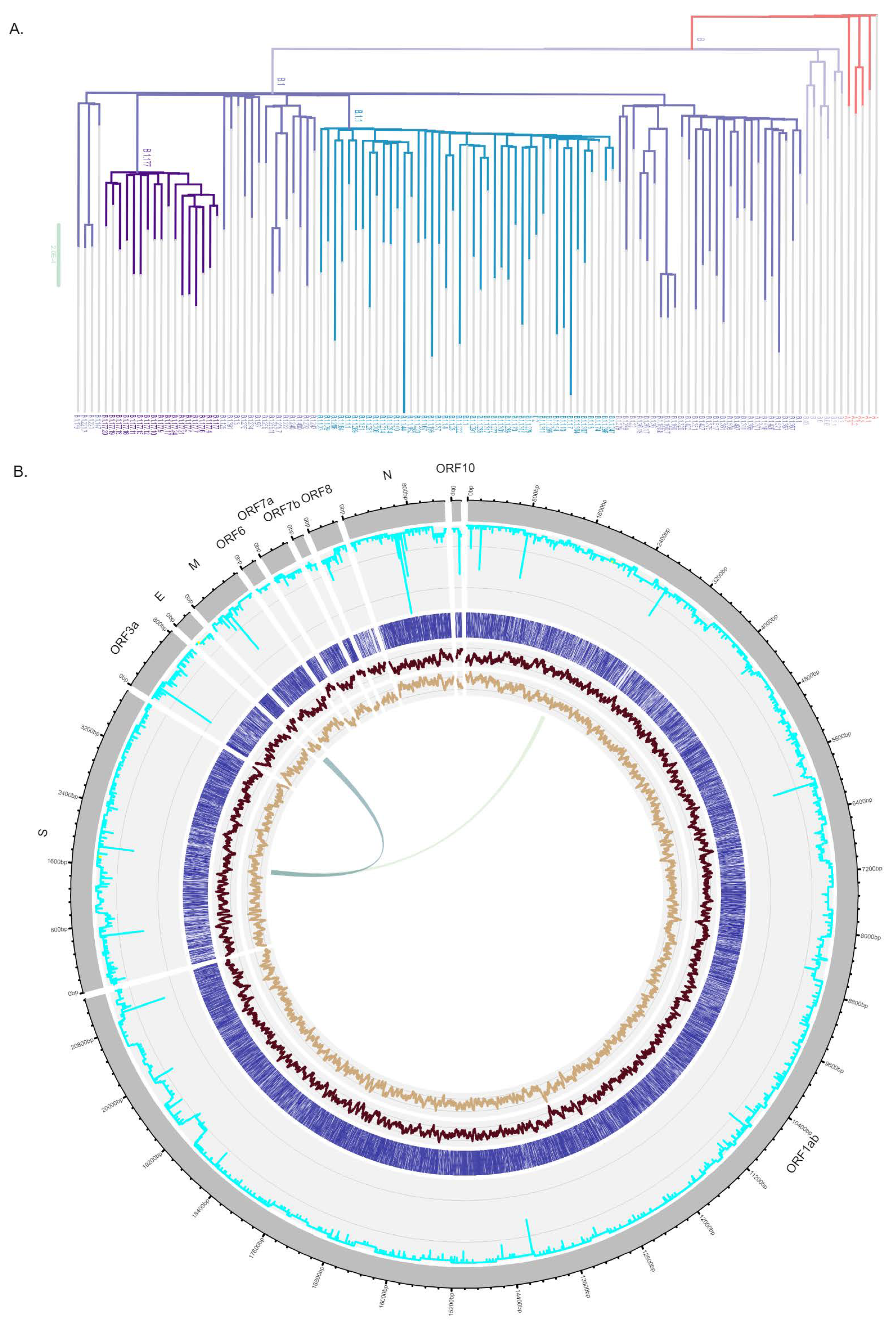
2.4. SARS-CoV-2 Genome Sequencing and Lineage Typing
2.5. Statistical Analysis
3. Results
3.1. In Silico Design of Oligonucleotide Sequences
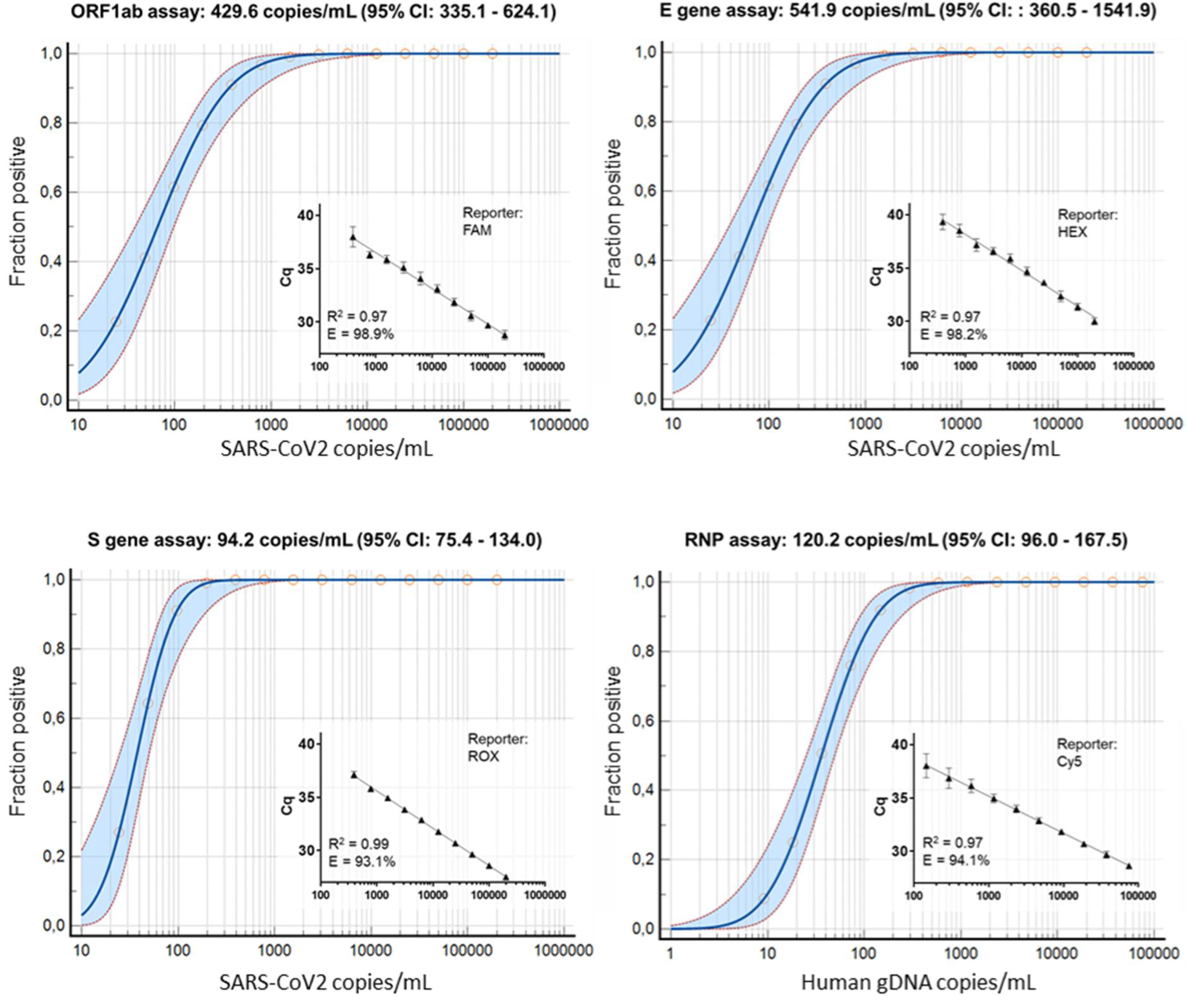
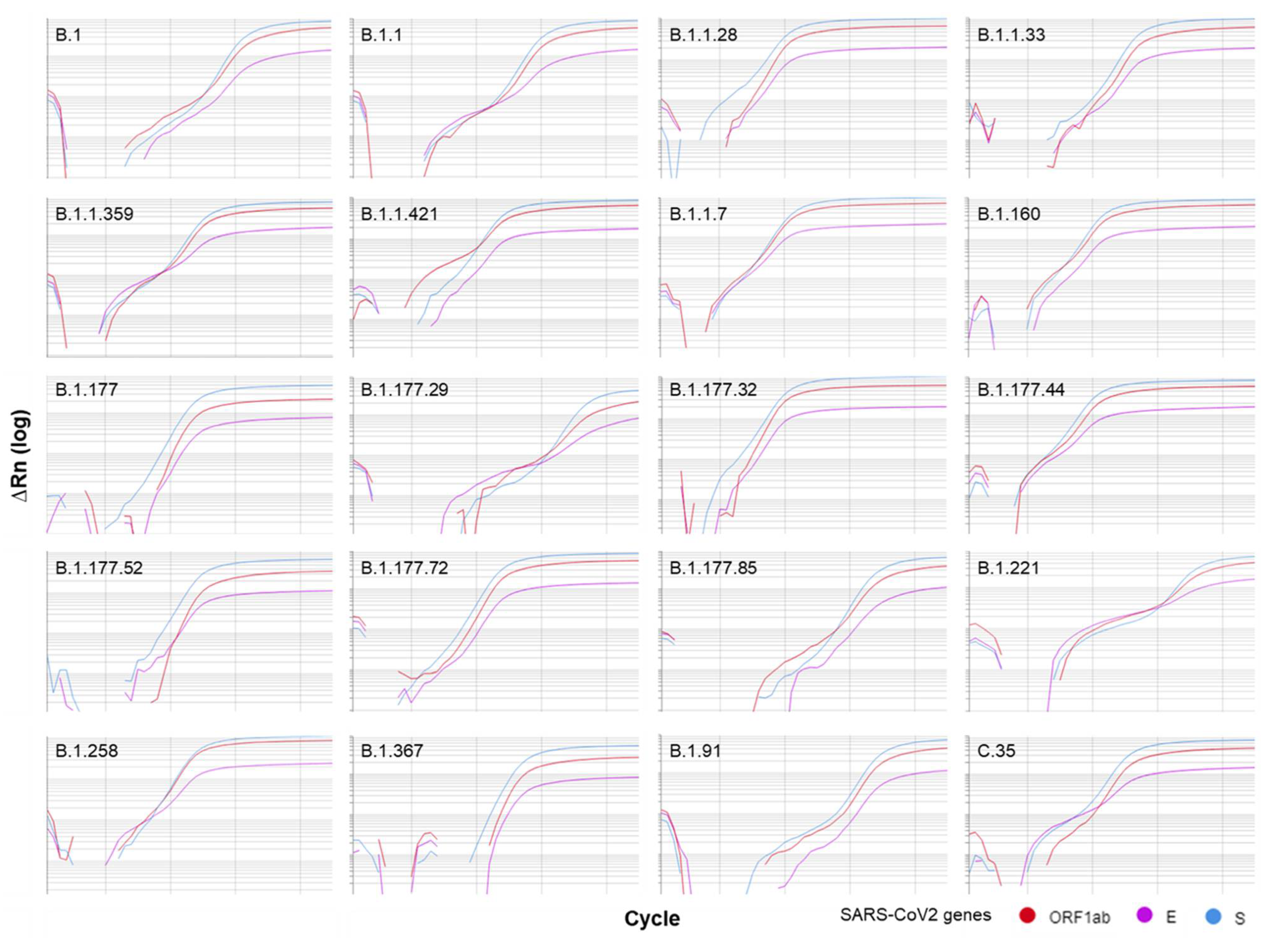
3.2. Wet-Lab Determination of the Analytical Sensitivity and Specificity
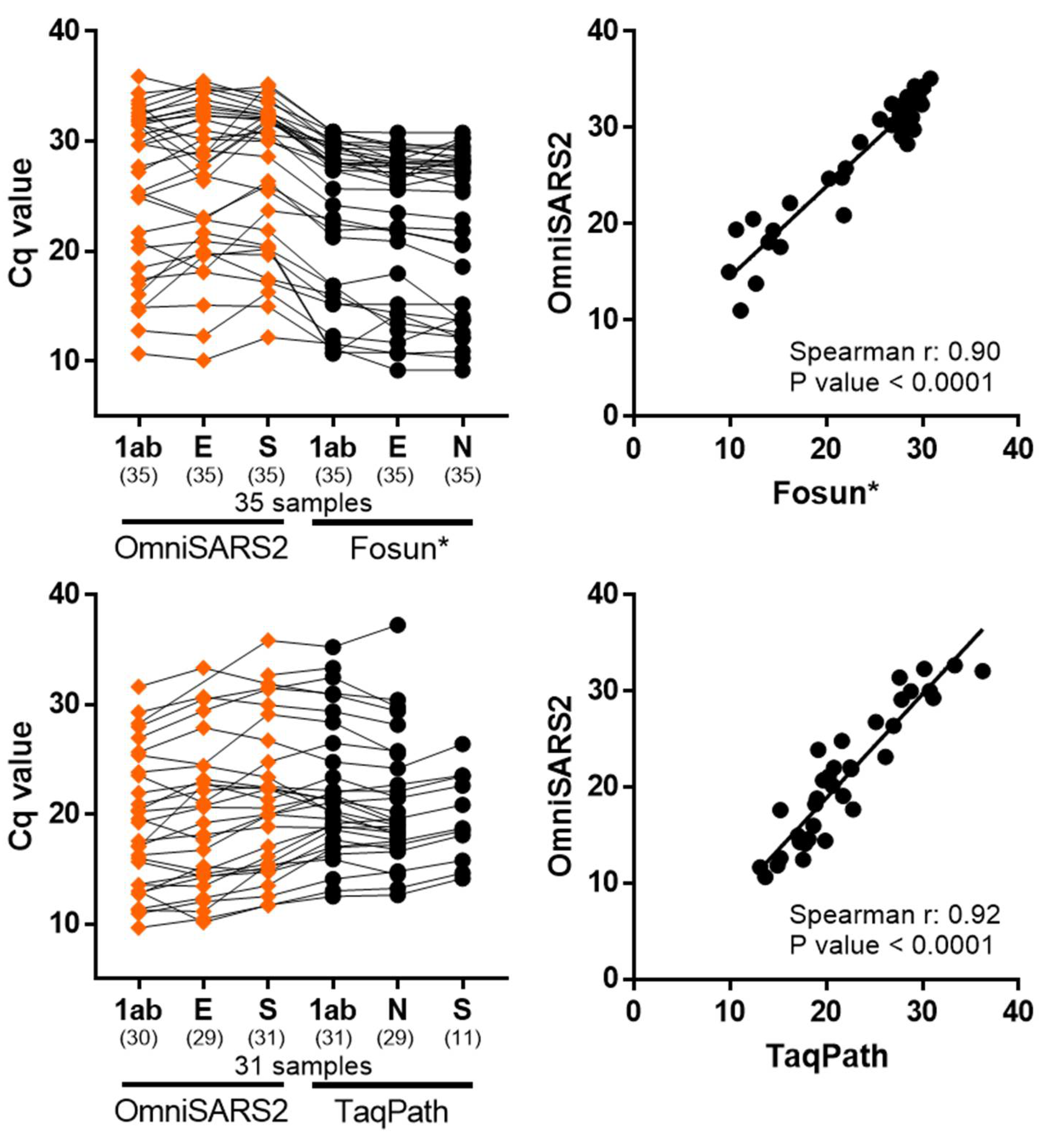
3.3. Clinical Validation of the Multiplex RT-qPCR Assay
3.4. Sensitivity Validation with Patient Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Stadhouders, R.; Pas, S.D.; Anber, J.; Voermans, J.; Mes, T.H.; Schutten, M. The effect of primer-template mismatches on the detection and quantification of nucleic acids using the 5’ nuclease assay. J. Mol. Diagn. 2010, 12, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Osório, N.S.; Correia-Neves, M. Implication of SARS-CoV-2 evolution in the sensitivity of RT-qPCR diagnostic assays. Lancet Infect. Dis. 2021, 21, 166–167. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Washington, N.L.; White, S.; Barrett, K.M.S.; Cirulli, E.T.; Bolze, A.; Lu, J.T. S gene dropout patterns in SARS-CoV-2 tests suggest spread of the H69del/V70del mutation in the US. medRxiv 2020. [Google Scholar] [CrossRef]
- Galloway, S.E.; Paul, P.; MacCannell, D.R.; Johansson, M.A.; Brooks, J.T.; MacNeil, A.; Slayton, R.B.; Tong, S.; Silk, B.J.; Armstrong, G.L.; et al. Emergence of SARS-CoV-2 B.1.1.7 Lineage—United States. Available online: http://dx.doi.org/10.15585/mmwr.mm7003e2 (accessed on 31 May 2021).
- England, P.H. Investigation of Novel SARS-CoV-2 Variant. Variant of Concern 202012/01. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/959360/Variant_of_Concern_VOC_202012_01_Technical_Briefing_3.pdf (accessed on 31 January 2021).
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv 2021. [Google Scholar] [CrossRef]
- Bal, A.; Destras, G.; Gaymard, A.; Regue, H.; Semanas, Q.; d’Aubarde, C.; Billaud, G.; Laurent, F.; Gonzales, C.; Valette, M.; et al. Screening of the H69 and V70 deletions in the SARS-CoV-2 spike protein with a RT-PCR diagnosis assay reveals low prevalence in Lyon, FRA. medRxiv 2020. [Google Scholar] [CrossRef]
- Kemp, S.; Datir, R.; Collier, D.; Ferreira, I.; Carabelli, A.; Harvey, W.; Robertson, D.; Gupta, R. Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion ΔH69/ΔV70. bioRxiv 2020. [Google Scholar] [CrossRef]
- Emery, S.L.; Erdman, D.D.; Bowen, M.D.; Newton, B.R.; Winchell, J.M.; Meyer, R.F.; Tong, S.; Cook, B.T.; Holloway, B.P.; McCaustland, K.A.; et al. Real-time reverse transcription-polymerase chain reaction assay for SARS-associated coronavirus. Emerg. Infect. Dis. 2004, 10, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- “Picard Tools.” Broad Institute, GitHub Repository. Picard Tools, Version 2.17.8. Available online: http://broadinstitute.github.io/picard/. (accessed on 21 February 2018).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. List of In-House-Developed Molecular Assays for SARS-CoV-2 Detection. Available online: https://www.who.int/docs/default-source/coronaviruse/whoinhouseassays.pdf (accessed on 15 May 2021).
- Rambaut, A. Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 13 May 2021).
- Lu, X.; Wang, L.; Sakthivel, S.; Whitaker, B.; Murray, J.; Kamili, S.; Lynch, B.; Malapati, L.; Burke, S.; Harcourt, J.; et al. US CDC Real-Time Reverse Transcription PCR Panel for Detection of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. J. 2020, 26, 1654. [Google Scholar] [CrossRef] [PubMed]
- FDA. SARS-CoV-2 Reference Panel Comparative Data. Available online: https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/sars-cov-2-reference-panel-comparative-data (accessed on 8 April 2021).
- Scohy, A.; Anantharajah, A.; Bodéus, M.; Kabamba-Mukadi, B.; Verroken, A.; Rodriguez-Villalobos, H. Low performance of rapid antigen detection test as frontline testing for COVID-19 diagnosis. J. Clin. Virol. 2020, 129, 104455. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Oligo Name | Sequence (5’-3’) | Amplicon Size |
|---|---|---|---|
| SARS-CoV-2 | 1ab1917F1 | AGAGTTTCTTAGAGACGGTTGG | 111 bp |
| ORF1ab | 1ab1977P1 | FAM-TGTCGGTGGACAAATTGTCACCTGT-BHQ | |
| 1ab2003R1 | TGAACACTCTCCTTAATTTCCTTTG | ||
| SARS-CoV-2 | S1703F1 | ACATTGCTGACACTACTGATGC | 119 bp |
| Spike (S) | S1725P1 | ROX-TGTCCGTGATCCACAGACACTTGAG-BHQ | |
| S1797R1 | CTGGTTAGAAGTATTTGTTCCTGGT | ||
| SARS-CoV-2 | E22F1 | AGACAGGTACGTTAATAGTTAATAGCG | 144 bp |
| Envelope (E) | E84P1 | HEX-AGTTACACTAGCCATCCTTACTGCGC | |
| E143R1 | AAGAAGGTTTTACAAGACTCACGT-BHQ | ||
| 1 Human | RNP_F | AGATTTGGACCTGCGAGCG | |
| Ribonuclease P | RNP_R | GAGCGGCTGTCTCCACAAGT | 65 bp |
| RNP_P | Cy5-TTCTGACCTGAAGGCTCTGCGCG-BBQ |
| Lineage 1 | Sample (n) | Sampling Dates | Common Countries# |
|---|---|---|---|
| B.1 | 13 | 16 April 2020–22 September 2020 | USA 46.0%, GBR 10.0%, DEU 5.0%, ESP 3.0%, IND 3.0% |
| B.1.1 | 88 | 03 April 2020–16 November 2020 | GBR 33.0%, USA 15.0%, JPN 8.0%, DEU 4.0%, RUS 3.0% |
| B.1.1.28 | 12 | 25 August 2020–13 January 2021 | BRA 60.0%, PHL 17.0%, USA 8.0%, URY 5.0%, JPN 2.0% |
| B.1.1.33 | 1 | 09 November 2020 | BRA 81.0%, USA 5.0%, CHL 4.0%, URY 1.0%, ARG 1.0% |
| B.1.1.359 | 1 | 04 November 2020 | GHA 70.0%, DNK 15.0%, BFA 7.0%, USA 4.0%, TGO 4.0% |
| B.1.1.421 | 2 | 17 June 2020 | PRT 74.0%, GBR 15.0%, USA 6.0%, RUS 3.0%, CHE 3.0% |
| B.1.1.7 | 2 | 13 January 2021 | GBR 30.0%, USA 20.0%, DEU 11.0%, DNK 6.0%, SWE 6.0% |
| B.1.160 | 1 | 20 November 2020 | DNK 16.0%, FRA 16.0%, CHE 11.0%, GBR 9.0%, DEU 8.0% |
| B.1.177 | 5 | 18 May 2020–13 January 2021 | GBR 62.0%, ESP 11.0%, DEU 5.0%, CHE 4.0%, ITA 4.0% |
| B.1.177.29 | 1 | 11 January 2021 | ESP 50.0%, ITA 31.0%, PRT 12.0%, GBR 6.0% |
| B.1.177.32 | 5 | 29 December 2020 | ESP 39.0%, PRT 38.0%, CHE 9.0%, LUX 3.0%, FRA 3.0% |
| B.1.177.44 | 5 | 01 October 2020–20 November 2020 | CHE 79.0%, DEU 5.0%, ITA 4.0%, GBR 3.0%, NLD 2.0% |
| B.1.177.52 | 9 | 03 November 2020–13 January 2021 | PRT 37.0%, GBR 26.0%, DEU 18.0%, LUX 4.0%, NLD 3.0% |
| B.1.177.72 | 13 | 22 October 2020–13 January 2021 | PRT 73.0%, FRA 8.0%, CHE 7.0%, LUX 5.0%, ESP 4.0% |
| B.1.177.85 | 1 | 08 January 2021 | PRT 61.0%, CHE 20.0%, LUX 13.0%, FRA 4.0%, GBR 1.0% |
| B.1.221 | 1 | 11 January 2021 | NLD 20.0%, DEU 17.0%, DNK 14.0%, BEL 11.0%, SWE 10.0% |
| B.1.258 | 1 | 13 January 2021 | DEU 20.0%, GBR 18.0%, DNK 16.0%, SWE 8.0%, CHE 6.0% |
| B.1.367 | 4 | 01 September 2020 | GBR 32.0%, FRA 17.0%, CHE 11.0%, NOR 10.0%, DNK 7.0% |
| B.1.91 | 8 | 14 April 2020–04 November 2020 | PRT 64.0%, GBR 13.0%, BRA 5.0%, NZL 5.0%, ITA 3.0% |
| C.35 (alias of B.1.1.1.35) | 1 | 28 July 2020 | DEU 19.0%, GBR 17.0%, CHE 12.0%, DNK 11.0%, GRC 7.0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carvalho-Correia, E.; Calçada, C.; Branca, F.; Estévez-Gómez, N.; De Chiara, L.; Varela, N.; Gallego-García, P.; Posada, D.; Sousa, H.; Sousa, J.; et al. OmniSARS2: A Highly Sensitive and Specific RT-qPCR-Based COVID-19 Diagnostic Method Designed to Withstand SARS-CoV-2 Lineage Evolution. Biomedicines 2021, 9, 1314. https://doi.org/10.3390/biomedicines9101314
Carvalho-Correia E, Calçada C, Branca F, Estévez-Gómez N, De Chiara L, Varela N, Gallego-García P, Posada D, Sousa H, Sousa J, et al. OmniSARS2: A Highly Sensitive and Specific RT-qPCR-Based COVID-19 Diagnostic Method Designed to Withstand SARS-CoV-2 Lineage Evolution. Biomedicines. 2021; 9(10):1314. https://doi.org/10.3390/biomedicines9101314
Chicago/Turabian StyleCarvalho-Correia, Eduarda, Carla Calçada, Fernando Branca, Nuria Estévez-Gómez, Loretta De Chiara, Nair Varela, Pilar Gallego-García, David Posada, Hugo Sousa, João Sousa, and et al. 2021. "OmniSARS2: A Highly Sensitive and Specific RT-qPCR-Based COVID-19 Diagnostic Method Designed to Withstand SARS-CoV-2 Lineage Evolution" Biomedicines 9, no. 10: 1314. https://doi.org/10.3390/biomedicines9101314