Metformin as an Adjuvant to Photodynamic Therapy in Resistant Basal Cell Carcinoma Cells

 , , ,
, , ,  ,
,  ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
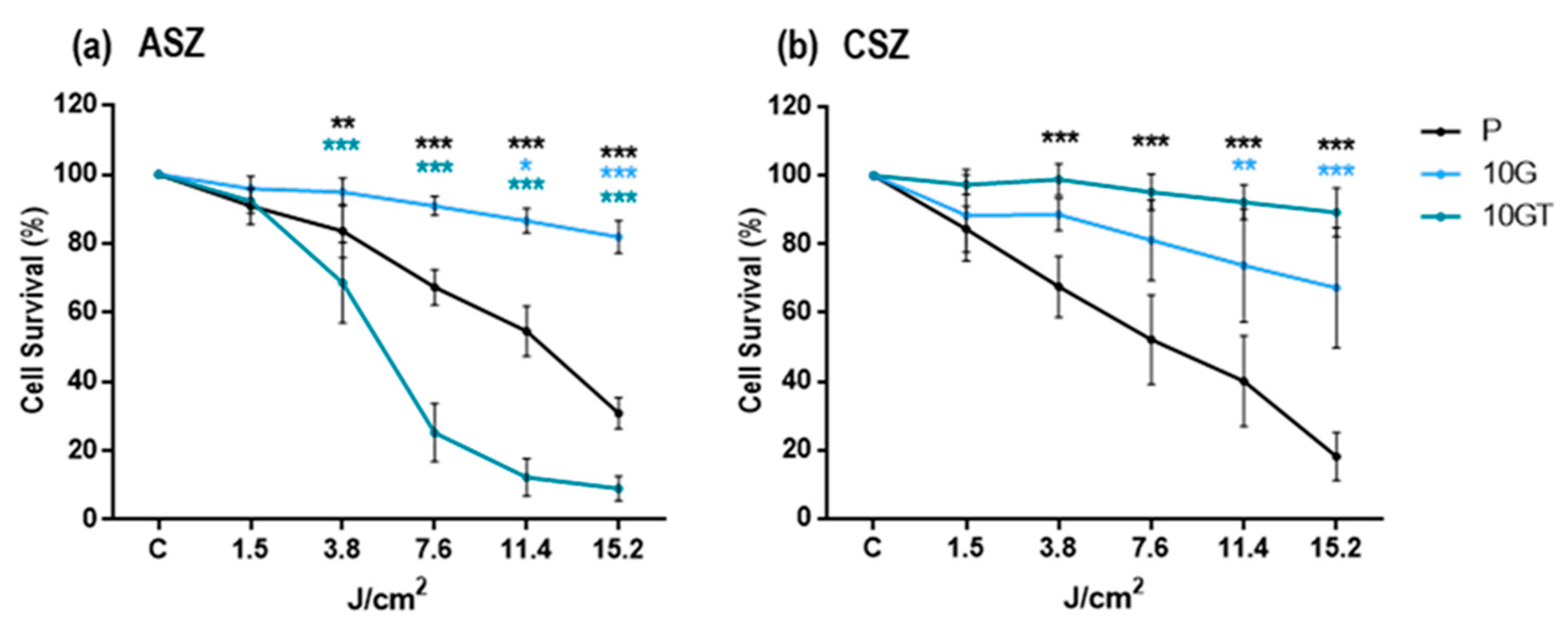
2.1. Cells Survival Evaluation after PDT
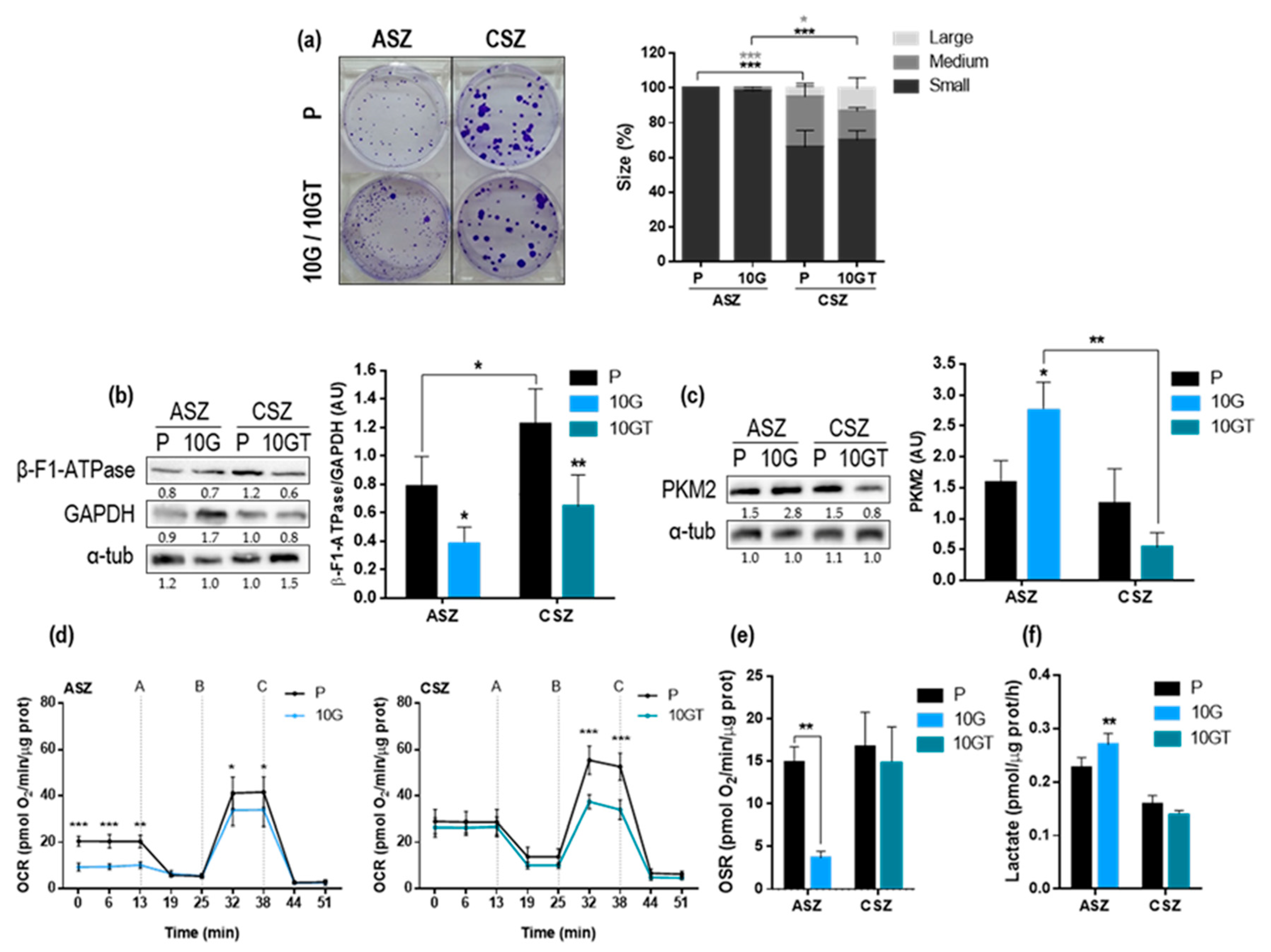
2.2. Proliferation Capacity and Metabolic Characterization
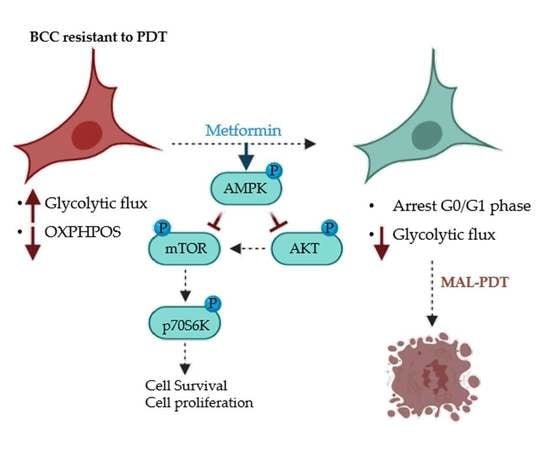
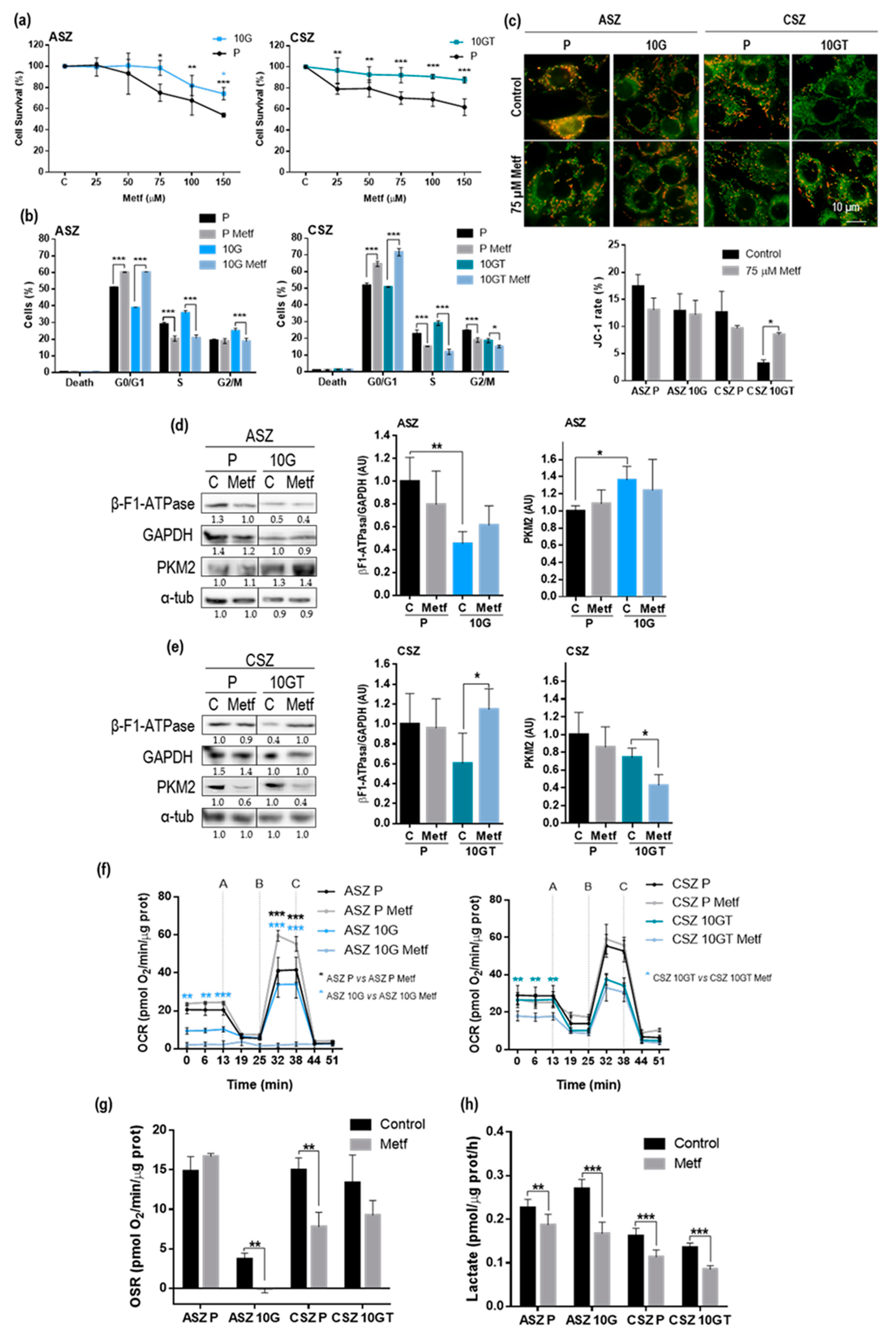
2.3. Metformin Treatment
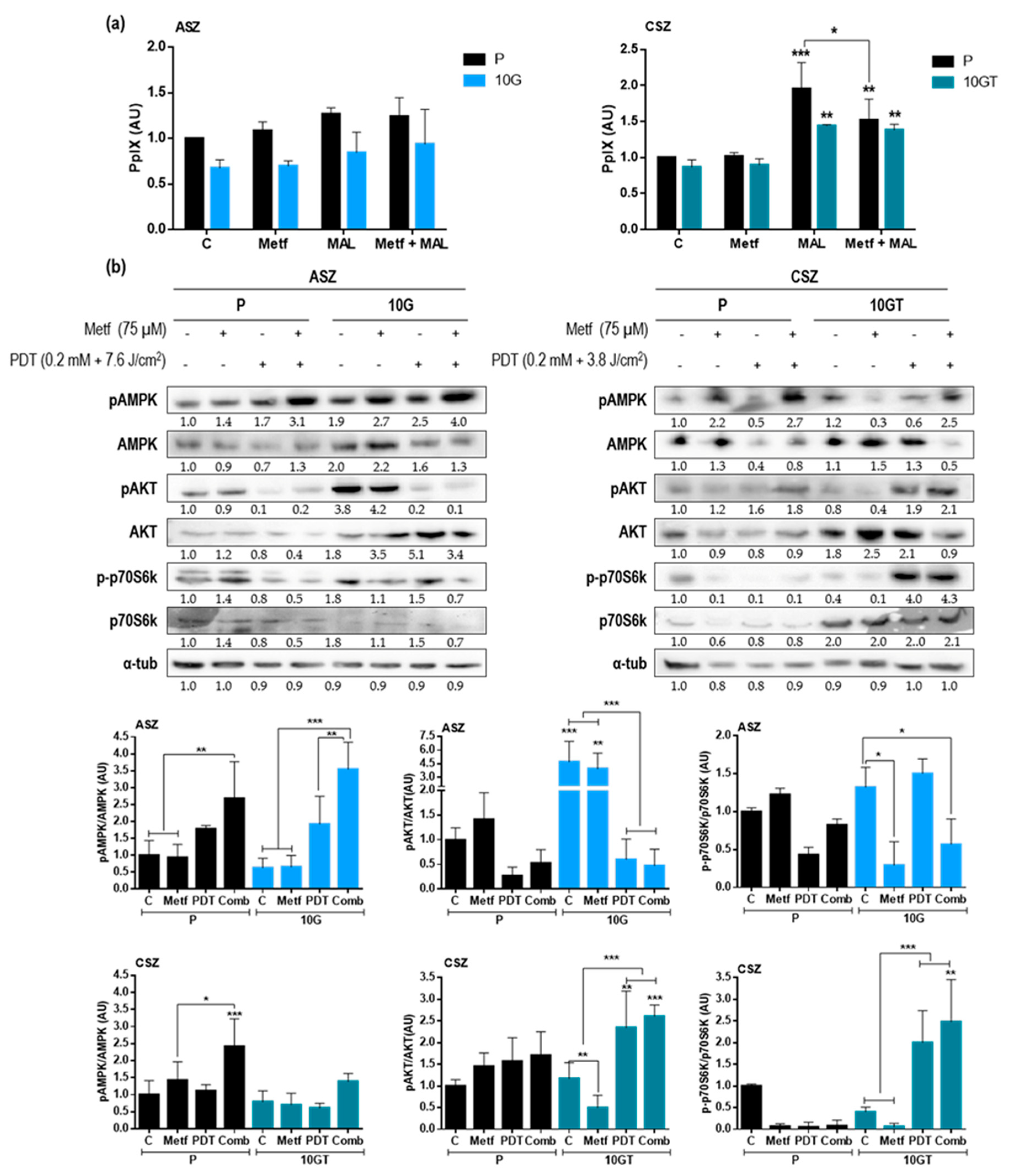
2.4. Combined Treatment of Metf and MAL-PDT on BCC Cell Lines
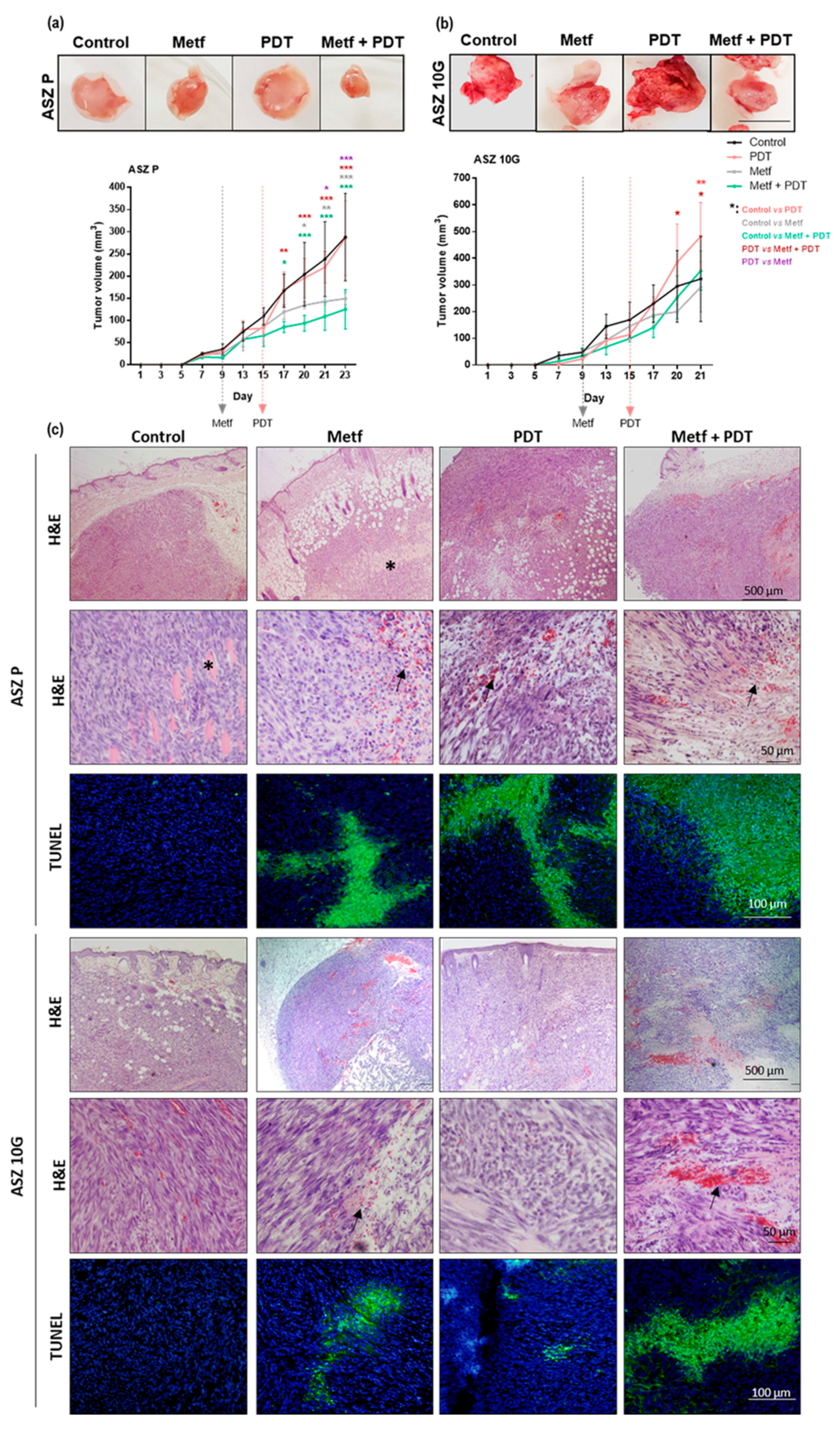
2.5. Effect of Metf and MAL-PDT on Tumor Growth Inhibition In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Treatments
4.3. Cellular Toxicity
4.4. Cell Proliferation
4.5. Western Blots
4.6. Determination of Cellular Respiration and Rates of Glycolysis
4.7. Detection of JC-1 Fluorescence
4.8. Cell Cycle
4.9. Production of PpIX
4.10. Evaluation of the Synergistic Effect after Combined Treatment
4.11. In Vivo Experiments
4.12. Optical Microscopy
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviations | Full Names |
| AMPK | AMP-activated protein kinase |
| β-F1-ATPase | β-catalytic subunit of the mitochondrial H+-ATP synthase |
| BCC | Basal Cell Carcinoma |
| CSP | Cell survival percentage |
| DL | Difference in logarithms |
| DMEM | Dulbecco’s modified Eagle’s medium high glucose |
| DMSO | Dimethyl sulfoxide |
| DNP | 2,4-Dinitrophenol |
| FBS | Fetal bovine serum |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| JC-1 | 5,5′6,6′- tetrachloro- 1,1′,3,3′- tetraethyl- benzamidazole carbo-cyanine iodide |
| MAL | Methyl-aminolevulinate |
| Metf | Metformin |
| mTOR | Mammalian target of rapamycin |
| MTT | 3-[4,5-dimethylthiazol-2-yl]-2,5- diphenyltetrazoliumbromide |
| OCR | Oxygen consumption rate |
| OSR | Oligomycin sensitive respiration |
| OXPHOS | Oxidative phosphorilation |
| PDT | Photodynamic therapy |
| PKM2 | Pyruvate kinase M2 |
| PpPIX | Protoporphytin IX |
| PS | Photosensitizer |
| ROS | Reactive oxygen species |
References
- Verkouteren, J.; Ramdas, K.; Wakkee, M.; Nijsten, T. Epidemiology of Basal Cell Carcinoma: Scholarly Review. Br. J. Dermatol. 2017, 177, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Lucena, S.R.; Zamarrón, A.; Carrasco, E.; Marigil, M.A.; Mascaraque, M.; Fernández-Guarino, M.; Gilaberte, Y.; González, S.; Juarranz, A. Characterisation of Resistance Mechanisms Developed by Basal Cell Carcinoma Cells in Response to Repeated Cycles of Photodynamic Therapy. Sci. Rep. 2019, 9, 4835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanoue, J.; Goldenberg, G. Basal Cell Carcinoma: A Comprehensive Review of Existing and Emerging Nonsurgical Therapies. J. Clin. Aesthet. Dermatol. 2016, 9, 26–36. [Google Scholar] [PubMed]
- Collier, N.; Haylett, A.; Wong, T.; Morton, C.; Ibbotson, S.; McKenna, K.; Mallipeddi, R.; Moseley, H.; Seukeran, D.; Ward, K. Conventional and Combination Topical Photodynamic Therapy for Basal Cell Carcinoma: Systematic Review and Meta-analysis. Br. J. Dermatol. 2018, 179, 1277–1296. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Xiong, L.; Chen, W.; Zhao, X.; He, J.; Zheng, Y.; Kong, F.; Liu, X.; Zhang, Z.; Miao, X. Photodynamic Therapy for Middle-Advanced Stage Upper Gastrointestinal Carcinomas: A Systematic Review and Meta-Analysis. World J. Clin. Cases 2018, 6, 650. [Google Scholar] [CrossRef]
- Wang, L.; Yang, H.; Li, B. Photodynamic Therapy for Prostate Cancer: A Systematic Review and Meta-Analysis. Prostate Int. 2018, 7, 83–90. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D. Photodynamic Therapy of Cancer: An Update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Ang, J.M.; Riaz, I.B.; Kamal, M.U.; Paragh, G.; Zeitouni, N.C. Photodynamic Therapy and Pain: A Systematic Review. Photodiagn. Photodyn. 2017, 19, 308–344. [Google Scholar] [CrossRef]
- Calixto, G.; Bernegossi, J.; de Freitas, L.; Fontana, C.; Chorilli, M. Nanotechnology-Based Drug Delivery Systems for Photodynamic Therapy of Cancer: A Review. Molecules 2016, 21, 342. [Google Scholar] [CrossRef]
- Cohen, D.; Lee, P. Photodynamic Therapy for Non-Melanoma Skin Cancers. Cancers 2016, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuezva, J.M.; Krajewska, M.; de Heredia, M.L.; Krajewski, S.; Santamaria, G.; Kim, H.; Zapata, J.M.; Marusawa, H.; Chamorro, M.; Reed, J.C. The Bioenergetic Signature of Cancer: A Marker of Tumor Progression. Cancer Res. 2002, 62, 6674–6681. [Google Scholar] [PubMed]
- Lopez-Rios, F.; Sanchez-Arago, M.; Garcia-Garcia, E.; Ortega, A.D.; Berrendero, J.R.; Pozo-Rodriguez, F.; Lopez-Encuentra, A.; Ballestin, C.; Cuezva, J.M. Loss of the Mitochondrial Bioenergetic Capacity Underlies the Glucose Avidity of Carcinomas. Cancer Res. 2007, 67, 9013–9017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, B.; Mohd Omar, M.F.; Soong, R. The Warburg Effect and Drug Resistance. Br. J. Pharmacol. 2016, 173, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Aragó, M.; Cuezva, J.M. The Bioenergetic Signature of Isogenic Colon Cancer Cells Predicts the Cell Death Response to Treatment with 3-Bromopyruvate, Iodoacetate Or 5-Fluorouracil. J. Transl. Med. 2011, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Santamaría, G.; Martínez-Diez, M.; Fabregat, I.; Cuezva, J.M. Efficient Execution of Cell Death in Non-Glycolytic Cells Requires the Generation of ROS Controlled by the Activity of Mitochondrial H -ATP Synthase. Carcinogenesis 2005, 27, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Dong, G.; Mao, Q.; Xia, W.; Xu, Y.; Wang, J.; Xu, L.; Jiang, F. PKM2 and Cancer: The Function of PKM2 Beyond Glycolysis. Oncol. Lett. 2016, 11, 1980–1986. [Google Scholar] [CrossRef] [Green Version]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer Metabolism and the Warburg Effect: The Role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef]
- Yecies, J.L.; Manning, B.D. mTOR Links Oncogenic Signaling to Tumor Cell Metabolism. J. Mol. Med. 2011, 89, 221–228. [Google Scholar] [CrossRef]
- Coyle, C.; Cafferty, F.; Vale, C.; Langley, R. Metformin as an Adjuvant Treatment for Cancer: A Systematic Review and Meta-Analysis. Ann. Oncol. 2016, 27, 2184–2195. [Google Scholar] [CrossRef]
- Heckman-Stoddard, B.M.; DeCensi, A.; Sahasrabuddhe, V.V.; Ford, L.G. Repurposing Metformin for the Prevention of Cancer and Cancer Recurrence. Diabetologia 2017, 60, 1639–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Checkley, L.A.; Rho, O.; Angel, J.M.; Cho, J.; Blando, J.; Beltran, L.; Hursting, S.D.; DiGiovanni, J. Metformin Inhibits Skin Tumor Promotion in Overweight and Obese Mice. Cancer Prev. Res. 2014, 7, 54–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osaki, T.; Yokoe, I.; Takahashi, K.; Inoue, K.; Ishizuka, M.; Tanaka, T.; Azuma, K.; Maruhata, Y.; Tsuka, T.; Itoh, N.; et al. Metformin enhances the cytotoxicity of 5-aminolevulinic acid-mediated photodynamic therapy in vitro. Oncol. Lett. 2017, 14, 1049–1053. [Google Scholar] [CrossRef]
- Nenu, I.; Popescu, T.; Aldea, M.D.; Craciun, L.; Olteanu, D.; Tatomir, C.; Bolfa, P.; Ion, R.M.; Muresan, A.; Filip, A.G. Metformin associated with photodynamic therapy–a novel oncological direction. J. Photochem. Photobiol. B 2014, 138, 80–91. [Google Scholar] [CrossRef]
- So, P.; Langston, A.W.; Daniallinia, N.; Hebert, J.L.; Fujimoto, M.A.; Khaimskiy, Y.; Aszterbaum, M.; Epstein, E.H., Jr. Long-term Establishment, Characterization and Manipulation of Cell Lines from Mouse Basal Cell Carcinoma Tumors. Exp. Dermatol. 2006, 15, 742–750. [Google Scholar] [CrossRef]
- Kuonen, F.; Surbeck, I.; Sarin, K.Y.; Dontenwill, M.; Rüegg, C.; Gilliet, M.; Oro, A.E.; Gaide, O. TGFβ, Fibronectin and Integrin α5β1 Promote Invasion in Basal Cell Carcinoma. J. Investig. Dermatol. 2018, 138, 2432–2442. [Google Scholar] [CrossRef] [Green Version]
- Morton, C.; Szeimies, R.; Sidoroff, A.; Wennberg, A.; Basset-Seguin, N.; Calzavara-Pinton, P.; Gilaberte, Y.; Hofbauer, G.; Hunger, R.; Karrer, S. European Dermatology Forum Guidelines on Topical Photodynamic Therapy. Eur. J. Dermatol. 2015, 25, 296–311. [Google Scholar] [CrossRef]
- Sun, L.; Suo, C.; Li, S.; Zhang, H.; Gao, P. Metabolic Reprogramming for Cancer Cells and their Microenvironment: Beyond the Warburg Effect. BBA Rev. Cancer 2018, 1870, 51–66. [Google Scholar] [CrossRef]
- Grasso, C.; Jansen, G.; Giovannetti, E. Drug Resistance in Pancreatic Cancer: Impact of Altered Energy Metabolism. Crit. Rev. Oncol. Hemat. 2017, 114, 139–152. [Google Scholar] [CrossRef]
- Morandi, A.; Indraccolo, S. Linking Metabolic Reprogramming to Therapy Resistance in Cancer. BBA Rev. Cancer 2017, 1868, 1–6. [Google Scholar] [CrossRef]
- Rosa, R.; Monteleone, F.; Zambrano, N.; Bianco, R. In Vitro and in Vivo Models for Analysis of Resistance to Anticancer Molecular Therapies. Curr. Med. Chem. 2014, 21, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, C.; Sfakianaki, M.; Lagoudaki, E.; Giagkas, G.; Ioannidis, G.; Trypaki, M.; Tsakalaki, E.; Voutsina, A.; Koutsopoulos, A.; Mavroudis, D. PKM2 as a Biomarker for Chemosensitivity to Front-Line Platinum-Based Chemotherapy in Patients with Metastatic Non-Small-Cell Lung Cancer. Br. J. Cancer 2014, 111, 1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Cenizo, L.; Formentini, L.; Aldea, M.; Ortega, A.D.; Garcia-Huerta, P.; Sanchez-Arago, M.; Cuezva, J.M. Up-Regulation of the ATPase Inhibitory Factor 1 (IF1) of the Mitochondrial H+-ATP Synthase in Human Tumors Mediates the Metabolic Shift of Cancer Cells to a Warburg Phenotype. J. Biol. Chem. 2010, 285, 25308–25313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Gu, J.; Zhou, Q. Review of Aerobic Glycolysis and its Key Enzymes–new Targets for Lung Cancer Therapy. Thorac. Cancer 2015, 6, 17–24. [Google Scholar] [CrossRef]
- Thirupathi, A.; Chang, Y. Role of AMPK and its Molecular Intermediates in Subjugating Cancer Survival Mechanism. Life Sci. 2019, 227, 30–38. [Google Scholar] [CrossRef]
- Bensinger, S.J.; Christofk, H.R. New Aspects of the Warburg Effect in Cancer Cell Biology. Semin. Cell Dev. Biol. 2012, 23, 352–361. [Google Scholar] [CrossRef]
- Faubert, B.; Vincent, E.E.; Poffenberger, M.C.; Jones, R.G. The AMP-Activated Protein Kinase (AMPK) and Cancer: Many Faces of a Metabolic Regulator. Cancer Lett. 2015, 356, 165–170. [Google Scholar] [CrossRef]
- Alimova, I.N.; Liu, B.; Fan, Z.; Edgerton, S.M.; Dillon, T.; Lind, S.E.; Thor, A.D. Metformin Inhibits Breast Cancer Cell Growth, Colony Formation and Induces Cell Cycle Arrest in Vitro. Cell Cycle 2009, 8, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Isakovic, A.; Harhaji, L.; Stevanovic, D.; Markovic, Z.; Sumarac-Dumanovic, M.; Starcevic, V.; Micic, D.; Trajkovic, V. Dual Antiglioma Action of Metformin: Cell Cycle Arrest and Mitochondria-Dependent Apoptosis. Cell. Mol. Life Sci. 2007, 64, 1290. [Google Scholar] [CrossRef]
- Vancura, A.; Bu, P.; Bhagwat, M.; Zeng, J.; Vancurova, I. Metformin as an Anticancer Agent. Trends Pharmacol. Sci. 2018, 39, 867–878. [Google Scholar] [CrossRef]
- Harada, K.; Ferdous, T.; Harada, T.; Ueyama, Y. Metformin in Combination with 5-Fluorouracil Suppresses Tumor Growth by Inhibiting the Warburg Effect in Human Oral Squamous Cell Carcinoma. Int. J. Oncol. 2016, 49, 276–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos Guimarães, I.; Ladislau-Magescky, T.; Tessarollo, N.G.; dos Santos, D.Z.; Gimba, E.R.P.; Sternberg, C.; Silva, I.V.; Rangel, L.B.A. Chemosensitizing Effects of Metformin on Cisplatin-and Paclitaxel-Resistant Ovarian Cancer Cell Lines. Pharmacol. Rep. 2018, 70, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Guo, X. Combinational Strategies of Metformin and Chemotherapy in Cancers. Cancer Chemother. Pharmacol. 2016, 78, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Rocha, G.Z.; Dias, M.M.; Ropelle, E.R.; Osorio-Costa, F.; Rossato, F.A.; Vercesi, A.E.; Saad, M.J.; Carvalheira, J.B. Metformin Amplifies Chemotherapy-Induced AMPK Activation and Antitumoral Growth. Clin. Cancer Res. 2011, 17, 3993–4005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honjo, S.; Ajani, J.A.; Scott, A.W.; Chen, Q.; Skinner, H.D.; Stroehlein, J.; Johnson, R.L.; Song, S. Metformin Sensitizes Chemotherapy by Targeting Cancer Stem Cells and the mTOR Pathway in Esophageal Cancer. Int. J. Oncol. 2014, 45, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. Metformin Decreases the Dose of Chemotherapy for Prolonging Tumor Remission in Mouse Xenografts Involving Multiple Cancer Cell Types. Cancer Res. 2011, 71, 3196–3201. [Google Scholar] [CrossRef] [Green Version]
- Luo, Q.; Hu, D.; Hu, S.; Yan, M.; Sun, Z.; Chen, F. In Vitro and in Vivo Anti-Tumor Effect of Metformin as a Novel Therapeutic Agent in Human Oral Squamous Cell Carcinoma. BMC Cancer 2012, 12, 517. [Google Scholar] [CrossRef] [Green Version]
- Tudor, D.; Nenu, I.; Filip, G.A.; Olteanu, D.; Cenariu, M.; Tabaran, F.; Ion, R.M.; Gligor, L.; Baldea, I. Combined Regimen of Photodynamic Therapy Mediated by Gallium Phthalocyanine Chloride and Metformin Enhances Anti-Melanoma Efficacy. PLoS ONE 2017, 12, e0173241. [Google Scholar] [CrossRef]
- Elefantova, K.; Lakatos, B.; Kubickova, J.; Sulova, Z.; Breier, A. Detection of the Mitochondrial Membrane Potential by the Cationic Dye JC-1 in L1210 Cells with Massive Overexpression of the Plasma Membrane ABCB1 Drug Transporter. Int. J. Mol. Sci. 2018, 19, 1985. [Google Scholar] [CrossRef] [Green Version]
- Foucquier, J.; Guedj, M. Analysis of Drug Combinations: Current Methodological Landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mascaraque, M.; Delgado-Wicke, P.; Nuevo-Tapioles, C.; Gracia-Cazaña, T.; Abarca-Lachen, E.; González, S.; Cuezva, J.M.; Gilaberte, Y.; Juarranz, Á. Metformin as an Adjuvant to Photodynamic Therapy in Resistant Basal Cell Carcinoma Cells. Cancers 2020, 12, 668. https://doi.org/10.3390/cancers12030668
Mascaraque M, Delgado-Wicke P, Nuevo-Tapioles C, Gracia-Cazaña T, Abarca-Lachen E, González S, Cuezva JM, Gilaberte Y, Juarranz Á. Metformin as an Adjuvant to Photodynamic Therapy in Resistant Basal Cell Carcinoma Cells. Cancers. 2020; 12(3):668. https://doi.org/10.3390/cancers12030668
Chicago/Turabian StyleMascaraque, Marta, Pablo Delgado-Wicke, Cristina Nuevo-Tapioles, Tamara Gracia-Cazaña, Edgar Abarca-Lachen, Salvador González, José M. Cuezva, Yolanda Gilaberte, and Ángeles Juarranz. 2020. "Metformin as an Adjuvant to Photodynamic Therapy in Resistant Basal Cell Carcinoma Cells" Cancers 12, no. 3: 668. https://doi.org/10.3390/cancers12030668