Integrative Analysis of Fecal Metagenomics and Metabolomics in Colorectal Cancer

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Results
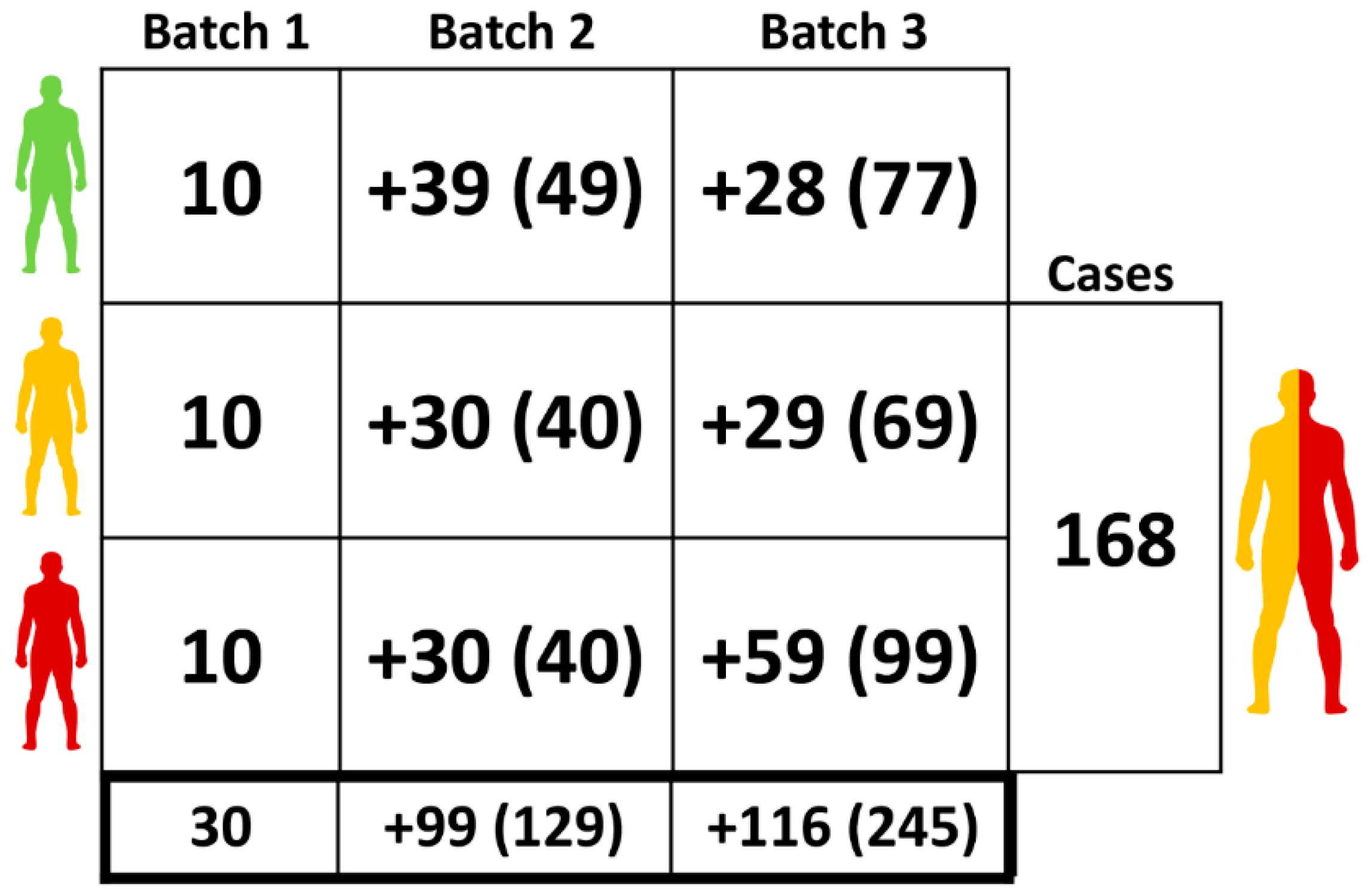
2.1. Clinical Samples
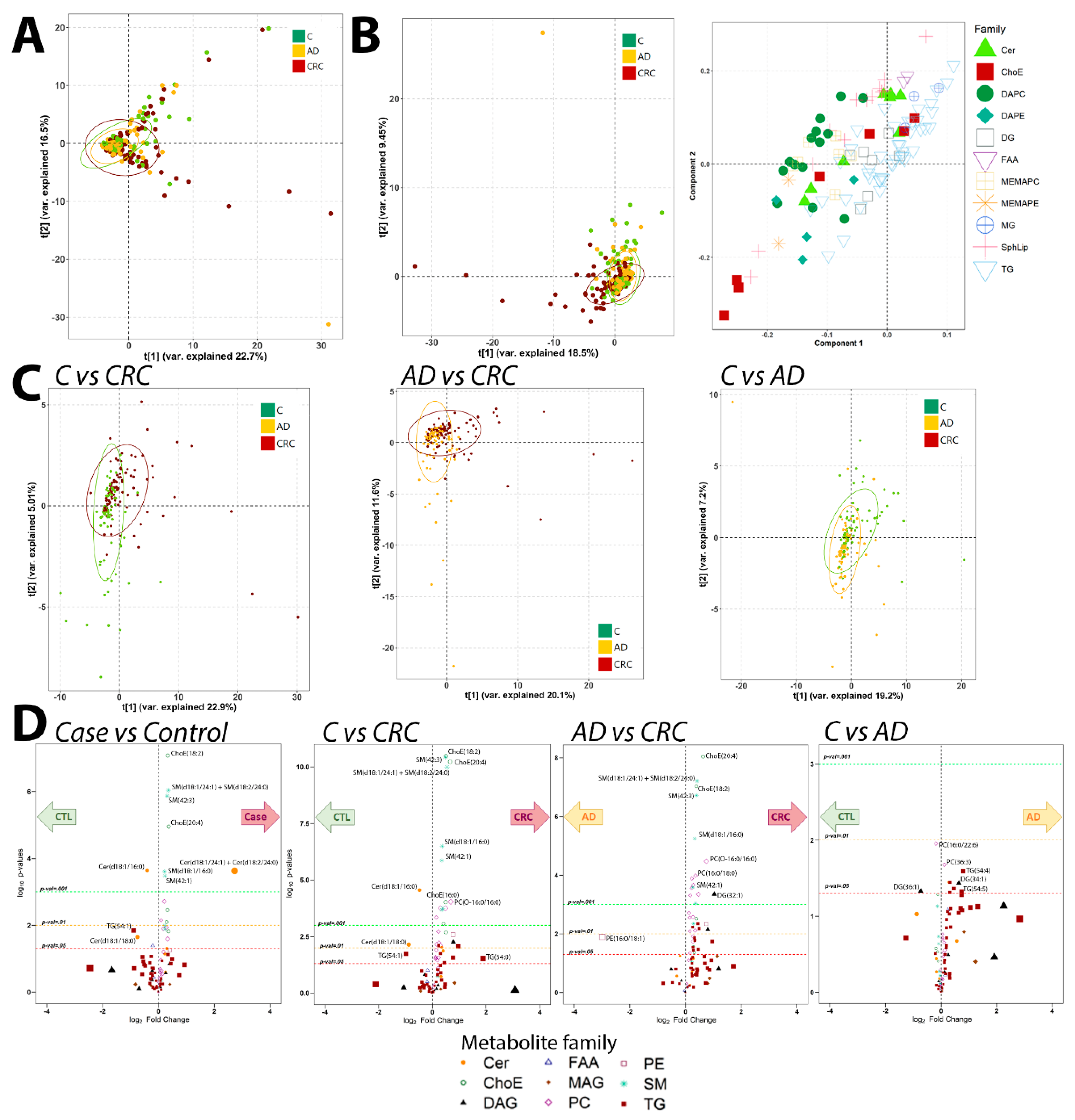
2.2. Metabolomics Analysis
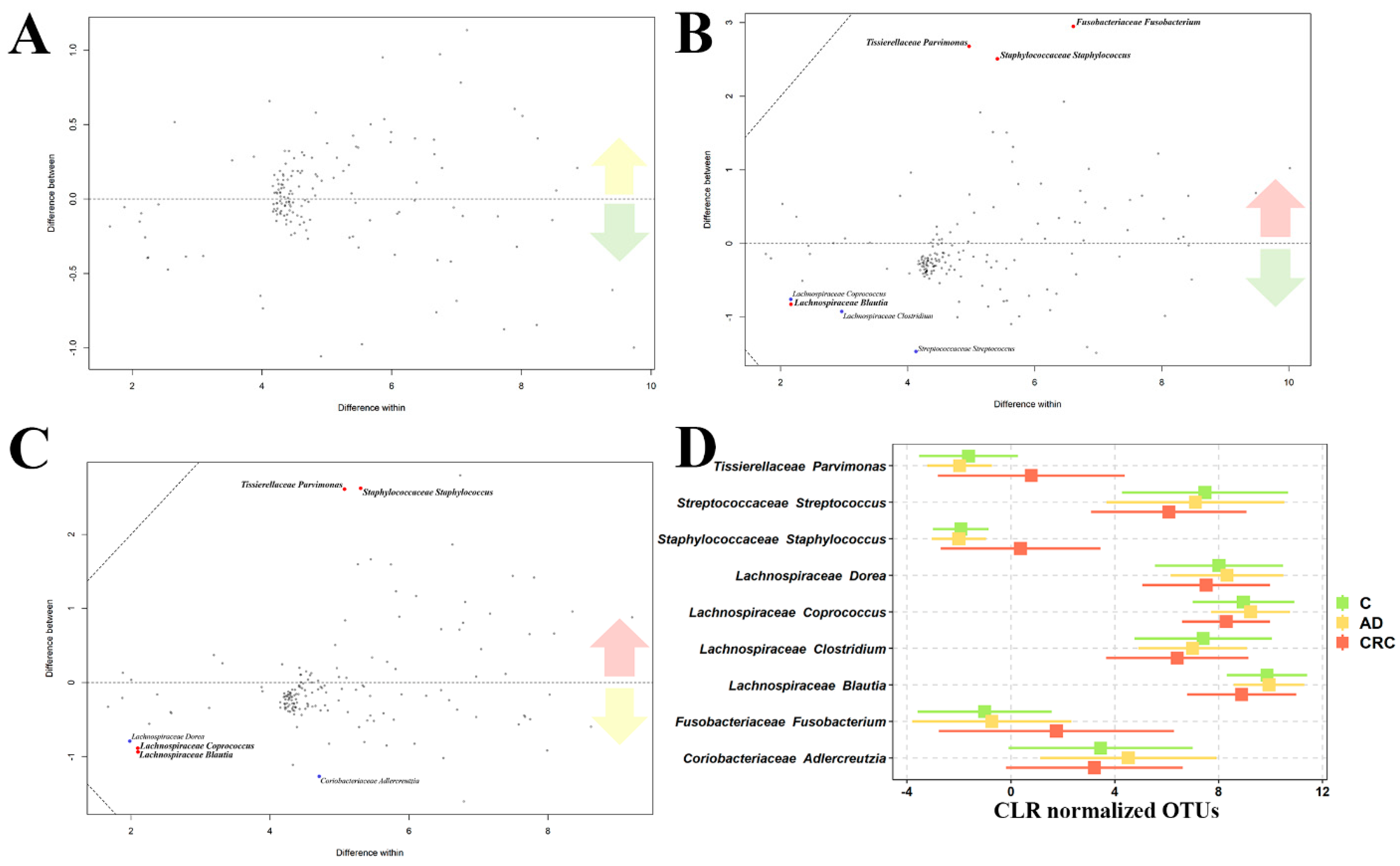
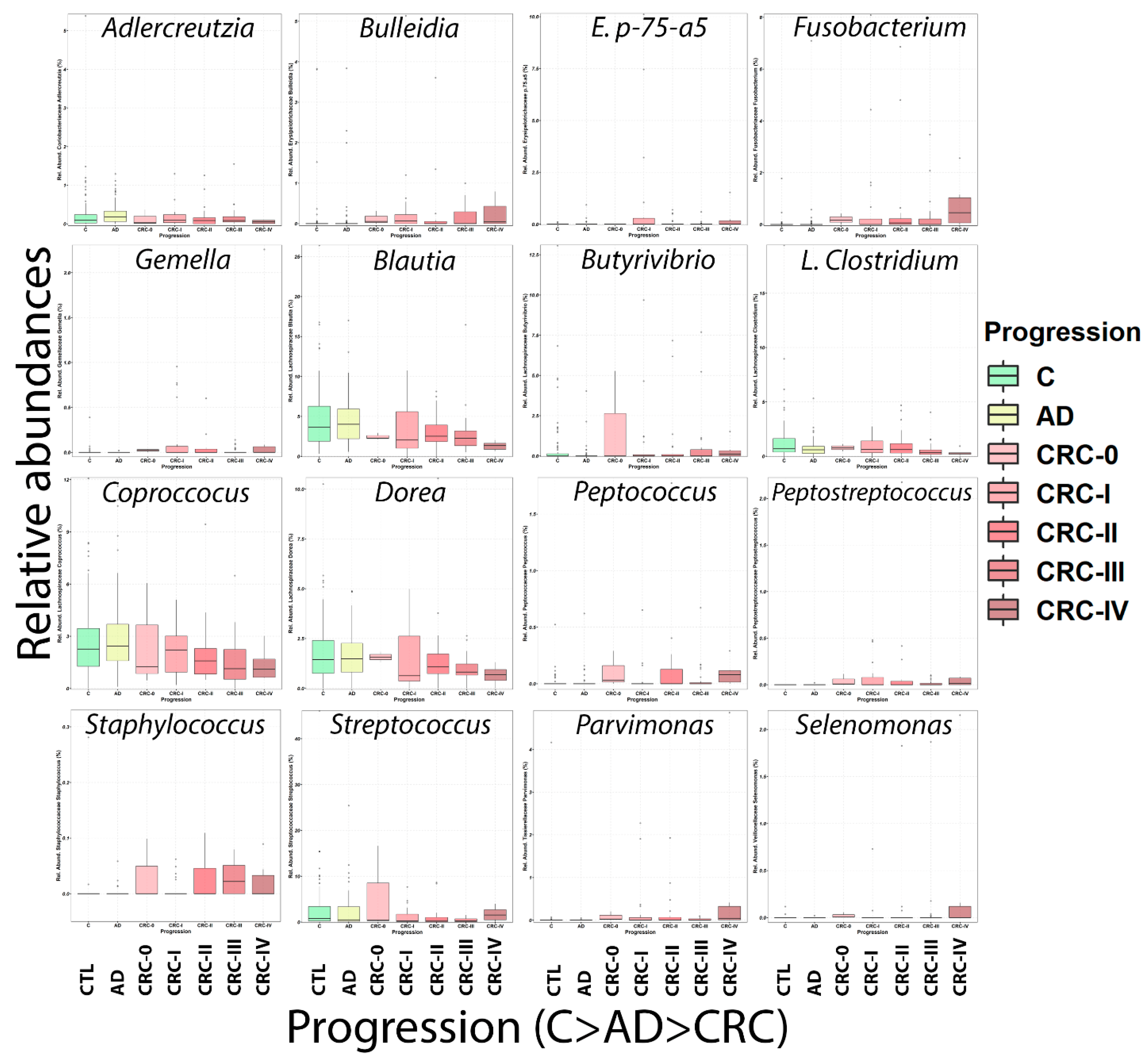
2.3. Microbiome Analysis
2.4. Combination of Microbiome and Metabolomics Data
2.4.1. MixOmics
2.4.2. Microbiome: Metabolomics Predictive Model
3. Discussion
4. Materials and Methods
4.1. Clinical Samples and Study Population
4.2. UHPLC-MS Metabolomics Analysis
4.3. Metabolomics Data Analysis
4.4. Fecal DNA Extraction
4.5. 16S rDNA Amplification and Sequencing
4.6. Metabolomics—Microbiome Data Integration
4.6.1. HAllA
4.6.2. Procrustes
4.6.3. MixOmics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stewart, B.W.; Wild, C.P. World Cancer Report 2014; World Health Organization: Lyon, France, 2014; pp. 1–2. [Google Scholar]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Lasry, A.; Zinger, A.; Ben-Neriah, Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016, 17, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.J.; Ferrucci, L.M.; Risch, A.; Graubard, B.I.; Ward, M.H.; Park, Y.; Hollenbeck, A.R.; Schatzkin, A.; Sinha, R. A large prospective study of meat consumption and colorectal cancer risk: An investigation of potential mechanisms underlying this association. Cancer Res. 2010, 70, 2406–2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Cancer Research Fund; American Institute for Cancer Research. Food, Nutrition, Physical Activity, and the Prevention of Cancer; American Institute for Cancer Research: Washington, DC, USA, 2007. [Google Scholar]
- Bénard, F.; Barkun, A.N.; Martel, M.; von Renteln, D. Systematic review of colorectal cancer screening guidelines for average-risk adults: Summarizing the current global recommendations. World J. Gastroenterol. 2018, 24, 124–138. [Google Scholar] [CrossRef]
- Cubiella, J.; Clos-Garcia, M.; Alonso, C.; Martinez-Arranz, I.; Perez-Cormenzana, M.; Barrenetxea, Z.; Berganza, J.; Rodríguez-Llopis, I.; D’Amato, M.; Bujanda, L.; et al. Targeted UPLC-MS Metabolic Analysis of Human Faeces Reveals Novel Low-Invasive Candidate Markers for Colorectal Cancer. Cancers 2018, 10, 300. [Google Scholar] [CrossRef] [Green Version]
- Silva, C.L.; Passos, M.; Cmara, J.S. Investigation of urinary volatile organic metabolites as potential cancer biomarkers by solid-phase microextraction in combination with gas chromatography-mass spectrometry. Br. J. Cancer 2011, 105, 1894–1904. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Ma, C.; Liu, C.; Wang, Z.; Yang, J.; Liu, X.; Shen, Z.; Wu, R. NMR-based fecal metabolomics fingerprinting as predictors of earlier diagnosis in patients with colorectal cancer. Oncotarget 2016, 7, 29454–29464. [Google Scholar] [CrossRef]
- Oliver, S.G.; Winson, M.K.; Kell, D.B.; Baganz, F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998, 16, 373–378. [Google Scholar] [CrossRef]
- Chen, C.; Gonzalez, F.J.; Idle, J.R. LC-MS-based metabolomics in drug metabolism. Drug Metab. Rev. 2007, 39, 581–597. [Google Scholar] [CrossRef] [Green Version]
- Clarke, C.J.; Haselden, J.N. Metabolic Profiling as a Tool for Understanding Mechanisms of Toxicity. Toxicol. Pathol. 2008, 36, 140–147. [Google Scholar] [CrossRef]
- Fernie, A.R.; Trethewey, R.N.; Krotzky, A.J.; Willmitzer, L. Metabolite profiling: From diganostics to systems biology. Nat. Rev. Mol. Cell Biol. 2004, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Wilson, I.D. Understanding ‘global’ systems biology: Metabonomics and the continuum of metabolism. Nat. Rev. Drug Discov. 2003, 2, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, I.; Amiot, A.; le Baleur, Y.; Levy, M.; Auriault, M.; van Nhieu, J.T.; Delchier, J.C. Microbial dysbiosis and colon carcinogenesis: Could colon cancer be considered a bacteria-related disease? Ther. Adv. Gastroenterol. 2013, 6, 215–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A bacterial driver-passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Dove, W.F.; Clipson, L.; Gould, K.A.; Luongo, C.; Marshall, D.J.; Moser, A.R.; Newton, M.A.; Jacoby, R.F. Intestinal neoplasia in the Apc(Min) mouse: Independence from the microbial and natural killer (beige locus) status. Cancer Res. 1997, 57, 812–814. [Google Scholar] [PubMed]
- Sellon, R.K.; Tonkonogy, S.; Schultz, M.; Dieleman, L.A.; Grenther, W.; Balish, E.; Rennick, D.M.; Sartor, R.B. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 1998, 66, 5224–5231. [Google Scholar] [CrossRef] [Green Version]
- Uronis, J.M.; Mühlbauer, M.; Herfarth, H.H.; Rubinas, T.C.; Jones, G.S.; Jobin, C. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [Green Version]
- Fearon, E.R.; Vogelstein, B. A Genetic Model for Colorectal Tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. The multistep nature of cancer. Trends Genet. 1993, 9, 138–141. [Google Scholar] [CrossRef]
- Cubiella, J.; Vega, P.; Salve, M.; Díaz-Ondina, M.; Alves, M.T.; Quintero, E.; Álvarez-Sánchez, V.; Fernández-Bañares, F.; Boadas, J.; Campo, R.; et al. Development and external validation of a faecal immunochemical test-based prediction model for colorectal cancer detection in symptomatic patients. BMC Med. 2016, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.Q.; Cao, H.-L.; Wang, W.-Q.; Wang, S.; Cao, X.-C.; Yan, F.; Wang, B.-M. Fecal microbiota transplantation broadening its application beyond intestinal disorders. World J. Gastroenterol. 2015, 21, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.D.; Reid, J.N.S.; Macklaim, J.M.; McMurrough, T.A.; Edgell, D.R.; Gloor, G.B. Unifying the analysis of high-throughput sequencing datasets: Characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2014, 2, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahnavard, G.; Franzosa, E.A.; McIver, L.J.; Schwager, E.; Lloyd-Price, J.; Weingart, G.; Moon, Y.S.; Morgan, X.C.; Waldron, L.; Huttenhower, C. High-Sensitivity Pattern Discovery in Large Multi’omic Datasets. Available online: http://huttenhower.sph.harvard.edu/halla (accessed on 7 July 2019).
- Gower, J.C. Statistical methods of comparing different multivariate analyses of the same data. In Mathematics in the Archaeological and Historical Science; Edinburgh University Press: Edinburgh, UK, 1971; pp. 138–149. [Google Scholar]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Jia, W.; Cai, S.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012, 6, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, A.N.; Araújo-Pérez, F.; Azcárate-Peril, A.; Yeh, J.J.; Sandler, R.S.; Keku, T.O. Fusobacterium Is Associated with Colorectal Adenoma. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Sinha, R.; Ahn, J.; Sampson, J.N.; Shi, J.; Yu, G.; Xiong, X.; Hayes, R.B.; Goedert, J.J. Fecal Microbiota, Fecal Metabolome, and Colorectal Cancer Interrelations. PLoS ONE 2016, 11, e0152126. [Google Scholar] [CrossRef] [Green Version]
- Weir, T.L.; Manter, D.K.; Sheflin, A.M.; Barnett, B.A.; Heuberger, A.L.; Ryan, E.P. Stool Microbiome and Metabolome Differences between Colorectal Cancer Patients and Healthy Adults. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Warren, R.L.; Freeman, D.J.; Pleasance, S.; Watson, P.; Moore, R.A.; Cochrane, K.; Allen-Vercoe, E.; Holt, R.A. Co-occurrence of anaerobic bacteria in colorectal carcinomas. Microbiome 2013, 1, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flemer, B.; Ng, O.; Brookes, M. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 2017, 66, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.S.; DeSantis, T.Z.; Weinmaier, T.; McMurdie, P.J.; Cope, J.L.; Altrichter, A.; Yamal, J.; Hollister, E.B. Leveraging sequence-based faecal microbial community survey data to identify a composite biomarker for colorectal cancer. Gut 2018, 67, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Song, Q.; Tang, X.; Liang, X.; Fan, H.; Peng, H.; Guo, Q.; Zhang, Z. Co-occurrence of driver and passenger bacteria in human colorectal cancer. Gut Pathog. 2014, 6, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 5240, eaal5240. [Google Scholar] [CrossRef] [Green Version]
- Vogtmann, E.; Hua, X.; Zeller, G.; Sunagawa, S.; Voigt, A.Y.; Hercog, R.; Goedert, J.J.; Shi, J.; Bork, P.; Sinha, R. Colorectal cancer and the human gut microbiome: Reproducibility with whole-genome shotgun sequencing. PLoS ONE 2016, 11, e0155362. [Google Scholar] [CrossRef] [Green Version]
- Gagnière, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef]
- Farshidfar, F.; Weljie, A.M.; Kopciuk, K.A.; Hilsden, R.; McGregor, S.E.; Buie, W.D.; MacLean, A.; Vogel, H.J.; Bathe, O.F. A validated metabolomic signature for colorectal cancer: Exploration of the clinical value of metabolomics. Br. J. Cancer 2016, 115, 848–857. [Google Scholar] [CrossRef] [Green Version]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Han, Y.W.; Shi, W.; Huang, G.T.-J.; Haake, S.K.; Park, N.; Kuramitsu, H.; Genco, R.J. Interactions between periodontal bacteria and human oral epithelial cells: Fusobacterium nucleatum adheres to and invades epithelial cells. Infect. Immun. 2000, 68, 3140–3146. [Google Scholar] [CrossRef] [Green Version]
- Weiss, E.I.; Shaniztki, B.; Dotan, M.; Ganeshkumar, N.; Kolenbrander, P.E.; Metzger, Z. Attachment of Fusobacterium nucleatum PK1594 to mammalian cells and its coaggregation with periodontopathogenic bacteria are mediated by the same galactose-binding adhesin. Oral Microbiol. Immunol. 2000, 15, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Krisanaprakornkit, S.; Kimball, J.R.; Weinberg, A.; Darveau, R.P.; Bainbridge, B.W.; Dale, B.A. Inducible expression of human β-defensin 2 by Fusobacterium nucleatum in oral epithelial cells: Multiple signaling pathways and role of commensal bacteria in innate immunity and the epithelial barrier. Infect. Immun. 2000, 68, 2907–2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, L.; Schmid, J.; Ebert, M.; Soucek, P.; Kunicka, T.; Liska, V.; Bruha, J.; Neary, P.; Dezeeuw, N.; Tommasino, M.; et al. Fusobacterium nucleatum associates with stages of colorectal neoplasia development, colorectal cancer and disease outcome. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Kanno, S.; Nosho, K.; Sukawa, Y.; Mitsuhashi, K.; Kurihara, H.; Igarashi, H.; Takahashi, T.; Tachibana, M.; Takahashi, H.; et al. Association of Fusobacterium nucleatum with clinical and molecular features in colorectal serrated pathway. Int. J. Cancer 2015, 137, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.A.; Yang, J.; Dou, R.; Masugi, Y.; Song, M.; et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 2016, 65, 1973–1980. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Hanor, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Sears, C.L.; Garrett, W.S. Microbes, microbiota, and colon cancer. Cell Host Microbe 2014, 15, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Dinh, D.M.; Volpe, G.E.; Duffalo, C.; Bhalchandra, S.; Tai, A.K.; Kane, A.V.; Wanke, C.A.; Ward, H.D. Intestinal Microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J. Infect. Dis. 2015, 211, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Fleissner, C.K.; Huebel, N.; El-Bary, M.M.A.; Loh, G.; Klaus, S.; Blaut, M. Absence of intestinal microbiota does not protect mice from diet-induced obesity. Br. J. Nutr. 2010, 104, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Martínez, I.; Wallace, G.; Zhang, C.; Legge, R.; Benson, A.K.; Carr, T.P.; Moriyama, E.N.; Walter, J. Diet-induced metabolic improvements in a hamster model of hypercholesterolemia are strongly linked to alterations of the gut microbiota. Appl. Environ. Microbiol. 2009, 75, 4175–4184. [Google Scholar] [CrossRef] [Green Version]
- Clavel, T.; Lepage, P.; Charrier, C. The Family Coriobacteriaceae. In The Prokaryotes: Actinobacteria; Springer-Verlag: Berlin/Heidelberg, Germany, 2014; pp. 1–1061. [Google Scholar]
- Marchesi, J.R.; Dutilh, B.E.; Hall, N.; Peters, W.H.M.; Roelofs, R.; Boleij, A.; Tjalsma, H. Towards the human colorectal cancer microbiome. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruo, T.; Sakamoto, M.; Ito, C.; Toda, T.; Benno, Y. Adlercreutzia equolifaciens gen. nov., sp. nov., an equol-producing bacterium isolated from human faeces, and emended description of the genus Eggerthella. Int. J. Syst. Evol. Microbiol. 2008, 58, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Ma, Y.; Zhao, A.; He, T.; Lyu, N.; Pan, Z.; Mao, G.; Liu, Y.; Li, J.; Wang, P.; et al. Compositional and functional differences in human gut microbiome with respect to equol production and its association with blood lipid level: A cross-sectional study. Gut Pathog. 2019, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Achaintre, D.; Zamora-Ros, R.; Jenab, M.; Boutron-Ruault, M.; Carbonnel, F.; Savoye, I.; Kaaks, R.; Kühn, T.; Boeing, H.; et al. A prospective evaluation of plasma polyphenol levels and colon cancer risk. Int. J. Cancer 2018, 143, 1620–1631. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, J.; Li, J.V.; Zhou, N.; Tang, H.; Wang, Y. Gut Microbiota Composition Modifies Fecal Metabolic Profiles in Mice. J. Proteome Res. 2013. [Google Scholar] [CrossRef]
- Han, S.; Gao, J.; Zhou, Q.; Liu, S.; Wen, C.; Yang, X. Role of intestinal flora in colorectal cancer from the metabolite perspective: A systematic review. Cancer Manag. Res. 2018, 10, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Buitenwerf, E.; Dullaart, R.P.F.; Kobold, A.C.M.; Links, T.P.; Sluiter, W.J.; Connelly, M.A.; Kerstens, M.N. Cholesterol delivery to the adrenal glands estimated by adrenal venous sampling: An in vivo model to determine the contribution of circulating lipoproteins to steroidogenesis in humans. J. Clin. Lipidol. 2017, 11, 733–738. [Google Scholar] [CrossRef]
- Farhana, L.; Nangia-Makker, P.; Arbit, E.; Shango, K.; Sarkar, S.; Mahmud, H.; Hadden, T.; Yu, Y.; Majumdar, A.P.N. Bile acid: A potential inducer of colon cancer stem cells. Stem Cell Res. Ther. 2016, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ajouz, H.; Mukherji, D.; Shamseddine, A. Secondary bile acids: An underrecognized cause of colon cancer. World J. Surg. Oncol. 2014, 12, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-Y, M.; Glickman, J.N.; Garrett, W.S. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, E.C.; Frick, I.M. Gram-positive anaerobic cocci—Commensals and opportunistic pathogens. FEMS Microbiol. Rev. 2013, 37, 520–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roccarina, D.; Lauritano, E.C.; Gabrielli, M.; Franceschi, F.; Ojetti, V.; Gasbarrini, A. The role of methane in intestinal diseases. Am. J. Gastroenterol. 2010, 105, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, P.D.; Shanahan, F.; Marchesi, J.R. Human methanogen diversity and incidence in healthy and diseased colonic groups using mcrA gene analysis. BMC Microbiol. 2008, 8, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishaq, S.L.; Moses, P.L.; Wright, A.-D.G. The Pathology of Methanogenic Archaea in Human Gastrointestinal Tract Disease. Gut Microbiome Implic. Hum. Dis. 2016. [Google Scholar] [CrossRef] [Green Version]
- Pausan, M.R.; Csorba, C.; Singer, G.; Till, H.; Schöpf, V.; Santigli, E.; Klug, B.; Högenauer, C.; Blohs, M.; Moissl-Eichinge, C. Exploring the Archaeome: Detection of Archaeal Signatures in the Human Body. Front. Microbiol. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Abell, G.C.J.; Conlon, M.A.; Mcorist, A.L. Methanogenic archaea in adult human faecal samples are inversely related to butyrate concentration. Microb. Ecol. Health Dis. 2006, 18, 154–160. [Google Scholar] [CrossRef]
- Wu, X.; Wu, Y.; He, L.; Wu, L.; Wang, X.; Liu, Z. Effects of the intestinal microbial metabolite butyrate on the development of colorectal cancer. J. Cancer 2018, 9, 2510–2517. [Google Scholar] [CrossRef]
- Kostic, A.D.; Catchpoole, E.M.; Runnegar, N.; Mapp, S.J.; Markey, K.A. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Mayo, R.; Crespo, J.; Martínez-Arranz, I.; Banales, J.M.; Arias, M.; Mincholé, I.; de la Fuente, R.A.; Jimenez-Agüero, R.; Alonso, C.; de Luis, D.A.; et al. Metabolomic-based noninvasive serum test to diagnose nonalcoholic steatohepatitis: Results from discovery and validation cohorts. Hepatol. Commun. 2018, 2, 807–820. [Google Scholar] [CrossRef]
- Cavill, R.; Kamburov, A.; Ellis, J.K.; Athersuch, T.J.; Blagrove, M.S.C.; Herwig, R.; Ebbels, T.M.D.; Keun, H.C. Consensus-phenotype integration of transcriptomic and metabolomic data implies a role for metabolism in the chemosensitivity of tumour cells. PLoS Comput. Biol. 2011, 7. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Picart-Armada, S.; Fernández-Albert, F.; Vinaixa, M.; Yanes, O.; Perera-Lluna, A. FELLA: An R package to enrich metabolomics data. BMC Bioinform. 2018, 19, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revelle, W. Psych: Procedures for Personality and Psychological Research, Version = 1.8.12. 2018. Available online: https://cran.r-project.org/package=psych (accessed on 1 May 2020).
- López-Ratón, M.; Rodríguez-Álvarez, M.X.; Suárez, C.C.; Sampedro, F.G. OptimalCutpoints: An R Package for Selecting Optimal Cutpoints in Diagnostic Tests. J. Stat. Softw. 2014, 61, 1–36. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.; Simko, V. R Package ‘corrplot’: Visualization of a Correlation Matrix (Version 0.84). 2017. Available online: https://cran.r-project.org/package=corrplot (accessed on 1 May 2020).
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high- throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME2. Nat. Biotechnol. 2019, 37, 848–857. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. The Vegan Package. 2018. Available online: https://cran.r-project.org/package=vegan (accessed on 1 May 2020).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zych, K.; Wirbel, J.; Essex, M.; Breuer, K.; Karcher, N.; Costea, P.I.; Sunagawa, S.; Bork, P.; Zeller, G. SIAMCAT: Statistical Inference of Associations between Microbial Communities And Host Phenotypes. 2018. Available online: https://siamcat.embl.de/ (accessed on 1 May 2020).
- Sing, T.; Sander, O.; Beerenwinkel, N.; Lengauer, T. ROCR: Visualizing the Performance of Scoring Classifiers. Bioinformatics 2005, 21, 3940–3941. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2: An improved and extensible approach for metagenome inference. bioRxiv 2019, 672295. [Google Scholar] [CrossRef] [Green Version]
- McHardy, I.H.; Goudarzi, M.; Tong, M.; Ruegger, P.M.; Schwager, E.; Weger, J.R.; Graeber, T.G.; Sonnenburg, J.L.; Horvath, S.; Huttenhower, C. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome 2013, 1, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Shannon, C.P.; Gautier, B.; Rohart, F.; Vacher, M.; Tebbutt, S.J.; Cao, K.L. DIABLO: From multi-omics assays to biomarker discovery, an integrative approach. bioRxiv 2018. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BACTERIAL PHYLUM | PR(>F) | C-AD | CRC-AD | CRC-C |
|---|---|---|---|---|
| K__BACTERIA.__ | 0.450 | 0.974 | 0.630 | 0.461 |
| K__BACTERIA.P__ACTINOBACTERIA | 0.577 | 0.827 | 0.916 | 0.548 |
| K__BACTERIA.P__BACTEROIDETES | 0.002 | 0.009 | 0.998 | 0.006 |
| K__BACTERIA.P__CYANOBACTERIA | 0.247 | 0.695 | 0.722 | 0.216 |
| K__BACTERIA.P__ELUSIMICROBIA | 0.396 | 0.528 | 0.994 | 0.418 |
| K__BACTERIA.P__FIRMICUTES | <0.001 | 0.002 | 0.449 | <0.001 |
| K__BACTERIA.P__FUSOBACTERIA | 0.036 | 0.634 | 0.285 | 0.030 |
| K__BACTERIA.P__LENTISPHAERAE | 0.086 | 0.865 | 0.283 | 0.085 |
| K__BACTERIA.P__OD1 | 0.096 | 1.000 | 0.168 | 0.145 |
| K__BACTERIA.P__PROTEOBACTERIA | 0.165 | 0.769 | 0.519 | 0.146 |
| K__BACTERIA.P__SR1 | 0.450 | 1.000 | 0.544 | 0.517 |
| K__BACTERIA.P__SPIROCHAETES | 0.951 | 0.957 | 0.958 | 1.000 |
| K__BACTERIA.P__SYNERGISTETES | 0.567 | 0.564 | 0.691 | 0.967 |
| K__BACTERIA.P__TM7 | 0.746 | 0.726 | 0.879 | 0.946 |
| K__BACTERIA.P__TENERICUTES | 0.920 | 0.990 | 0.967 | 0.915 |
| K__BACTERIA.P__VERRUCOMICROBIA | 0.549 | 0.965 | 0.559 | 0.705 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clos-Garcia, M.; Garcia, K.; Alonso, C.; Iruarrizaga-Lejarreta, M.; D’Amato, M.; Crespo, A.; Iglesias, A.; Cubiella, J.; Bujanda, L.; Falcón-Pérez, J.M. Integrative Analysis of Fecal Metagenomics and Metabolomics in Colorectal Cancer. Cancers 2020, 12, 1142. https://doi.org/10.3390/cancers12051142
Clos-Garcia M, Garcia K, Alonso C, Iruarrizaga-Lejarreta M, D’Amato M, Crespo A, Iglesias A, Cubiella J, Bujanda L, Falcón-Pérez JM. Integrative Analysis of Fecal Metagenomics and Metabolomics in Colorectal Cancer. Cancers. 2020; 12(5):1142. https://doi.org/10.3390/cancers12051142
Chicago/Turabian StyleClos-Garcia, Marc, Koldo Garcia, Cristina Alonso, Marta Iruarrizaga-Lejarreta, Mauro D’Amato, Anais Crespo, Agueda Iglesias, Joaquín Cubiella, Luis Bujanda, and Juan Manuel Falcón-Pérez. 2020. "Integrative Analysis of Fecal Metagenomics and Metabolomics in Colorectal Cancer" Cancers 12, no. 5: 1142. https://doi.org/10.3390/cancers12051142