Reduced SLIT2 is Associated with Increased Cell Proliferation and Arsenic Trioxide Resistance in Acute Promyelocytic Leukemia

 , , , , , , , ,
, , , , , , , ,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
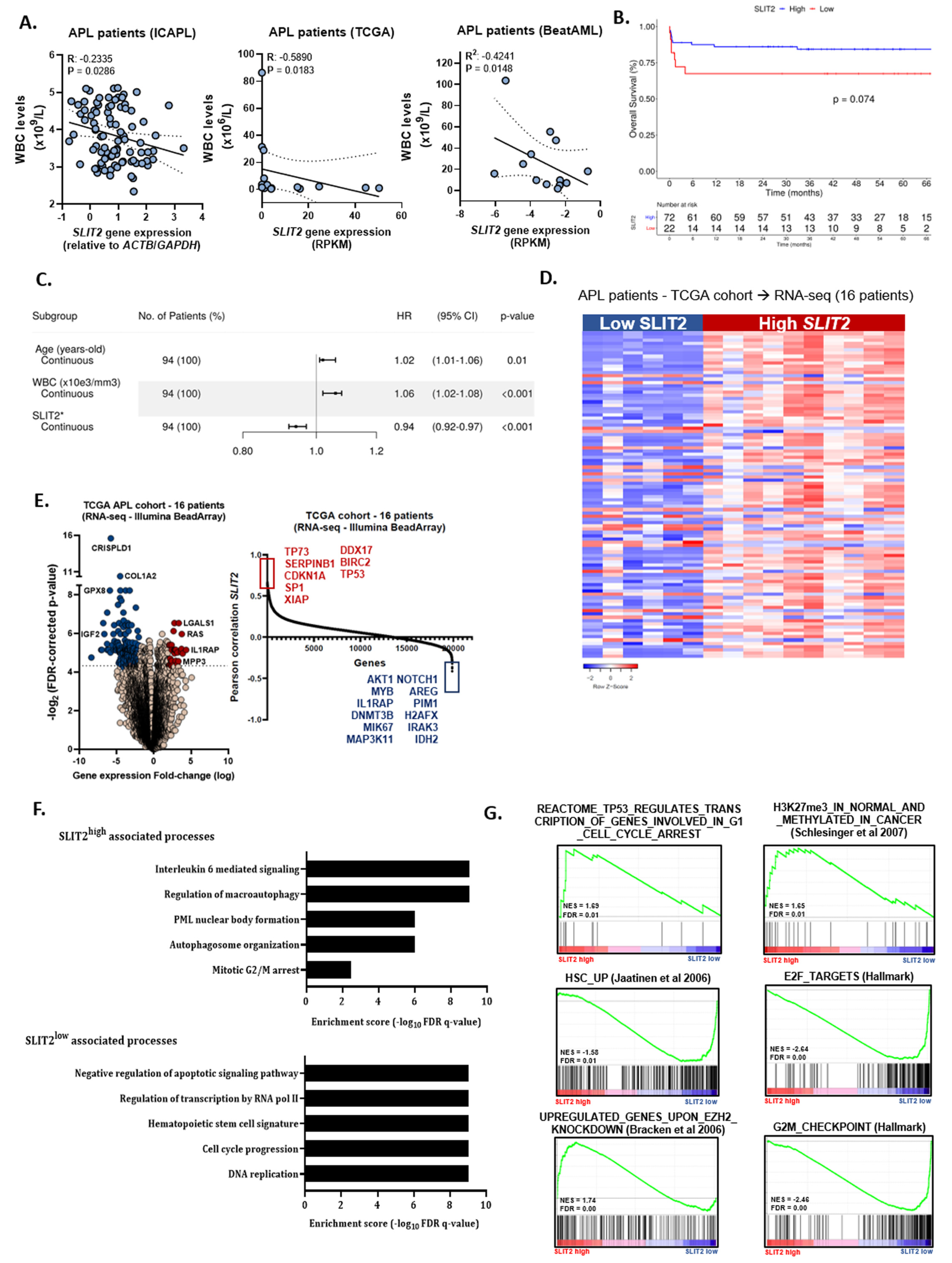
2.1. Low SLIT2 Transcript Levels Predict Lower Overall Survival in APL Characterized by a More Aggressive Course of Disease Progression
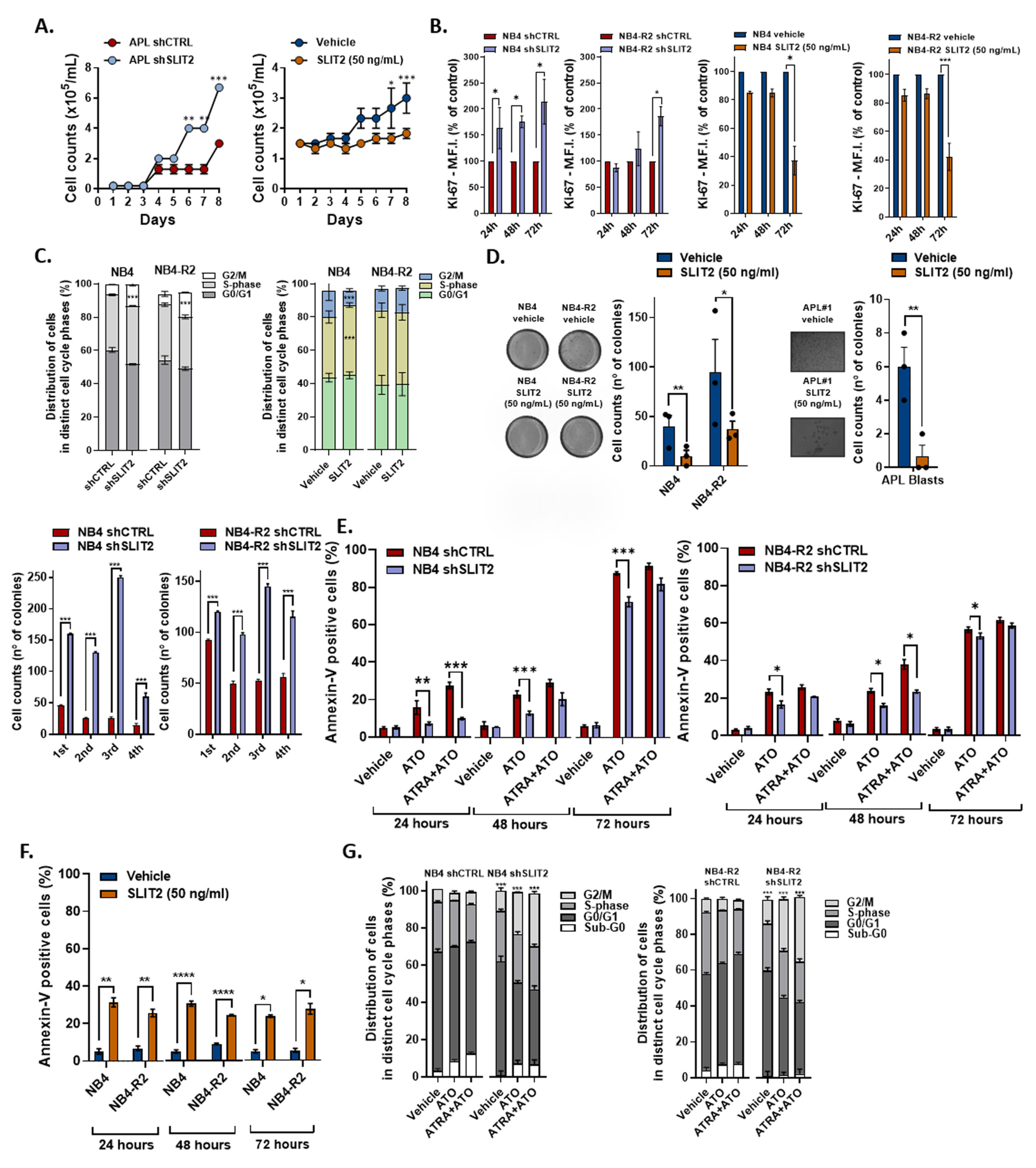
2.2. SLIT2 Significantly Impacts APL Cell Proliferation and Cell Cycle Progression
2.3. SLIT2 Impacts on APL Cell Viability and APL Drug Induced Apoptosis
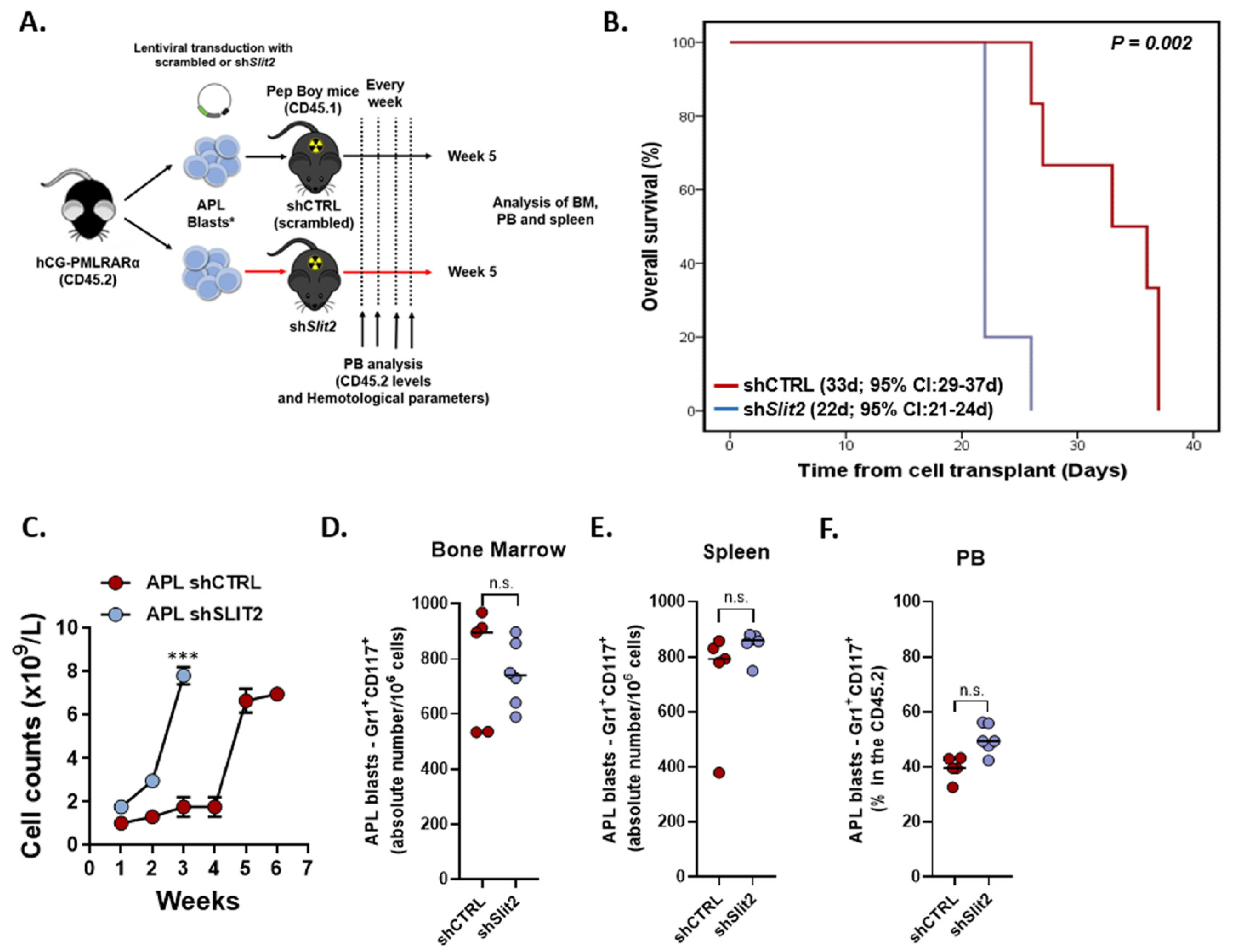
2.4. Slit2 Knockdown was Associated with Increased Leukocyte Count and Decreased OS in an APL Knockin Murine Model
3. Discussion
4. Materials and Methods
4.1. Patients Samples
4.2. Gene Expression Profile Using Public Datasets
4.3. Gene Set Enrichment Analysis (GSEA) for SLIT2 Biological Pathways in APL
4.4. Differential Expression Analysis
4.5. Gene Expression Profile of SLIT2 in APL Patients
4.6. Cell Lines and Drugs
4.7. Lentiviral Vectors and Lentivirus Production
4.8. In Vitro Assays
4.9. Quantitative PCR
4.10. Generation of PML-RARA Knockin-Induced APL Mice by Bone Marrow Transplantation
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhou, B.B.S.; Zhang, H.; Damelin, M.; Geles, K.G.; Grindley, J.C.; Dirks, P.B. Tumour-initiating cells: Challenges and opportunities for anticancer drug discovery. Nat. Rev. Drug Discov. 2009, 8, 806–823. [Google Scholar] [CrossRef]
- Jiang, Z.; Liang, G.; Xiao, Y.; Qin, T.; Chen, X.; Wu, E.; Ma, Q.; Wang, Z. Targeting the SLIT/ROBO pathway in tumor progression: Molecular mechanisms and therapeutic perspectives. Ther. Adv. Med. Oncol. 2019, 11, 1–14. [Google Scholar] [CrossRef]
- Huang, T.; Kang, W.; Cheng, A.S.L.; Yu, J.; To, K.F. The emerging role of Slit-Robo pathway in gastric and other gastro intestinal cancers. BMC Cancer 2015, 15, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Gu, F.; Ma, Y.; Zhang, J.; Qin, F.; Fu, L. Function of Slit/Robo signaling in breast cancer. Front. Med. 2015, 9, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Blockus, H.; Chédotal, A. Slit-robo signaling. Development 2016, 143, 3037–3044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gołos, A.; Jesionek-Kupnicka, D.; Gil, L.; Braun, M.; Komarnicki, M.; Robak, T.; Wierzbowska, A. The Expression of the SLIT–ROBO Family in Adult Patients with Acute Myeloid Leukemia. Arch. Immunol. Ther. Exp. (Warsz.) 2019, 67, 109–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N. Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.J.; et al. Management of acute promyelocytic leukemia: Updated recommendations from an expert panel of the European Leukemia. Net. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amodeo, V.; Deli, A.; Betts, J.; Bartesaghi, S.; Zhang, Y.; Richard-Londt, A.; Ellis, M.; Roshani, R.; Vouri, M.; Galavotti, S.; et al. A PML/Slit Axis Controls Physiological Cell Migration and Cancer Invasion in the CNS. Cell Rep. 2017, 20, 411–426. [Google Scholar] [CrossRef] [Green Version]
- Ablain, J.; Rice, K.; Soilihi, H.; De Reynies, A.; Minucci, S.; De Thé, H. Activation of a promyelocytic leukemia-tumor protein 53 axis underlies acute promyelocytic leukemia cure. Nat. Med. 2014, 20, 167–174. [Google Scholar] [CrossRef]
- Bagger, F.O.; Kinalis, S.; Rapin, N. BloodSpot: A database of healthy and malignant haematopoiesis updated with purified and single cell mRNA sequencing profiles. Nucleic Acids Res. 2019, 47, D881–D885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rego, E.M.; Kim, H.T.; Ruiz-Argüelles, G.J.; Undurraga, M.S.; Del Rosario Uriarte, M.; Jacomo, R.H.; Gutiérrez-Aguirre, H.; Melo, R.A.M.; Bittencourt, R.; Pasquini, R.; et al. Improving acute promyelocytic leukemia (APL) outcome in developing countries through networking, results of the International Consortium on APL. Blood 2013, 121, 1935–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gara, R.K.; Kumari, S.; Ganju, A.; Yallapu, M.M.; Jaggi, M.; Chauhan, S.C. Slit/Robo pathway: A promising therapeutic target for cancer. Drug Discov. Today 2015, 20, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Rego, E.M.; Ruggero, D.; Tribioli, C.; Cattoretti, G.; Kogan, S.; Redner, R.L.; Pandolfi, P.P. Leukemia with distinct phenotypes in transgenic mice expressing PML/RARα, PLZF/RARα or NPM/RARα. Oncogene 2006, 25, 1974–1979. [Google Scholar] [CrossRef] [Green Version]
- Mukai, M.; Kishima, K.; Fukumitsu, H.; Sekido, Y.; Izumi, H.; Hoshikawa, T.; Tajima, T.; Tobita, K.; Sadahiro, S.; Yasuda, S.; et al. Is the T1/2N1 (≤3 nodes) category actually stage III (TNM)/IIIa (Japanese classification) in patients with primary colorectal cancer? Oncol. Rep. 2011, 26, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Yang, X.; Deng, X.; Zhang, X.; Li, P.; Tao, J.; Lu, Q. MicroRNA-218 inhibits bladder cancer cell proliferation, migration, and invasion by targeting BMI-1. Tumor Biol. 2015, 36, 8015–8023. [Google Scholar] [CrossRef] [PubMed]
- Callens, C.; Chevret, S.; Cayuela, J.M.; Cassinat, B.; Raffoux, E.; de Botton, S.; Thomas, X.; Guerci, A.; Fegueux, N.; Pigneux, A.; et al. Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): A retrospective study from the European APL Group. Leukemia 2005, 19, 1153–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, J.H.A.; Brinkman, A.B.; Simmer, F.; Francoijs, K.J.; Nebbioso, A.; Ferrara, F.; Altucci, L.; Stunnenberg, H.G. PML-RARα/RXR Alters the Epigenetic Landscape in Acute Promyelocytic Leukemia. Cancer Cell 2010, 17, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Zhang, B.; Fan, W.; Zhao, Q.; Yang, L.; Xin, W.; Fu, D. Identification of prognostic genes in the acute myeloid leukemia microenvironment. Aging 2019, 11, 10557–10580. [Google Scholar] [CrossRef]
- Kim, J.; Lee, Y.; Lu, X.; Song, B.; Fong, K.W.; Cao, Q.; Licht, J.D.; Zhao, J.C.; Yu, J. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep. 2018, 25, 2808–2820.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.A.; Petraglia, F.; Nebbioso, A.; Yi, G.; Conte, M.; Valente, S.; Mandoli, A.; Scisciola, L.; Lindeboom, R.; Kerstens, H.; et al. Multi-omics profiling reveals a distinctive epigenome signature for high-risk acute promyelocytic leukemia. Oncotarget 2018, 9, 25647–25660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alajez, N.M.; Lenarduzzi, M.; Ito, E.; Hui, A.B.Y.; Shi, W.; Bruce, J.; Yue, S.; Huang, S.H.; Xu, W.; Waldron, J.; et al. miR-218 suppresses nasopharyngeal cancer progression through downregulation of survivin and the SLIT2-ROBO1 pathway. Cancer Res. 2011, 71, 2381–2391. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sun, H.H.; Sui, M.H.; Ma, J.J. miR-218 inhibits acute promyelocytic leukemia cell growth by targeting BMI-1. Oncol. Lett. 2017, 14, 8078–8083. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Jiang, S.X.; Yang, Y.M.; Xu, H.; Liu, J.L.; Wang, X.S. USP22 Acts as an Oncogene by the Activation of BMI-1-Mediated INK4a/ARF Pathway and Akt Pathway. Cell Biochem. Biophys. 2012, 62, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Ojo, D.; Wei, F.; Wong, N.; Gu, Y.; Tang, D. A novel aspect of tumorigenesis—BMI1 functions in regulating DNA damage response. Biomolecules 2015, 5, 3396–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, S.; Luo, M.; Sun, H.; Yu, X.; Shen, M.; Zhang, Q.; Zhou, R.; Ju, X.; Tao, W.; Liu, D.; et al. Identification and characterization of Bmi-1-responding element within the human p16 promoter. J. Biol. Chem. 2010, 285, 33219–33229. [Google Scholar] [CrossRef] [Green Version]
- Cells, S.; Rizo, A.; Olthof, S.; Han, L.; Vellenga, E.; Haan, G.; De Schuringa, J.J.; Biology, C.; Stem, S.; Biology, C.; et al. Repression of BMI1 in normal and leukemic human CD34. Blood 2016, 114, 1498–1506. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Yang, Z.; Liu, W.; Liu, B.; Xu, Z.; Zhang, Z. Knockdown of Slit2 promotes growth and motility in gastric cancer cells via activation of AKT/β-catenin. Oncol. Rep. 2014, 31, 812–818. [Google Scholar] [CrossRef]
- Prasad, A.; Paruchuri, V.; Preet, A.; Latif, F.; Ganju, R.K. Slit-2 induces a tumor-suppressive effect by regulating β-catenin in breast cancer cells. J. Biol. Chem. 2008, 283, 26624–26633. [Google Scholar] [CrossRef] [Green Version]
- Thomé, C.H.; Ferreira, G.A.; Pereira-Martins, D.A.; dos Santos, G.A.; Ortiz, C.A.; de Souza, L.E.B.; Sobral, L.M.; Silva, C.L.A.; Scheucher, P.S.; Gil, C.D.; et al. NTAL is associated with treatment outcome, cell proliferation and differentiation in acute promyelocytic leukemia. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, G.; Hoadley, K.; Triche, T.J.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.W.; Wouters, B.J.; Erpelinck, C.A.J.; Abbas, S.; Beverloo, H.B.; Lugthart, S.; Löwenberg, B.; Delwel, R.; Valk, P.J.M. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica 2009, 94, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucena-Araujo, A.R.; Pereira-Martins, D.A.; Koury, L.C.; Franca-Neto, P.L.; Coelho-Silva, J.L.; de Deus Wagatsuma, V.M.; Melo, R.A.M.; Bittencourt, R.; Pagnano, K.; Pasquini, R.; et al. Clinical impact of BAALC expression in high-risk acute promyelocytic leukemia. Blood Adv. 2017, 1, 1807–1814. [Google Scholar] [CrossRef] [Green Version]
- Lucena-Araujo, A.R.; Coelho-Silva, J.L.; Pereira-Martins, D.A.; Thome, C.; Scheucher, P.S.; Lange, A.P.; Paiva, H.H.; Hemmelgarn, B.T.; Morais-Sobral, M.C.; Azevedo, E.A.; et al. DeltaNp73 overexpression promotes resistance to apoptosis but does not cooperate with PML/RARA in the induction of an APL-leukemic phenotype. Oncotarget 2017, 8, 8475–8483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Root, D.E.; Hacohen, N.; Hahn, W.C.; Lander, E.S.; Sabatini, D.M. Genome-scale loss-of-function screening with a lentiviral RNAi library. Nat. Methods 2006, 3, 715–719. [Google Scholar] [CrossRef]
- Gaillard, C.; Surianarayanan, S.; Bentley, T.; Warr, M.R.; Fitch, B.; Geng, H.; Passegué, E.; De Thé, H.; Kogan, S.C. Identification of IRF8 as a potent tumor suppressor in murine acute promyelocytic leukemia. Blood Adv. 2018, 2, 2462–2466. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Hsieh, M.K.; Chang, W.Y.; Chiang, A.J.; Chen, J. Determining the optimal number and location of cutoff points with application to data of cervical cancer. PLoS ONE 2017, 12, e0176231. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Characteristic | Characteristic | All Patients | SLIT2 Expression | p Value | ||||
|---|---|---|---|---|---|---|---|---|
| Low Expression | High Expression | |||||||
| No. | % | No. | % | No. | % | |||
| Gender | Female | 47 | 50 | 14 | 60.9 | 33 | 46.5 | 0.337 |
| Male | 47 | 50 | 9 | 39.1 | 38 | 53.5 | ||
| Age, median | 35.7 | 39.4 | 33.6 | 0.482 | ||||
| (range) | (18.9, 73.6) | (19, 65.43) | (18.9, 73.6) | |||||
| ECOG performance status | 0 | 59 | 62.8 | 11 | 47.8 | 48 | 67.6 | 0.243 |
| 1 | 14 | 14.9 | 6 | 26.1 | 8 | 11.3 | ||
| 2 | 12 | 12.8 | 4 | 17.4 | 8 | 11.3 | ||
| ≥3 | 9 | 9.6 | 2 | 8.7 | 7 | 9.9 | ||
| Relapse-risk group | Unknown | - | - | - | - | - | - | |
| Low risk | 16 | 17 | 2 | 8.7 | 14 | 19.7 | 0.307 | |
| Intermediate risk | 40 | 42.6 | 9 | 39.1 | 31 | 43.7 | ||
| High risk | 38 | 40.4 | 12 | 52.2 | 26 | 36.6 | ||
| FLT3-mutational status | Mutated | 14 | 19.7 | 5 | 29.4 | 9 | 16.7 | 0.299 |
| Non-mutated | 57 | 80.3 | 12 | 70.6 | 45 | 83.3 | ||
| Unknown | 23 | - | 6 | - | 17 | - | ||
| PML breakpoint | BCR1 | 55 | 65.5 | 10 | 47.6 | 45 | 71.4 | 0.061 |
| BCR2 | 2 | 2.4 | - | - | 2 | 3.2 | ||
| BCR3 | 27 | 32.1 | 11 | 52.4 | 16 | 25.4 | ||
| Unknown | 10 | - | 2 | - | 8 | - | ||
| WBC counts (×109/L), median | 6.78 | 15.02 | 3.3 | 0.024 * | ||||
| (range) | (0.22, 132.5) | (0.89, 126.8) | (0.22, 132.5) | |||||
| Platelet counts (×109/L), median | 26.7 | 28 | 24 | 0.329 | ||||
| (range) | (4, 128) | (4, 92) | (4, 128) | |||||
| Hemoglobin (g/dL), median | 8.8 | 8.6 | 8.8 | 0.898 | ||||
| (range) | (3.4, 14.1) | (4.7, 13.3) | (3.4, 14.1) | |||||
| Creatinine (mg/dL), median | 0.8 | 0.9 | 0.8 | 0.738 | ||||
| (range) | (0.42, 2.2) | (0.42, 1.88) | (0.5, 2.2) | |||||
| Uric acid (mg/dL), median | 3.9 | 4.2 | 3.7 | 0.099 | ||||
| (range) | (1.1, 9) | (2.5, 7.1) | (1.1, 9) | |||||
| Fibrinogen (mg/dL), median | 159.5 | 148 | 165 | 0.082 | ||||
| (range) | (10, 605) | (48, 271) | (10, 605) | |||||
| Albumin, g/dL | 4 | 4.5 | 4 | 0.115 | ||||
| (range) | (2.2, 5.3) | (2.9, 5.2) | (2.2, 5.3) | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weinhäuser, I.; Pereira-Martins, D.A.; Ortiz, C.; Silveira, D.R.; Simões, L.A.A.; Bianco, T.M.; Araujo, C.L.; Koury, L.C.; Melo, R.A.M.; Bittencourt, R.I.; et al. Reduced SLIT2 is Associated with Increased Cell Proliferation and Arsenic Trioxide Resistance in Acute Promyelocytic Leukemia. Cancers 2020, 12, 3134. https://doi.org/10.3390/cancers12113134
Weinhäuser I, Pereira-Martins DA, Ortiz C, Silveira DR, Simões LAA, Bianco TM, Araujo CL, Koury LC, Melo RAM, Bittencourt RI, et al. Reduced SLIT2 is Associated with Increased Cell Proliferation and Arsenic Trioxide Resistance in Acute Promyelocytic Leukemia. Cancers. 2020; 12(11):3134. https://doi.org/10.3390/cancers12113134
Chicago/Turabian StyleWeinhäuser, Isabel, Diego A. Pereira-Martins, Cesar Ortiz, Douglas R. Silveira, Luíse A. A. Simões, Thiago M. Bianco, Cleide L. Araujo, Luisa C. Koury, Raul A. M. Melo, Rosane I. Bittencourt, and et al. 2020. "Reduced SLIT2 is Associated with Increased Cell Proliferation and Arsenic Trioxide Resistance in Acute Promyelocytic Leukemia" Cancers 12, no. 11: 3134. https://doi.org/10.3390/cancers12113134