Phase 1 Dose Escalation Study of the Allosteric AKT Inhibitor BAY 1125976 in Advanced Solid Cancer—Lack of Association between Activating AKT Mutation and AKT Inhibition-Derived Efficacy

 , ,
, , 
 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Baseline Patient Demographics
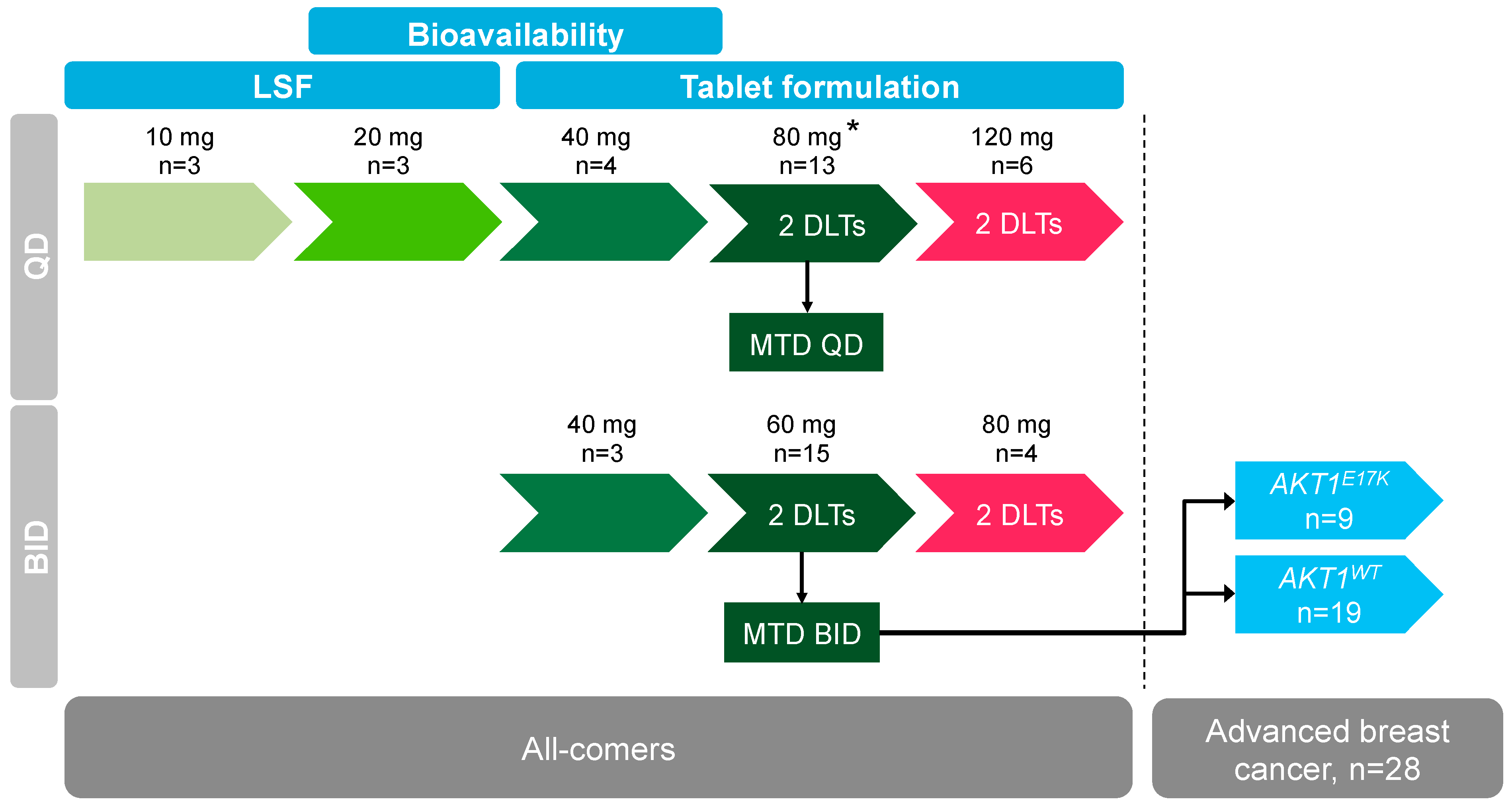
2.2. Dose Escalation and Maximum Tolerated Dose
2.3. Safety
2.4. Pharmacokinetic Evaluation
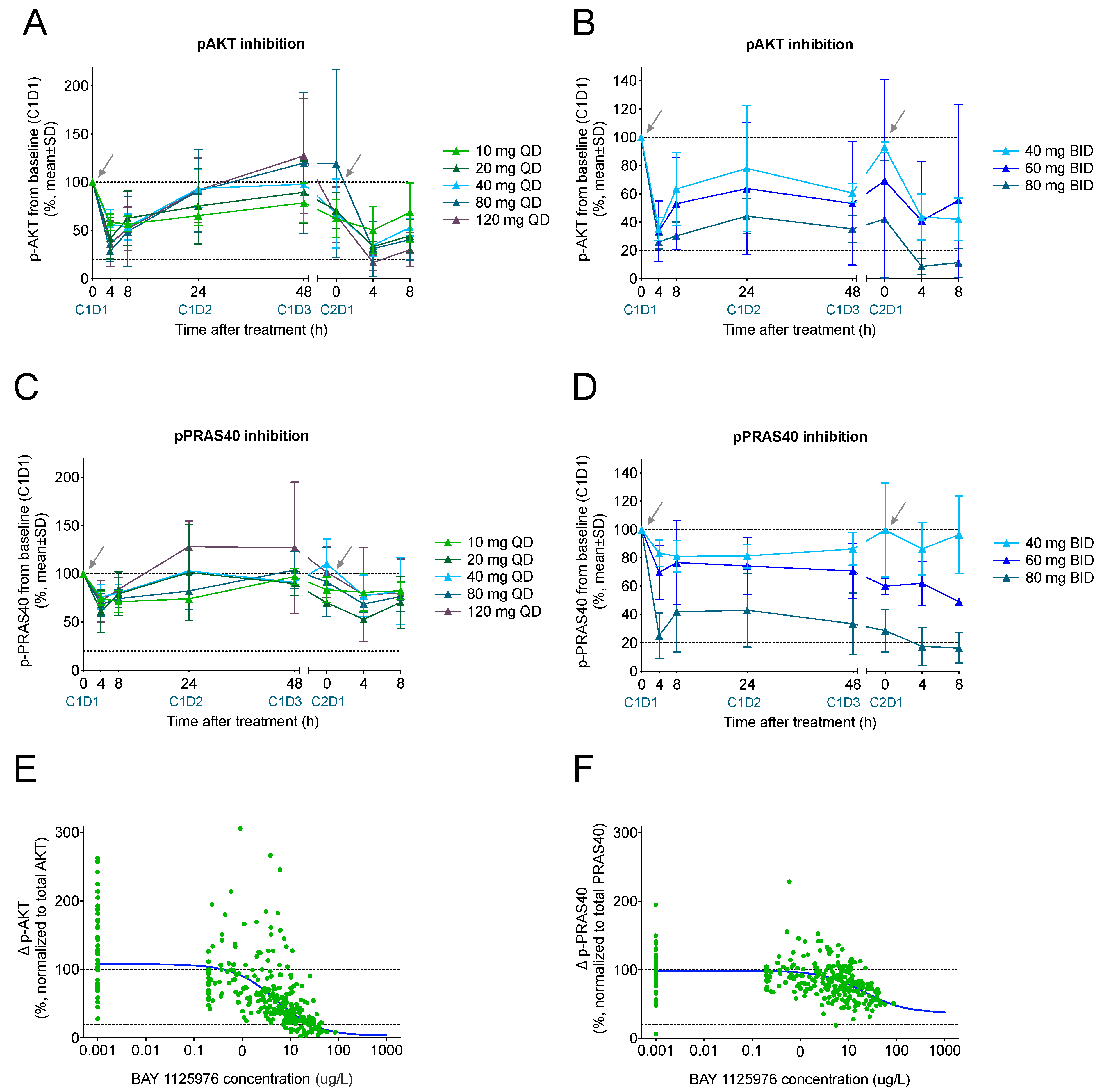
2.5. Pharmacodynamic Biomarkers
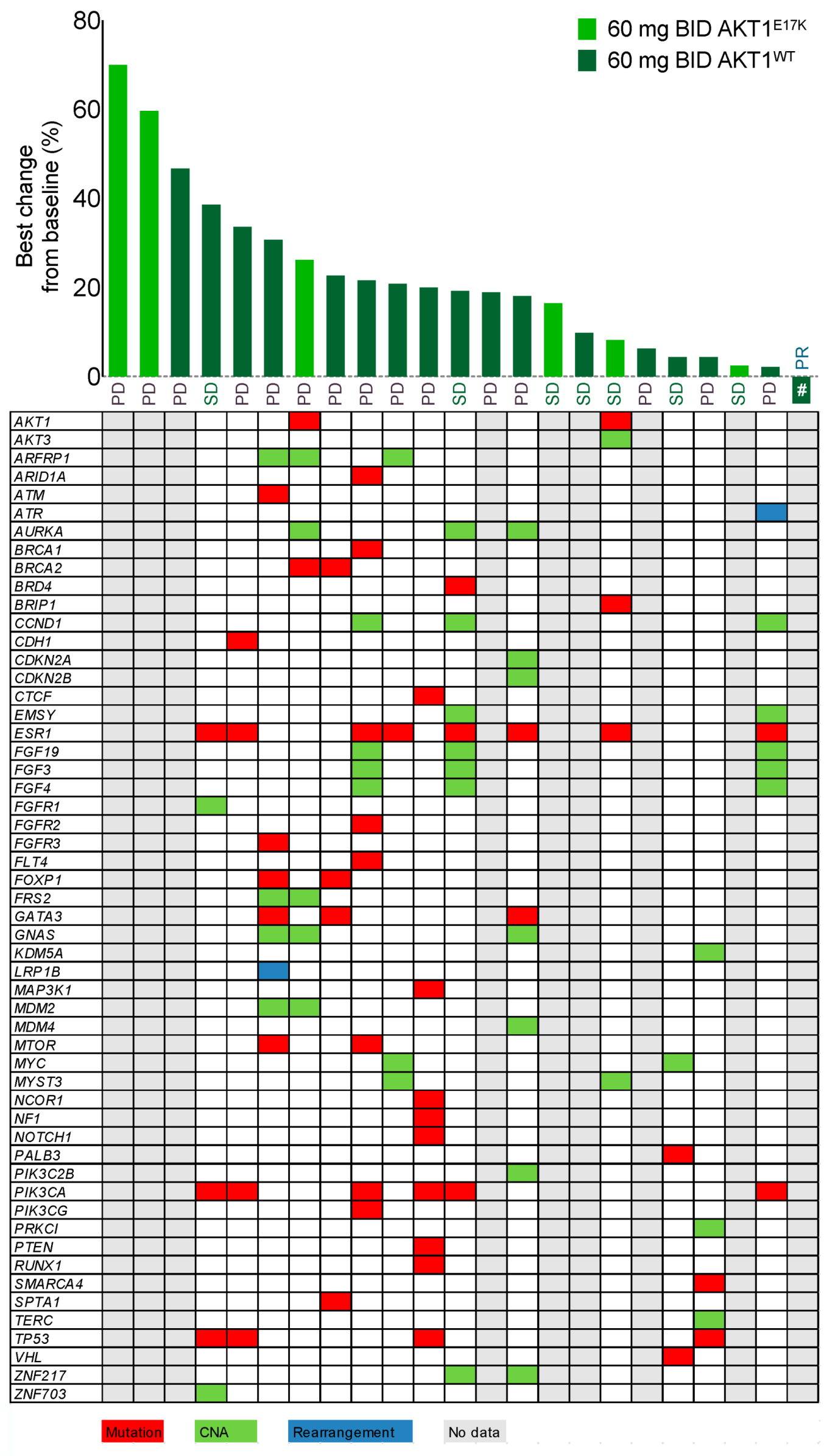
2.6. AKT1E17K Status and Mutational Profiling
2.7. Tumor Response Evaluation
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Treatment
4.3. Inclusion Criteria
4.4. Safety, Response, and Pharmacokinetics Assessments
4.5. Biomarker Assessments
4.6. PK/PD analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Bleeker, F.E.; Felicioni, L.; Buttitta, F.; Lamba, S.; Cardone, L.; Rodolfo, M.; Scarpa, A.; Leenstra, S.; Frattini, M.; Barbareschi, M.; et al. AKT1(E17K) in human solid tumours. Oncogene 2008, 27, 5648–5650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, M.; Anzeneder, T.; Schulz, A.; Beckmann, G.; Byrne, A.T.; Jeffers, M.; Pena, C.; Politz, O.; Kochert, K.; Vonk, R.; et al. AKT1 (E17K) mutation profiling in breast cancer: Prevalence, concurrent oncogenic alterations, and blood-based detection. BMC Cancer 2016, 16, 622. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Davies, B.R.; Greenwood, H.; Dudley, P.; Crafter, C.; Yu, D.H.; Zhang, J.; Li, J.; Gao, B.; Ji, Q.; Maynard, J.; et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: Pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol. Cancer Ther. 2012, 11, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.S.; et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef] [Green Version]
- Politz, O.; Siegel, F.; Barfacker, L.; Bomer, U.; Hagebarth, A.; Scott, W.J.; Michels, M.; Ince, S.; Neuhaus, R.; Meyer, K.; et al. BAY 1125976, a selective allosteric AKT1/2 inhibitor, exhibits high efficacy on AKT signaling-dependent tumor growth in mouse models. Int. J. Cancer 2017, 140, 449–459. [Google Scholar] [CrossRef]
- Patnaik, A.; Appleman, L.J.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Weiss, G.J.; Sachdev, J.C.; Chadha, M.; Fulk, M.; et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann. Oncol. 2016, 27, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; de Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; De Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Juric, D.; Rodon, J.; Tabernero, J.; Janku, F.; Burris, H.A.; Schellens, J.H.M.; Middleton, M.R.; Berlin, J.; Schuler, M.; Gil-Martin, M.; et al. Phosphatidylinositol 3-Kinase alpha-Selective Inhibition With Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results From the First-in-Human Study. J. Clin. Oncol. 2018, 36, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Scheulen, M.E.; Johnston, S.; Mross, K.; Cardoso, F.; Dittrich, C.; Eiermann, W.; Hess, D.; Morant, R.; Semiglazov, V.; et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J. Clin. Oncol. 2005, 23, 5314–5322. [Google Scholar] [CrossRef]
- Fleming, G.F.; Ma, C.X.; Huo, D.; Sattar, H.; Tretiakova, M.; Lin, L.; Hahn, O.M.; Olopade, F.O.; Nanda, R.; Hoffman, P.C.; et al. Phase II trial of temsirolimus in patients with metastatic breast cancer. Breast Cancer Res. Treat. 2012, 136, 355–363. [Google Scholar] [CrossRef] [Green Version]
- Ellard, S.L.; Clemons, M.; Gelmon, K.A.; Norris, B.; Kennecke, H.; Chia, S.; Pritchard, K.; Eisen, A.; Vandenberg, T.; Taylor, M.; et al. Randomized phase II study comparing two schedules of everolimus in patients with recurrent/metastatic breast cancer: NCIC Clinical Trials Group IND.163. J. Clin. Oncol. 2009, 27, 4536–4541. [Google Scholar] [CrossRef]
- Maass, N.; Harbeck, N.; Mundhenke, C.; Lerchenmuller, C.; Barinoff, J.; Luck, H.J.; Ettl, J.; Aktas, B.; Kummel, S.; Rosel, S.; et al. Everolimus as treatment for breast cancer patients with bone metastases only: Results of the phase II RADAR study. J. Cancer Res. Clin. Oncol. 2013, 139, 2047–2056. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Zeng, T.; Zhang, C.L.; Zhao, N.; Guan, M.J.; Xiao, M.; Yang, R.; Zhao, X.L.; Yu, L.H.; Zhu, Z.P.; Xie, K.Q. Impairment of Akt activity by CYP2E1 mediated oxidative stress is involved in chronic ethanol-induced fatty liver. Redox Biol. 2018, 14, 295–304. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Gradinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt pathway in oncology therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yan, H.; Xu, Z.; Yang, B.; Luo, P.; He, Q. Molecular basis for class side effects associated with PI3K/AKT/mTOR pathway inhibitors. Expert Opin. Drug Metab. Toxicol. 2019, 15, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.B.; Goh, B.C.; Lim, W.T.; Hui, E.P.; Tan, E.H.; Lopes Gde, L.; Lo, K.W.; Li, L.; Loong, H.; Foster, N.R.; et al. Multicenter phase II study of the AKT inhibitor MK-2206 in recurrent or metastatic nasopharyngeal carcinoma from patients in the mayo phase II consortium and the cancer therapeutics research group (MC1079). Investig. New Drugs 2015, 33, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.H.; Li, J.; Wei, L.; Doyle, A.; Marshall, J.L.; Schaaf, L.J.; Phelps, M.A.; Villalona-Calero, M.A.; Bekaii-Saab, T. Results of an abbreviated phase-II study with the Akt Inhibitor MK-2206 in Patients with Advanced Biliary Cancer. Sci. Rep. 2015, 5, 12122. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.L.; Kennecke, H.F.; Iqbal, S.; Baranda, J.C.; Seery, T.E.; Lim, H.J.; Hezel, A.F.; Vaccaro, G.M.; Blanke, C.D. Phase 2 study of MK-2206, an allosteric inhibitor of AKT, as second-line therapy for advanced gastric and gastroesophageal junction cancer: A SWOG cooperative group trial (S1005). Cancer 2015, 121, 2193–2197. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Yan, L.; Patnaik, A.; Fearen, I.; Olmos, D.; Papadopoulos, K.; Baird, R.D.; Delgado, L.; Taylor, A.; Lupinacci, L.; et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J. Clin. Oncol. 2011, 29, 4688–4695. [Google Scholar] [CrossRef]
- Yap, T.A.; Yan, L.; Patnaik, A.; Tunariu, N.; Biondo, A.; Fearen, I.; Papadopoulos, K.P.; Olmos, D.; Baird, R.; Delgado, L.; et al. Interrogating two schedules of the AKT inhibitor MK-2206 in patients with advanced solid tumors incorporating novel pharmacodynamic and functional imaging biomarkers. Clin. Cancer Res. 2014, 20, 5672–5685. [Google Scholar] [CrossRef] [Green Version]
- Hyman, D.M.; Smyth, L.M.; Donoghue, M.T.A.; Westin, S.N.; Bedard, P.L.; Dean, E.J.; Bando, H.; El-Khoueiry, A.B.; Perez-Fidalgo, J.A.; Mita, A.; et al. AKT Inhibition in Solid Tumors with AKT1 Mutations. J. Clin. Oncol. 2017, 35, 2251–2259. [Google Scholar] [CrossRef]
- Sangai, T.; Akcakanat, A.; Chen, H.; Tarco, E.; Wu, Y.; Do, K.A.; Miller, T.W.; Arteaga, C.L.; Mills, G.B.; Gonzalez-Angulo, A.M.; et al. Biomarkers of response to Akt inhibitor MK-2206 in breast cancer. Clin. Cancer Res. 2012, 18, 5816–5828. [Google Scholar] [CrossRef] [Green Version]
- Jonasch, E.; Hasanov, E.; Corn, P.G.; Moss, T.; Shaw, K.R.; Stovall, S.; Marcott, V.; Gan, B.; Bird, S.; Wang, X.; et al. A randomized phase 2 study of MK-2206 versus everolimus in refractory renal cell carcinoma. Ann Oncol 2017, 28, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.T.; Hortobagyi, G.N.; Campone, M.; Lebrun, F.; Deleu, I.; Rugo, H.S.; Pistilli, B.; Masuda, N.; Hart, L.; Melichar, B.; et al. Everolimus plus exemestane as first-line therapy in HR(+), HER2(-) advanced breast cancer in BOLERO-2. Breast Cancer Res. Treat. 2014, 143, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Im, S.A.; Iwata, H.; Cortes, J.; De Laurentiis, M.; Jiang, Z.; Arteaga, C.L.; Jonat, W.; Clemons, M.; Ito, Y.; et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 904–916. [Google Scholar] [CrossRef]
- Mayer, I.A.; Abramson, V.G.; Formisano, L.; Balko, J.M.; Estrada, M.V.; Sanders, M.E.; Juric, D.; Solit, D.; Berger, M.F.; Won, H.H.; et al. A Phase Ib Study of Alpelisib (BYL719), a PI3Kalpha-Specific Inhibitor, with Letrozole in ER+/HER2- Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Shah, A.N.; Santa-Maria, C.A.; Siziopikou, K.; Rademaker, A.; Helenowski, I.; Cristofanilli, M.; Gradishar, W.J. Phase I study of alpelisib (BYL-719) and trastuzumab emtansine (T-DM1) in HER2-positive metastatic breast cancer (MBC) after trastuzumab and taxane therapy. Breast Cancer Res. Treat. 2018, 171, 371–381. [Google Scholar] [CrossRef]
- Davies, B.R.; Guan, N.; Logie, A.; Crafter, C.; Hanson, L.; Jacobs, V.; James, N.; Dudley, P.; Jacques, K.; Ladd, B.; et al. Tumors with AKT1E17K Mutations Are Rational Targets for Single Agent or Combination Therapy with AKT Inhibitors. Mol. Cancer Ther. 2015, 14, 2441–2451. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.X.; Suman, V.; Goetz, M.P.; Northfelt, D.; Burkard, M.E.; Ademuyiwa, F.; Naughton, M.; Margenthaler, J.; Aft, R.; Gray, R.; et al. A Phase II Trial of Neoadjuvant MK-2206, an AKT Inhibitor, with Anastrozole in Clinical Stage II or III PIK3CA-Mutant ER-Positive and HER2-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 6823–6832. [Google Scholar] [CrossRef] [Green Version]
- Andre, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Costa, C.; Bosch, A. The Strategy of PIKing a Target: What Is AKTually Most Effective? Clin. Cancer Res. 2018, 24, 2029–2031. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 10 mg LSF QD | 20 mg LSF QD | 40 mg Tablet QD | 80 mg Tablet QD | 120 mg Tablet QD | 40 mg Tablet BID | 60 mg Tablet BID | 80 mg Tablet BID | 60 mg Expansion | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tablet (BID) | |||||||||||
| WT | AKT1E17K | ||||||||||
| N | 3 | 3 | 4 | 13 | 6 | 3 | 15 | 4 | 19 | 9 | 79 |
| Mean age, years (range) | 53.3 (44–67) | 49.7 (37–64) | 59.0 (47–75) | 61.2 (48–75) | 53.5 (37–69) | 66.3 (50–82) | 55.1 (40–74) | 56.3 (38–70) | 55.3 (31–76) | 57.3 (41–73) | 56.7 (31–82) |
| Females, n (%) | 2 (66.7) | 2 (66.7) | 2 (50.0) | 10 (76.9) | 4 (66.7) | 2 (66.7) | 8 (53.3) | 3 (75.0) | 19 (100.0) | 9 (100.0) | 61 (77.2) |
| ECOG performance status, n (%) | |||||||||||
| 0 | 0 | 1 (33.3) | 2 (50.0) | 7 (53.8) | 2 (33.3) | 2 (66.7) | 10 (66.7) | 2 (50.0) | 14 (73.7) | 7 (77.8) | 47 (59.5) |
| 1 | 3 (100.0) | 2 (66.7) | 2 (50.0) | 5 (38.5) | 4 (66.7) | 1 (33.3) | 5 (33.3) | 1 (25.0) | 5 (26.3) | 2 (22.2) | 30 (38.0) |
| 2 | 0 | 0 | 0 | 1 (7.7) | 0 | 0 | 0 | 1 (25.0) | 0 | 0 | 2 (2.5) |
| Prior systemic anticancer therapy, n (%) | 3 (100.0) | 3 (100.0) | 4 (100.0) | 13 (100.0) | 6 (100.0) | 2 (66.7) | 15 (100.0) | 4 (100.0) | 19 (100.0) | 9 (100.0) | 78 (98.7) |
| Prior radiotherapy, n (%) | 3 (100.0) | 3 (100.0) | 2 (50.0) | 8 (61.5) | 5 (83.3) | 2 (66.7) | 12 (80.0) | 1 (25.0) | 18 (94.7) | 6 (66.7) | 60 (75.9) |
| Adverse Events | 10 mg LSF QD | 20 mg LSF QD | 40 mg Tablet QD | 80 mg Tablet QD | 120 mg Tablet QD | 40 mg Tablet BID | 60 mg Tablet BID | 80 mg Tablet BID | 60 mg Expansion Tablet (BID) | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | AKT1E17K | ||||||||||
| N | 3 | 3 | 4 | 13 | 6 | 3 | 15 | 4 | 19 | 9 | 79 |
| All, n (%) | |||||||||||
| Grade 3 | - | - | 1 (25.0) | 5 (38.5) | 4 (66.7) | 2 (66.7) | 6 (40.0) | 4 (100.0) | 12 (61.1) | 8 (88.9) | 42 (53.2) |
| Grade 4 | - | - | - | - | 1 (16.7) | - | - | - | - | - | 1 (1.3) |
| ALT increased, n (%) | |||||||||||
| Grade 3 | - | - | - | 2 (15.4) | 2 (33.3) | 1 (33.3) | 2 (13.3) | 3 (75.0) | 4 (21.0) | 3 (33.3) | 17 (21.5) |
| AST increased, n (%) | |||||||||||
| Grade 3 | - | - | - | 2 (15.4) | 3 (50.0) | - | 3 (20.0) | 2 (50.0) | 7 (36.8) | 3 (33.3) | 20 (25.3) |
| AP increased, n (%) | |||||||||||
| Grade 3 | - | - | 1 (25.0) | 3 (23.1) | 2 (33.3) | - | 3 (20.0) | 3 (75.0) | 11 (57.9) | 4 (44.4) | 27 (34.2) |
| γ-GT increased, n (%) | |||||||||||
| Grade 3 | - | - | - | - | - | - | 1 (6.7) | - | - | - | 1 (1.3) |
| Grade 4 | - | - | - | - | 1 (16.7) | - | - | - | - | - | 1 (1.3) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneeweiss, A.; Hess, D.; Joerger, M.; Varga, A.; Moulder, S.; Tsimberidou, A.M.; Ma, C.; Hurvitz, S.A.; Rentzsch, C.; Rudolph, M.; et al. Phase 1 Dose Escalation Study of the Allosteric AKT Inhibitor BAY 1125976 in Advanced Solid Cancer—Lack of Association between Activating AKT Mutation and AKT Inhibition-Derived Efficacy. Cancers 2019, 11, 1987. https://doi.org/10.3390/cancers11121987
Schneeweiss A, Hess D, Joerger M, Varga A, Moulder S, Tsimberidou AM, Ma C, Hurvitz SA, Rentzsch C, Rudolph M, et al. Phase 1 Dose Escalation Study of the Allosteric AKT Inhibitor BAY 1125976 in Advanced Solid Cancer—Lack of Association between Activating AKT Mutation and AKT Inhibition-Derived Efficacy. Cancers. 2019; 11(12):1987. https://doi.org/10.3390/cancers11121987
Chicago/Turabian StyleSchneeweiss, Andreas, Dagmar Hess, Markus Joerger, Andrea Varga, Stacy Moulder, Apostolia M. Tsimberidou, Cynthia Ma, Sara A. Hurvitz, Christine Rentzsch, Marion Rudolph, and et al. 2019. "Phase 1 Dose Escalation Study of the Allosteric AKT Inhibitor BAY 1125976 in Advanced Solid Cancer—Lack of Association between Activating AKT Mutation and AKT Inhibition-Derived Efficacy" Cancers 11, no. 12: 1987. https://doi.org/10.3390/cancers11121987