Risk of Cancer in Family Members of Patients with Lynch-Like Syndrome

 , , , ,
, , , ,  , , , , ,
, , , , ,  add
Show full author list
add
Show full author list
Abstract
:1. Introduction
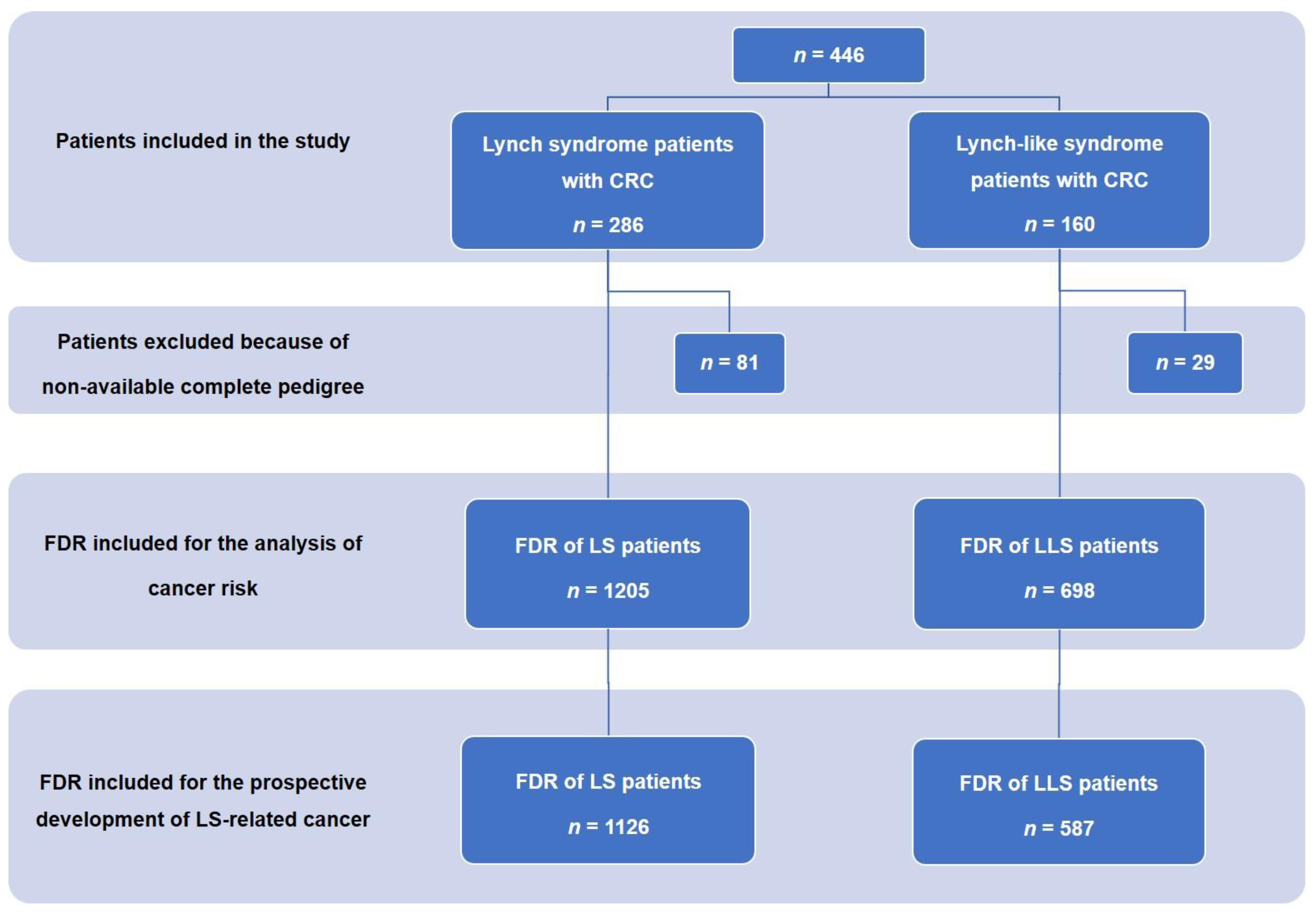
2. Material and Methods
2.1. Patients and Data Collection
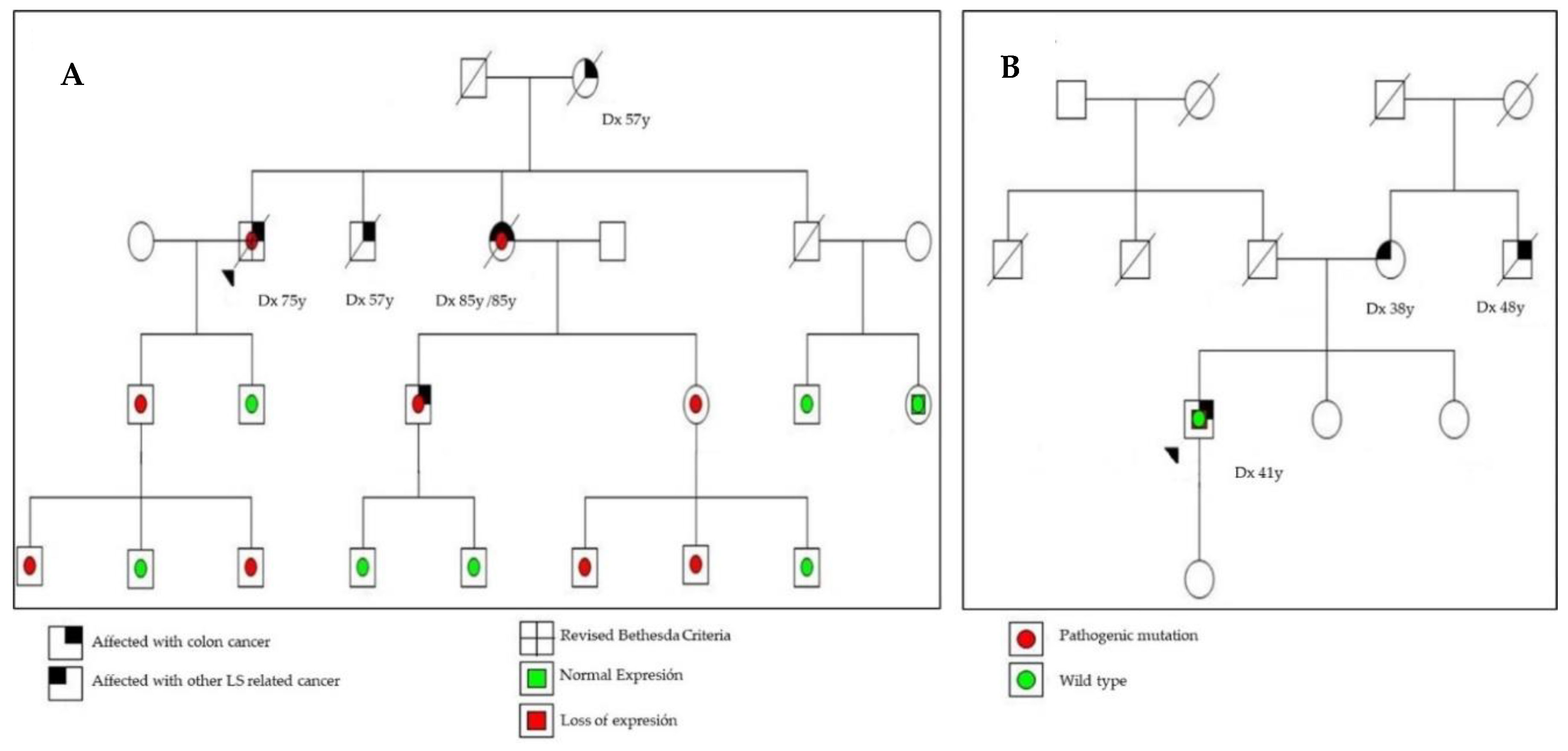
2.2. MSI, IHC Staining, and Detection of Germline Mutations
2.3. Calculation of Standardized Incidence Ratio
2.4. Statistical Analysis
3. Results
3.1. Clinical and Pathology Differences in CRC between LS and LLS
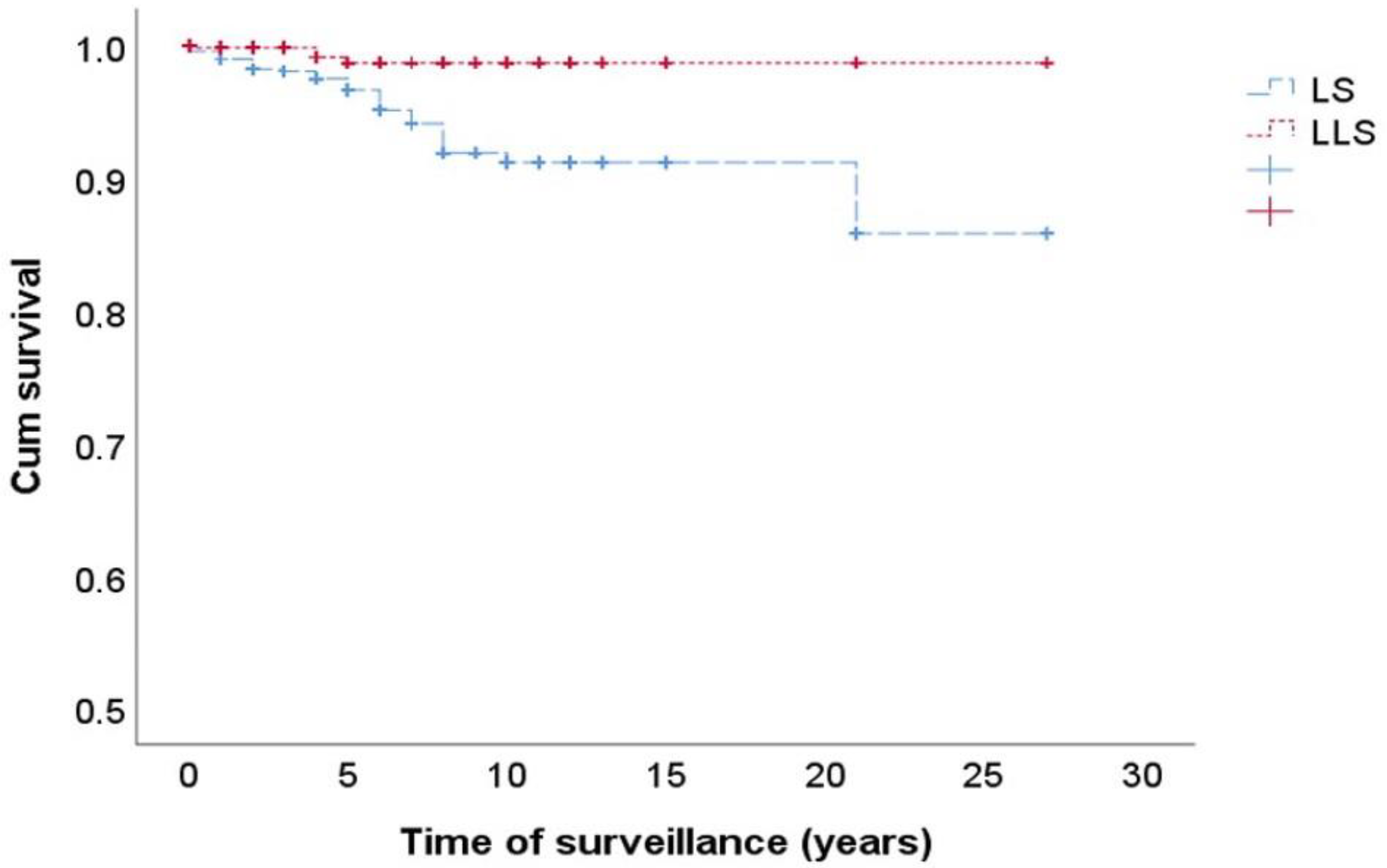
3.2. Risk of CRC in FDRs of LS and LLS Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CI | confidence interval |
| CRC | colorectal cancer |
| FDR | first-degree relatives |
| IHC | Immunohistochemistry |
| IQR | interquartile range |
| LLS | Lynch-like syndrome |
| LS | Lynch syndrome |
| MMR | mismatch repair |
| MSI | microsatellite instability |
| SD | standard deviation |
| SIR | standardized incidence ratios |
| TNM | Tumor, lymph Nodes, Metastasis |
References
- Lynch, H.; De La Chapelle, A. Hereditary Colorectal Cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Bonadona, V.; Bonaiti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.-J.; Caron, O.; et al. Cancer Risks Associated With Germline Mutations in MLH1, MSH2, and MSH6 Genes in Lynch Syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef]
- Boland, C.R.; Shike, M. Report From the Jerusalem Workshop on Lynch Syndrome-Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 2010, 138, 2197.e1–2197.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Carbonell, L.; Ruiz-Ponte, C.; Guarinos, C.; Alenda, C.; Payá, A.; Brea, A.; Egoavil, C.M.; Castillejo, A.; Bessa, X.; Xicola, R.M.; et al. Comparison between universal molecular screening for Lynch syndrome and revised Bethesda guidelines in a large population-based cohort of patients with colorectal cancer. Gut 2011, 61, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Niessen, R.C.; Vonk, J.; Westers, H.; Hofstra, R.M.; Sijmons, R.H. A database to support the interpretation of human mismatch repair gene variants. Hum. Mutat. 2008, 29, 1337–1341. [Google Scholar] [CrossRef] [PubMed]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.; Tavtigian, S.V.; et al. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, B.A.; Insight, O.B.O.; Spurdle, A.; Plazzer, J.-P.; Greenblatt, M.S.; Akagi, K.; Al-Mulla, F.; Bapat, B.; Bernstein, I.; Capella, G.; et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat. Genet. 2013, 46, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Tricarico, R.; Kasela, M.; Mareni, C.; Thompson, B.A.; Drouet, A.; Staderini, L.; Gorelli, G.; Crucianelli, F.; Ingrosso, V.; Kantelinen, J.; et al. Assessment of the InSiGHT Interpretation Criteria for the Clinical Classification of 24MLH1andMSH2Gene Variants. Hum. Mutat. 2016, 38, 64–77. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez–Soler, M.; Pérez–Carbonell, L.; Guarinos, C.; Zapater, P.; Castillejo, A.; Barbera, V.M.; Juarez, M.; Bessa, X.; Xicola, R.M.; Clofent, J.; et al. Risk of Cancer in Cases of Suspected Lynch Syndrome Without Germline Mutation. Gastroenterology 2013, 144, 926–932.e1. [Google Scholar] [CrossRef] [Green Version]
- Picó, M.D.; Castillejo, A.; Murcia, O.; Giner-Calabuig, M.; Alustiza, M.; Sánchez, A.; Moreira, L.; Pellise, M.; Castells, A.; Carrillo-Palau, M.; et al. Clinical and Pathological Characterization of Lynch-Like Syndrome. Clin. Gastroenterol. Hepatol. 2020, 18, 368–374.e1. [Google Scholar] [CrossRef]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; Chapelle, A.D.L.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and Microsatellite Instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Marzo, M.; Mascort, J.; Quintero, E.; García-Alfonso, P.; López-Ibor, C.; Castells, A.; Segura, P.P. Prevención del cáncer colorrectal. Gastroenterología y Hepatología 2009, 32, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, N.; Duval, A.; Reperant, M.; Vaury, C.; Furlan, D.; Leroy, K.; Seruca, R.; Iacopetta, B.; Hamelin, R. Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Gastroenterology 2002, 123, 1804–1811. [Google Scholar] [CrossRef] [PubMed]
- Xicola, R.M.; Llor, X.; Pons, E.; Castells, A.; Alenda, C.; Piñol, V.; Andreu, M.; Castellvi-Bel, S.; Payá, A.; Jover, R.; et al. Performance of Different Microsatellite Marker Panels for Detection of Mismatch Repair–Deficient Colorectal Tumors. J. Natl. Cancer Inst. 2007, 99, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Jover, R.; Alenda, C.; Poveda, M.J.; Peiró, G.; López, F.I.A.; Pérez-Mateo, M.; Payá, A. Defective Mismatch-Repair Colorectal Cancer Clinicopathologic Characteristics and Usefulness of Immunohistochemical Analysis for Diagnosis. Am. J. Clin. Pathol. 2004, 122, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Carbonell, L.; Alenda, C.; Payá, A.; Castillejo, A.; Barberá, V.M.; Guillen, C.; Rojas, E.; Acame, N.; Gutiérrez-Aviñó, F.J.; Castells, A.; et al. Methylation Analysis of MLH1 Improves the Selection of Patients for Genetic Testing in Lynch Syndrome. J. Mol. Diagn. 2010, 12, 498–504. [Google Scholar] [CrossRef]
- Benlloch, S.; Payá, A.; Alenda, C.; Bessa, X.; Andreu, M.; Jover, R.; Castells, A.; Llor, X.; Aranda, F.; Massutí, B. Detection of BRAF V600E Mutation in Colorectal Cancer. J. Mol. Diagn. 2006, 8, 540–543. [Google Scholar] [CrossRef] [Green Version]
- Chirlaque-Lopez, M.D. Murciasalud. Available online: https://www.murciasalud.es/ (accessed on 31 July 2019). (In Spanish).
- Breslow, N.E.; Day, N.E. Statistical methods in cancer research. IARC Workshop 25–27 May 1983. IARC Sci. Publ. 1987, 82, 1–406. [Google Scholar]
- Haraldsdottir, S.; Hampel, H.; Tomsic, J.; Frankel, W.L.; Pearlman, R.; De La Chapelle, A.; Pritchard, C.C. Colon and Endometrial Cancers With Mismatch Repair Deficiency Can Arise From Somatic, Rather Than Germline, Mutations. Gastroenterology 2014, 147, 1308–1316.e1. [Google Scholar] [CrossRef] [Green Version]
- Sijmons, R.H.; Greenblatt, M.S.; Genuardi, M. Gene variants of unknown clinical significance in Lynch syndrome. An introduction for clinicians. Fam. Cancer 2013, 12, 181–187. [Google Scholar] [CrossRef]
- Sourrouille, I.; Coulet, F.; Lefevre, J.H.; Colas, C.; Eyries, M.; Svrcek, M.; Bardier-Dupas, A.; Parc, Y.; Soubrier, F. Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Fam. Cancer 2012, 12, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Mensenkamp, A.R.; Vogelaar, I.; Van Zelst–Stams, W.A.; Goossens, M.; Ouchene, H.; Hendriks–Cornelissen, S.J.; Kwint, M.P.; Hoogerbrugge, N.; Nagtegaal, I.; Ligtenberg, M.J. Somatic Mutations in MLH1 and MSH2 Are a Frequent Cause of Mismatch-Repair Deficiency in Lynch Syndrome-Like Tumors. Gastroenterology 2014, 146, 643–646.e8. [Google Scholar] [CrossRef] [PubMed]
- Geurts-Giele, W.R.; Leenen, C.H.; Dubbink, H.J.; Meijssen, I.C.; Post, E.; Sleddens, H.F.; Kuipers, E.J.; Goverde, A.; Ouweland, A.M.V.D.; Van Lier, M.G.; et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J. Pathol. 2014, 234, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Pearlman, R.; Beightol, M.; Zhao, W.J.; Jones, D.; Frankel, W.L.; Goodfellow, P.J.; Yilmaz, A.; Miller, K.; Bacher, J.; et al. Assessment of Tumor Sequencing as a Replacement for Lynch Syndrome Screening and Current Molecular Tests for Patients With Colorectal Cancer. JAMA Oncol. 2018, 4, 806–813. [Google Scholar] [CrossRef]
- Pearlman, R.; Haraldsdottir, S.; De La Chapelle, A.; Jonasson, J.G.; Liyanarachchi, S.; Frankel, W.L.; Rafnar, T.; Stefansson, K.; Pritchard, C.C.; Hampel, H. Clinical characteristics of patients with colorectal cancer with double somatic mismatch repair mutations compared with Lynch syndrome. J. Med. Genet. 2019, 56, 462–470. [Google Scholar] [CrossRef]
- Hemminger, J.A.; Pearlman, R.; Haraldsdottir, S.; Knight, D.; Jónasson, J.G.; Pritchard, C.C.; Hampel, H.; Frankel, W.L. Histology of colorectal adenocarcinoma with double somatic mismatch-repair mutations is indistinguishable from those caused by Lynch syndrome. Hum. Pathol. 2018, 78, 125–130. [Google Scholar] [CrossRef]
- Mas-Moya, J.; Dudley, B.; Brand, R.E.; Thull, D.; Bahary, N.; Nikiforova, M.N.; Pai, R.K. Clinicopathological comparison of colorectal and endometrial carcinomas in patients with Lynch-like syndrome versus patients with Lynch syndrome. Hum. Pathol. 2015, 46, 1616–1625. [Google Scholar] [CrossRef]
- Win, A.K.; Buchanan, D.D.; Rosty, C.; Maclnnis, R.; Dowty, J.G.; Dite, G.; Giles, G.G.; Southey, M.C.; Young, J.P.; Clendenning, M.; et al. Role of tumour molecular and pathology features to estimate colorectal cancer risk for first-degree relatives. Gut 2014, 64, 101–110. [Google Scholar] [CrossRef]
- Carayol, J.; Khlat, M.; Maccario, J.; Bonaïti-Pellié, C. Hereditary non-polyposis colorectal cancer: Current risks of colorectal cancer largely overestimated. J. Med. Genet. 2002, 39, 335–339. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristics of Patients | LS, n = 286 | LLS, n = 160 |
|---|---|---|
| Female sex | 132 (46.1%) | 66 (41.3%) |
| Age at CRC diagnosis, median (SD) | 48.1 (SD 12.9) | 54.9 (SD 14.2) * |
| Reason for IHC | ||
| Amsterdam I and II criteria | 206 (72.1%) | 18 (11.2%) * |
| Revised Bethesda guidelines | 234 (84.2%) | 103 (64.4%) * |
| Immunohistochemistry (IHC), n (%) | ||
| Loss of MLH1 and PMS2 | 98 (34.3) | 77 (48.1) * |
| Loss of MSH2 and MSH6 | 122 (42.6) | 43 (26.9) * |
| Isolated loss of MSH6 | 40 (14) | 20 (12.5) |
| Isolated loss of PMS2 | 18 (6.3) | 14 (8.8) |
| IHC non-available; MSI-H | 8 (2.8) | 6 (3.7) |
| Location | ||
| Right colon | 165 (62.7%) | 89 (61.4%) |
| Left colon and rectum | 98 (37.3%) | 56 (38.6%) |
| TNM | ||
| Stage I and II | 159 (55.6%) | 80 (60.1%) |
| Histology | ||
| Poor differentiation | 50 (17.5%) | 33 (20.6%) |
| Lymphocytic infiltration | 41 (14.3%) | 37 (23.1%) * |
| Mucinous tumor | 88 (30.8%) | 46 (28.7%) |
| Vascular invasion | 35 (12.2%) | 18 (11.3%) |
| Personal history | ||
| metachronous CRC | 37 (12.9%) | 5 (3.1%) * |
| synchronous CRC | 26 (9.1%) | 2 (1.3%) * |
| non-CRC LS tumor | 84 (29.4%) | 5 (3.1%) * |
| Neoplasms Associated with LS | LS n = 1205 | LLS n = 698 | p Value | ||
|---|---|---|---|---|---|
| Number of Tumors | SIR (95% CI) | Number of Tumors | SIR (95% CI) | ||
| CRC | 191 | 4.25 (3.67–4.90) | 54 | 2.08 (1.56–2.71) | 0.0000 |
| Non-CRC LS-associated tumor | 161 | 5.01 (4.26–5.84) | 38 | 2.04 (1.44–2.80) | 0.0000 |
| TOTAL | 352 | 4.57 (4.10–5.07) | 92 | 2.06 (1.66–2.53) | 0.0000 |
| Location of the Tumors | LS Families | LLS Families | p Value |
|---|---|---|---|
| Number of Tumors: 184 | Number of Tumors: 40 | ||
| Ovary | 20 (10.9%) | 5 (12.5%) | n.s. |
| Endometrium | 89 (48.4%) | 8 (20.0%) | 0.001 |
| Pancreas | 6 (3.3%) | 6 (15.0%) | 0.003 |
| Stomach | 37 (20.1%) | 11 (27.5%) | n.s. |
| Urinary tract | 10 (5.4%) | 4 (10%) | n.s. |
| Skin | 3 (1.6%) | 2 (5%) | n.s. |
| Small intestine | 5 (2.7%) | 0 (0%) | n.s. |
| Brain | 6 (3.3%) | 2 (5%) | n.s. |
| Biliary tract | 8 (4.3%) | 2 (5%) | n.s. |
| Neoplasms Associated with LS | LS n = 1126 | LLS n = 587 | p Value |
|---|---|---|---|
| No. of Tumors | No. of Tumors | ||
| CRC | 22 (1.9%) | 3 (0.5%) | 0.019 |
| Non-CRC LS-associated tumor | 23 (2%) | 2 (0.3%) | 0.006 |
| TOTAL | 45 (3.9%) | 5 (0.8%) | 0.000 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picó, M.D.; Sánchez-Heras, A.B.; Castillejo, A.; Giner-Calabuig, M.; Alustiza, M.; Sánchez, A.; Moreira, L.; Pellise, M.; Castells, A.; Llort, G.; et al. Risk of Cancer in Family Members of Patients with Lynch-Like Syndrome. Cancers 2020, 12, 2225. https://doi.org/10.3390/cancers12082225
Picó MD, Sánchez-Heras AB, Castillejo A, Giner-Calabuig M, Alustiza M, Sánchez A, Moreira L, Pellise M, Castells A, Llort G, et al. Risk of Cancer in Family Members of Patients with Lynch-Like Syndrome. Cancers. 2020; 12(8):2225. https://doi.org/10.3390/cancers12082225
Chicago/Turabian StylePicó, María Dolores, Ana Beatriz Sánchez-Heras, Adela Castillejo, Mar Giner-Calabuig, Miren Alustiza, Ariadna Sánchez, Leticia Moreira, María Pellise, Antoni Castells, Gemma Llort, and et al. 2020. "Risk of Cancer in Family Members of Patients with Lynch-Like Syndrome" Cancers 12, no. 8: 2225. https://doi.org/10.3390/cancers12082225