Cellular and Synaptic Dysfunctions in Parkinson’s Disease: Stepping Out of the Striatum
by
 and
and
Nicolas Mallet
1,2, 
Lorena Delgado
1,2,3,
Marine Chazalon
4,
Cristina Miguelez
3 and
Jérôme Baufreton
1,2,* 1
Université de Bordeaux, Institut des Maladies Neurodégénératives, F-33000 Bordeaux, France
2
CNRS UMR 5293, Institut des Maladies Neurodégénératives, F-33000 Bordeaux, France
3
Department of Pharmacology, University of the Basque Country (UPV/EHU), 48940 Leioa, Spain
4
Laboratory of Neurophysiology, ULB Neuroscience Institute, Université Libre de Bruxelles, 1070 Brussels, Belgium
*
Author to whom correspondence should be addressed.
Cells 2019, 8(9), 1005; https://doi.org/10.3390/cells8091005
Submission received: 31 July 2019
/
Revised: 28 August 2019
/
Accepted: 29 August 2019
/
Published: 29 August 2019
(This article belongs to the Special Issue The Molecular and Cellular Basis for Parkinson's Disease 2019)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The basal ganglia (BG) are a collection of interconnected subcortical nuclei that participate in a great variety of functions, ranging from motor programming and execution to procedural learning, cognition, and emotions. This network is also the region primarily affected by the degeneration of midbrain dopaminergic neurons localized in the substantia nigra pars compacta (SNc). This degeneration causes cellular and synaptic dysfunctions in the BG network, which are responsible for the appearance of the motor symptoms of Parkinson’s disease. Dopamine (DA) modulation and the consequences of its loss on the striatal microcircuit have been extensively studied, and because of the discrete nature of DA innervation of other BG nuclei, its action outside the striatum has been considered negligible. However, there is a growing body of evidence supporting functional extrastriatal DA modulation of both cellular excitability and synaptic transmission. In this review, the functional relevance of DA modulation outside the striatum in both normal and pathological conditions will be discussed.
1. Introduction
The basal ganglia (BG) participate in a great variety of functions including motor programming and execution, procedural learning, cognition, and emotions [1,2]. Dopaminergic innervation provided by mesencephalic dopamine (DA) neurons from the substantia nigra pars compacta (SNc) plays an essential role in the control of BG functions by modulating cellular and synaptic properties at each stage of the BG network. DA modulation of the striatal microcircuit has been extensively studied over the past decades and has been instrumental to better understand striatal function in health and disease states (for reviews, see [3,4]). The degeneration of midbrain DA neurons and the subsequent loss of DA in the BG trigger cellular and synaptic alterations, which are believed to be responsible for the appearance of the motor symptoms of Parkinson’s disease (PD). Dopaminergic innervation of the striatum (STR) is acknowledged to be by far denser than in the rest of the BG. Nevertheless, there is a growing body of evidences supporting the existence of discrete, functional dopaminergic innervation of extra-striatal nuclei (ESN), namely the subthalamic nucleus (STN), the internal and external segments of the globus pallidus (GPi and GPe, respectively) and the substantia nigra pars reticulata (SNr) [5]. In line with this evidence, the action exerted by DA on ESN has been investigated only recently and is the focus of the present review. After a brief description of BG network organization, we will discuss recent discoveries supporting a functional role of DA outside the striatum in both healthy and disease conditions.
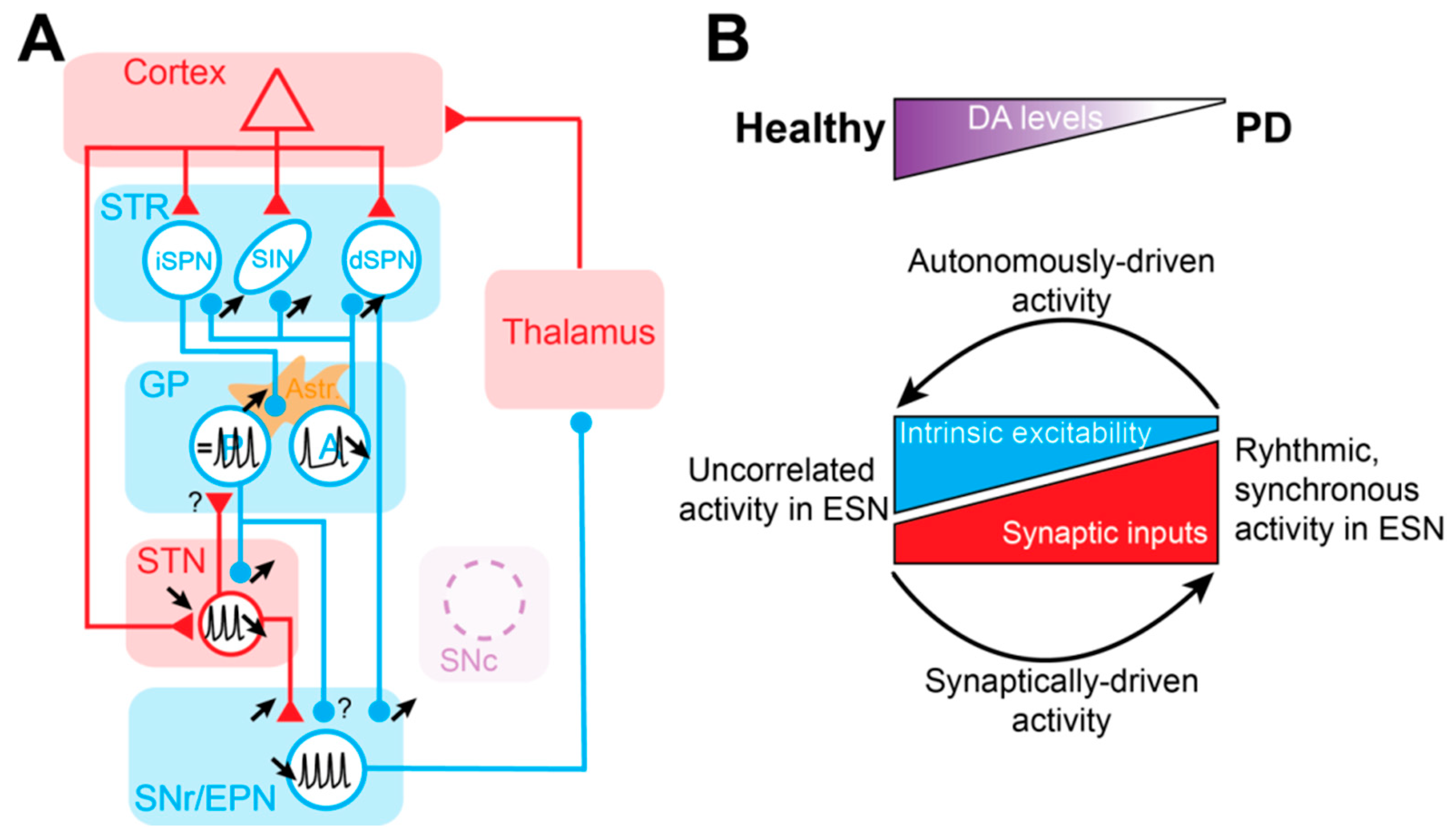
The STR is the main entry structure of the BG and the principal recipient of cortical inputs. This GABAergic nucleus consists of 95% projection neurons, recognizable by their small soma and their dendrites covered by spines—therefore called spiny projection neurons (SPN). The remaining 5% of the STR is composed of several classes of GABAergic and cholinergic interneurons [6,7]. SPNs give rise to two pathways that propagate cortical information directly or indirectly to the output of the BG. SPNs of the direct pathway (dSPNs) express dopamine D1 receptors (D1Rs) and primarily innervate BG output neurons of the SNr and GPi (called the entopeduncular nucleus in rodents, EPN) in a monosynaptic manner. On the other hand, SPNs of the indirect pathway (iSPNs) express dopamine D2 receptors (D2Rs) and project to BG outputs via a polysynaptic route involving the GPe (called the globus pallidus in rodents, GP) and the STN. As predicted by the anatomical-functional model of the BG [8], it has been shown experimentally that these two pathways exert opposite control over motor execution [9] (Figure 1). However, this view of opposite functions of the direct and the indirect pathways has been challenged by a study showing that both iSPNs and dSPNs are co-activated before movement initiation [10]. This coordinated activity has been interpreted as dSPN activation representing a process of selection/activation of a desired movement, while simultaneously activating iSPNs provides global suppression of alternative/unwanted actions [11]. In addition to these two pathways, multiple loops within the BG network, such as the hyperdirect cortico-subthalamic pathway [12], the reciprocally-connected GP-STN loop [13], the pallido-striatal pathway [14,15], and bridging collaterals from dSPNs in the GP [16,17], certainly contribute to normal and pathological operations of the BG network (Figure 1).
The aim and scope of the present review are to provide a comprehensive overview of the current literature regarding the role of DA in the modulation of ESN activity in both healthy conditions and in PD.
2. Extra-Striatal Nuclei Neurons Share Specific Features
2.1. ESN Neurons are Fast-Spiking Autonomous Pacemakers
A striking feature of ESN neurons compared to SPN is their ability to fire action potentials (APs) in a completely autonomous manner (i.e., in absence of excitatory synaptic inputs). While several voltage-dependent and voltage-independent conductances contribute to maintaining the membrane potential of ESN neurons depolarized, persistent sodium and Kv3-family potassium channels appear to play a critical role in the capability of GP, STN, EPN, and SNr neurons to perpetually fire APs at a high rate [18,19,20,21,22,23]. Because of their very hyperpolarized potential [24,25], SPNs need to receive convergent excitatory inputs to fire APs and convey information downwards through the BG network [26,27,28,29]. In contrast, because ESN are autonomous pacemakers, their spiking activity is instead independent of excitatory inputs [30]. Therefore, information encoding and transfer by these neurons relies more on changes in their firing rate and pattern rather than on spiking activity per se.
2.2. GABAergic Transmission Efficiently Sculpts the Activity of ESN Neurons
Another interesting advantage of pacemaking for neurons is the emerging properties related to AP timing relative to incoming afferent inputs [31,32,33]. Most of the ESN except for the STN, the only glutamatergic nucleus of the network, are inhibitory nuclei. Therefore, GABAergic transmission, through the activation of ionotropic GABAA and metabotropic GABAB receptors, is certainly the most efficient system to control the firing of neurons and induces profound changes in their firing rate and pattern [34,35]. Hyperpolarization of the membrane potential resets ESN neuron pacemaking activity and engages voltage-gated channels. Activation or deactivation of those channels will transiently modify the intrinsic excitability of ESN neurons following the inhibition [21,36,37,38], leading to complex sequences of activity. These interactions between synaptic inhibitory inputs and voltage-gated channels can contribute to the emergence of transiently correlated and synchronous activity within and among the different nuclei, both in healthy and pathological states (see Section 7).
2.3. ESN Receive Functional Dopaminergic Innervation
In the brain, the STR is by far the principal recipient of dopaminergic inputs arising from the SNc [39]. The dopaminergic modulation of the striatal microcircuit has been well characterized and is the subject of many reviews [40,41], so it will not be covered here. Even if, in comparison, dopaminergic innervation of the ESN can appear negligible compared to the STR, there are several lines of evidence supporting functional extra-striatal dopaminergic innervation. Dopaminergic fibers and synapses have been detected in the GP and the STN using light and electron microscopy [42,43,44,45]. DA release has also been measured in the STN upon electrical stimulation of dopaminergic inputs using fast-scan voltammetry [43]. In addition, DA receptors are expressed in all ESN (for a review, see [46]), and local applications of dopaminergic receptor agonists and antagonists locally in various ESN affect the neuronal activity of these nuclei [47,48,49,50]. All these data suggest that DA participates in the normal function of the ESN by regulating both cellular excitability and synaptic transmission.
3. The Globus Pallidus
3.1. Neuronal Diversity in the GP
The GP has long been considered as a simple relay nucleus in the indirect pathway of the BG despite anatomical/immunohistochemical and functional/electrophysiological studies suggesting a certain degree of diversity in this nucleus [51,52,53,54,55,56,57]. More recent investigations have revealed the molecular [58,59] and functional complexity [60,61,62] of the GP. Several cell types have been identified based on the expression of specific molecular markers and electrophysiological properties [63,64,65,66]. Even if a unified nomenclature is still missing [67], two main clusters of neurons are emerging from single-cell RNA profiling [59]. The first cluster is composed of the so-called prototypic GP neurons which project to downstream nuclei (the STN, the SNr, and the EPN) but also to the STR [63,68,69]. These neurons express the transcription factor (TF) Nkx2.1 and show a regular fast-spiking discharge ex vivo [63,64]. This population represents more than 70% of the total number of neurons in the mouse and the rat GP and include of a subgroup, representing approximately ¾ of prototypic GP neurons, which also express the calcium binding protein parvalbumin (PV) in addition to Nkx2.1 [63,70]. The second cluster is made of arkypallidal GP neurons, which were described in the seminal study of Mallet and colleagues [14]. This cell type, which represents between 15 to 28% of GP neurons depending on the species studied [63,70], is characterized by the selective expression of the TF FoxP2 and a low and irregular firing discharge. The main distinguishing feature of arkypallidal neurons is their exclusive and massive axonal projections to the STR, which certainly constitutes the main external source of GABA of this nucleus [14]. It is interesting to note here, that all arkypallidal neurons co-express the TF NPas-1, but this marker is not specific to this neuronal population, as it is also found in a small proportion of prototypic GP neurons [63,65,70].
The activity of prototypic and arkypallidal neurons has been characterized in vivo under anesthesia both in the DA-intact (DI) and the DA-depleted (DD) rat [63,71]. In anesthetized rodents, prototypic neurons are the population most affected by DD, with the appearance of pauses in their activity leading to a reduced mean firing rate compared to the DI rat [63]. Several studies suggest that the emergence of these pauses is due to an exaggerated drive from iSPNs [72,73,74], maybe because iSPN preferentially send projections to prototypic neurons [75]. Together, these observations fit with the hypoactivity of GP neurons predicted by the anatomo-functional model of the BG [8]. In contrast, arkypallidal neuron activity seems less affected by DD, as only their phase-coupling increases without significantly changing their firing rate or pattern [63]. These alterations in GP neuron activity in vivo can be due either to changes in the intrinsic excitability and/or to changes in their afferent synaptic input drive (discussed in Section 3.4 and Section 3.5).
3.2. Ionic Conductances Underlying Pacemaking in GP Neurons
GP neurons exhibit a large range of firing activity and rates (between 0 to 100 Hz) in rodents and non-human primates in vivo [57,76,77,78]. Recordings of GP neurons in dissociated neuron cultures and in brain slices have demonstrated that this variability in firing is correlated with the type and density of voltage-gated conductances expressed by individual GP neurons [53,79]. Despite these individual differences, a subset of voltage-gated channels plays a key role in the autonomous pacemaking of GP neurons. Repetitive emission of APs by GP neurons relies primarily on persistent sodium and hyperpolarization-activated, cyclic nucleotide-gated cation (HCN) channels [21,80], which depolarize the membrane potential at subthreshold potentials. The regularity and precision of GP neuron firing is governed by both calcium-activated SK channels and HCN channels [81], while Kv3-family potassium channels promote spike repolarization in these neurons, enabling high frequencies of discharges [22].
3.3. Dopamine Modulation of Intrinsic Excitability of GP Neurons
DA modulation of the excitability of GP neurons has been suggested by various techniques, showing either the presence of dopaminergic fibers and receptors in the nucleus [42,44,82], the release of DA [83,84,85], dopaminergic modulation of voltage-gated ion channels [86], and neuronal excitability in vivo [87]. Interestingly, D2 receptor mRNA is present in all classes of pallidal neurons, with the pallido-striatal neurons expressing higher levels of D2 transcripts than pallido-subthalamic cells [54,55]. This suggests a differential modulation by DA of prototypic and arkypallidal GP neurons. Thus, it is likely that DD preferentially affects the intrinsic excitability of one population of GP neurons. DA modulation of GP neuron excitability is poorly understood, and so far, only one study has shown a direct action of DA on GP neuron voltage-gated conductance. In this study, the authors showed that D2Rs inhibit CaV2.2 (N-type) channels in a protein kinase C (PKC)-dependent manner [86]. This result suggests that D2R activation reduces GP neuron excitability (but see Section 4.1).
Moreover, some GP neurons lose their autonomous pacemaking after DD [88]. The mechanism underlying GP neuron pacemaking loss involves a CaV1.3 (L-type) calcium channel-dependent downregulation of HCN2 channels [88]. It also seems that this alteration of excitability of GP neurons preferentially affects NPas1-expressing pallido-striatal neurons. Indeed, the firing rate of the main subpopulation of prototypic neurons, PV+ neurons, remains unaffected in DD mice [89]. As a consequence, these results suggest that the change in the pattern of activity of PV+ GP neurons observed in vivo [63,71] relies on changes in the activity of iSPNs. Indeed, the hyperactivity of iSPNs promotes pauses in activity of prototypic cells [72,73,74]. It is interesting to note that, while excitability of Npas1 GP neurons is reduced, the pallido-striatal GABAergic inhibition provided by these neurons is increased in all subtypes of striatal neurons [89], suggesting the existence of a compensatory mechanism.
3.4. Dopamine Modulation of Gabaergic Transmission in the GP
GP neurons receive two main sources of GABA: extrinsic inputs coming from iSPNs and some collaterals of dSPNs [16,17,90] and intrinsic inputs from local axon collaterals [91]. Striato-pallidal inputs are mainly localized on proximal and distal dendrites of GP neurons [92], while pallido-pallidal inhibitory synapses are found on the soma and proximal dendrites of GP neurons [91]. Regarding their properties, striato-pallidal synapses are characterized by short-term facilitation (STF) [93,94] and their strength is modulated by a plethora of G protein–coupled receptors (GPCR) [95,96,97,98,99,100,101,102] including presynaptic D2Rs, which decrease the probability of GABA release [93,103]. In contrast, pallido-pallidal synapses are characterized by short-term depression (STD) [93,94,104] and are not modulated by presynaptic D2-like receptors [93]. GABAergic transmission is also regulated by postsynaptic D4Rs, which reduce the amplitude of GABAA-mediated current through the suppression of protein kinase A (PKA) activity [105]. The paired-pulse facilitation observed at striato-pallidal connection suggests that this synapse has a low initial release probability. However, a full characterization of unitary iSPN-GP connections will be required to better understand the release dynamics of these synapses. On the other hand, paired-recordings of GP neurons have revealed that despite the sparse connectivity (~1%; [91,104]) and STD, unitary pallido-pallidal transmission is able to reduce the postsynaptic firing rate through a combination of chloride driving force, synaptic summation, and incomplete STD [104]. The sparse connectivity combined with the relative efficacy of axon local collaterals certainly contributes to uncorrelated activity of GP neurons recorded in vivo under healthy conditions [78].
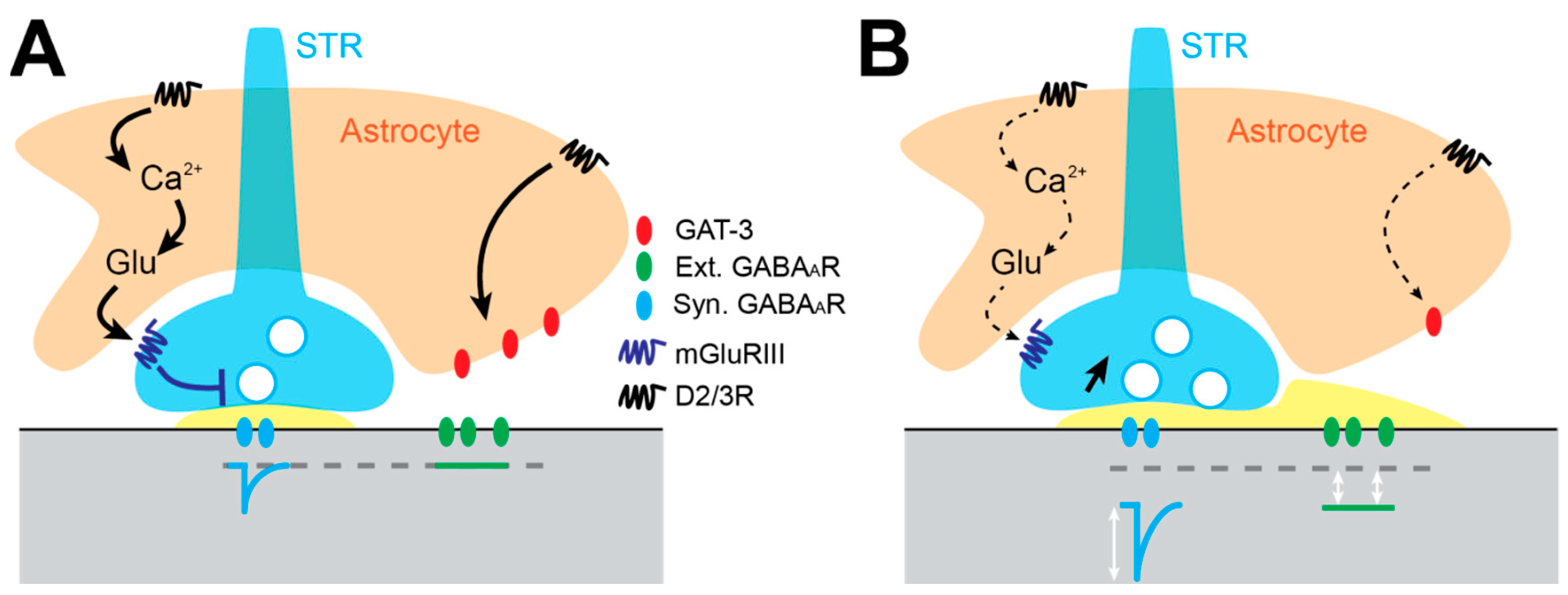
3.5. Astrocyte-Dependent Alteration of Gabaergic Inhibition in the GP
The GP is the nucleus in the BG with the highest astrocyte density [106], suggesting an important role of these glial cells in regulating GP motor function. Indeed, voluntary exercise in mice triggers astrocyte structural plasticity. This consists of elaboration of perisynaptic astrocyte processes (PAP) [107], which are dynamic elements thought to regulate synaptic transmission by clearing neurotransmitter and releasing gliotransmitter into the synaptic clefts [108,109,110]. GP astrocytes express GAT-3 GABA transporters [111,112], which are primarily localized on PAPs [107]. They also express a variety of ionotropic and GPCR receptors including DARs, suggesting that DA somehow modulates intracellular signaling. Calcium imaging experiments have shown that D3R activation triggers a reduction in intracellular Ca2+ waves under control conditions, while DD exerts the opposite effect [113]. We also observed that DD is responsible for GP astrocytosis [106], which suggests astrocyte dysfunction in PD. Two recent studies have elucidated distinct astrocyte signaling pathway impairments that promote an increase in GABAergic transmission in the GP. In the first study, the authors demonstrated that after DD, glutamate release from GP astrocyte is reduced, which dampens the activity of pre-synaptic mGluR3 and in turn increases GABA release at striato-pallidal synapses (Figure 2) [113]. A second study showed that DD also induces a reduction of the expression of GAT-3 in GP astrocytes, leading to reduced uptake and elevation of ambient extracellular GABA levels promoting the activation of extrasynaptic GABAAR-mediated tonic inhibition (Figure 2) [114]. Both astrocyte glutamate release and GAT-3 uptake activity seem to be regulated by D2-like family receptors, suggesting that reduced stimulation of these receptors in DD conditions is responsible for the increase of extracellular GABA concentrations [115] and GABAergic transmission in GP neurons. Overall, GP neuron hypoactivity in vivo seems to be the consequence of several convergent mechanisms—at the circuit level, with the hyperactivity of iSPNs [72,73], but also locally, with the reduction of GP excitability and the dysregulation of GABAergic synaptic and extrasynaptic transmission [92,113,114] (Figure 4). Restoring autonomous pacemaking in the GP by overexpression of HCN2 fails to re-establish motor function in DD animals [88]. Interestingly, because GP astrocytes play a key role in the regulation of GABAergic transmission, restoring normal levels of inhibition in the GP by manipulating astrocyte function appears to be an attractive and promising therapeutic strategy to reduce PD motor symptoms.
4. The Subthalamic Nucleus
4.1. Dopamine Modulation of STN Neuron Excitability
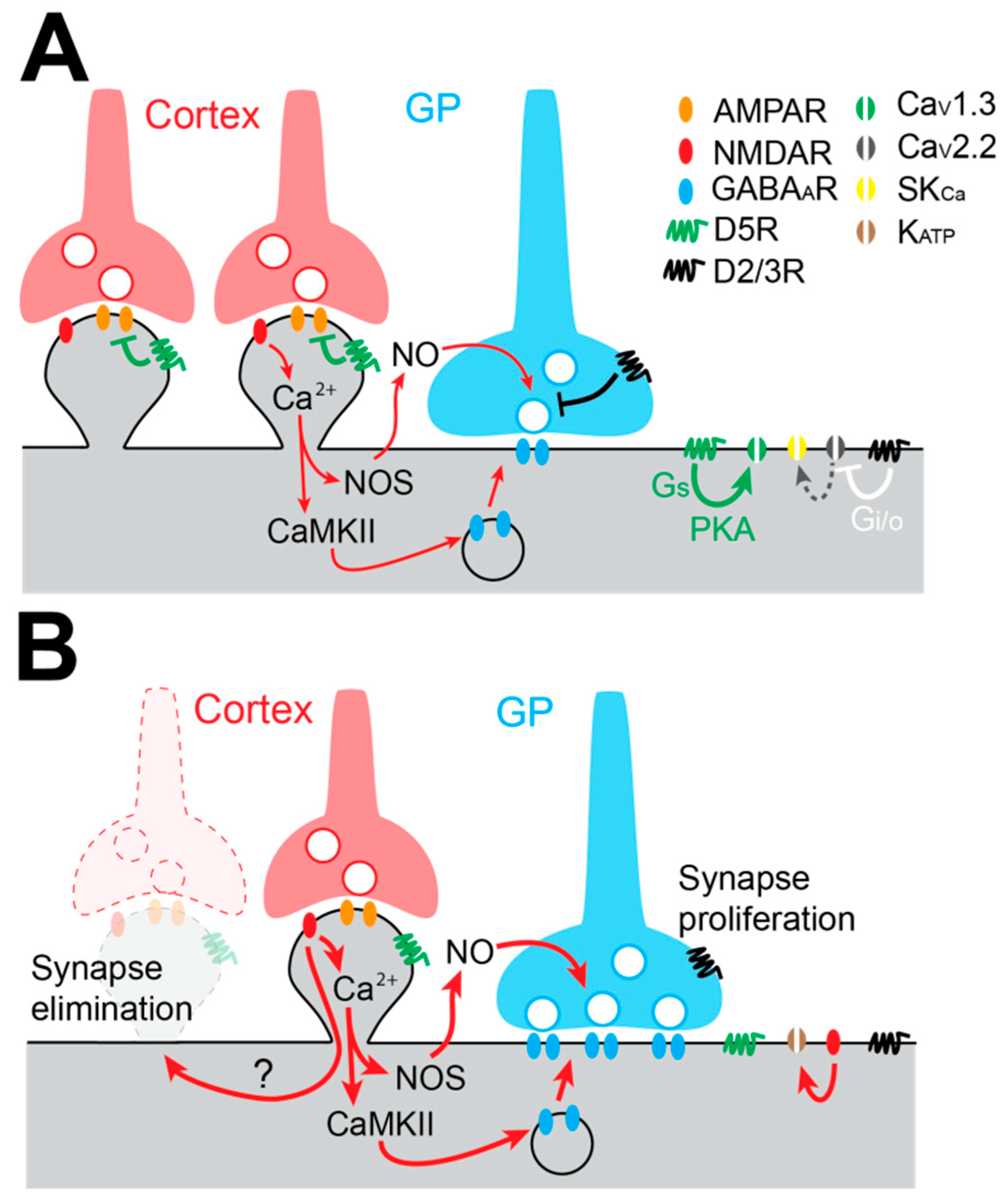
Because the change in the firing pattern of STN neurons is considered an electrophysiological hallmark of PD, the modulation of the activity of STN neurons by DA has been the focus of many studies. STN neurons mainly express D2/3Rs and D5Rs on their membrane [116,117]. Most of our current knowledge about DA action on STN neurons comes from exogenous application of DA itself or agonists of the D1- and D2-like receptor families in acute brain slices. Application of low concentrations of DA or the D2-like receptor agonist quinpirole induces a depolarization of the membrane potential and an increase in the spontaneous firing rate of STN neurons [43,116,118]. This excitatory action of D2-like dopaminergic receptors is mediated by inhibition of CaV2.2 (N-type) calcium channel currents by Gi/o βγ subunits. This inhibition reduces CaV2.2 channel functional coupling with small-conductance Ca2+-dependent K+ (SKCa) channels, promotes membrane depolarization, and increases firing discharge of STN neurons [116] (Figure 3A). On the other hand, D5R activation targets several conductances depending on the mode of discharge of STN neurons [119]. When STN neurons’ membrane potential is hyperpolarized, they fire bursts of APs and D5R activation potentiates CaV1.3 calcium channel currents, prolonging burst duration [120] (Figure 3A). When STN neurons’ membrane potential is depolarized, these neurons fire single APs in a tonic fashion, and D5R increases their firing rate via the activation of cyclic nucleotide gated non-cationic channels [121]. Therefore, it has been proposed that D2Rs control the pattern of activity (tonic or burst) by setting the level of depolarization of STN, while D5Rs reinforce each mode of discharge [118].
4.2. Alteration of STN Autonomous Pacemaking in DA Depleted Rodents
It is widely accepted that the STN is hyperactive in PD and that STN neuron activity is composed of rhythmic and synchronous bursts of APs [122]. This pathological activity can have several origins, such as DA loss in the STN, leading to altered excitability of STN neurons or alterations in the BG network. Ex vivo studies suggested that the excitability of STN neurons is strongly downregulated in DD rodents [123,124], but the underlying mechanisms were unknown until recent work confirmed that STN neuron autonomous pacemaking is lost in neurotoxic and genetic mouse models of PD [125]. This loss of excitability seems to have a network origin, as it is triggered by the increased drive of iSPNs onto GP neurons, leading to disinhibition of STN neurons and excessive activation of NMDAR, which finally increases KATP channels [125]. Indeed, the expression of these channels has been reported to be increased after DD [126], which produces sufficient hyperpolarization of the membrane potential to silence STN neurons [125]. Another study suggests that DD alters the expression profile of HCN2 channels in the STN and also contributes to pathological activity in the BG in experimental PD [127].
4.3. Augmentation of Pallido-Subthalamic Transmission in Experimental Parkinson’s Disease Models
The GP provides the STN’s principal source of GABA. Single GP axons make sparse clusters of synaptic boutons distributed in distinct functional domains of the STN [128]. On average, a GP axon makes at least six synaptic contacts on the soma and the proximal dendrites of STN neurons. Thus, each GP neuron provides potent somatic inhibition, which is highly efficient to reset the autonomous activity of STN neurons [128]. GP neurons fire APs at a high rate [70,129] and this elevated activity influences, in an activity-dependent manner, the efficacy of GP-STN inputs that are characterized by STD [130,131]. GP-STN inputs are modulated by presynaptic D2/3Rs, which decrease the release probability of GABA and thus reduce the strength of this connection. It is also interesting to note that DA converts GP-STN synapses from low-pass filters to band-pass filters [131] favoring information transfer [132]. Under DD conditions, GP-STN GABAergic transmission is greatly augmented due to structural modifications of GP-STN synapses, which make more active zones per bouton [133] (Figure 3B). Recently, Chu and colleagues [134] have unraveled the mechanisms underlying this proliferation of GABAergic synapses. In this study, they showed that GABAergic long term potentiation (LTP) involves the excessive activation of NMDA receptors, which triggers 1) nitric oxide synthesis and release by STN neurons, which activates presynaptic protein kinase G (PKG) and increases GABA release probability, and 2) an increase in intracellular Ca2+ concentrations that activate a CaMKIIα pathway, which augments GABAA receptor insertion on the post-synaptic side of GABAergic GP-STN synapses [134] (Figure 3B).
4.4. Loss of the Cortico-Subthalamic Pathway in Experimental Parkinson’s Disease Models
The cortico-subthalamic (Cx-STN) projection is well described anatomically. Cx-STN synapses are mainly found on the distal part of STN neurons’ dendrites [135,136]. The functional properties of Cx-STN glutamatergic synapses have been described recently. Around the resting potential of STN neurons, Cx-STN post-synaptic excitatory currents (EPSC) are mainly composed of AMPAR-mediated currents with a small NMDA component [134,137]. Interestingly, it has been shown that the strength of the Cx-STN inputs is under the control of DA and more specifically, of post-synaptic D5Rs which depress AMPAR-mediated EPSC through a PKA-dependent intracellular pathway [137]. Another study has also shown that pre-synaptic D2Rs modulate AMPA currents by decreasing glutamate release [138]. Together, these data suggest that Cx-STN synaptic transmission is modulated by DA, and hence, this pathway may be altered in PD. Indeed, it has been shown that, in DD rodents, Cx-STN innervation is reduced [139,140,141] but the mechanisms underlying this loss of Cx-STN glutamatergic inputs remain to be determined. It has been proposed that the alleviation of motor symptoms by M1 motor cortex deep brain stimulation (DBS) [142,143] relies on the restoration of Cx-STN functional connectivity of the remaining Cx-STN afferent inputs [141].
5. The Substantia Nigra Pars Reticulata
5.1. Neuronal Diversity in the Substantia Nigra Pars Reticulata
Together with the EPN, the SNr is one of the output nuclei in the basal ganglia. Like other BG nuclei, the SNr has been classically considered a homogeneous nucleus, and cell differences were based more on topographical innervation patterns than on neuronal diversity. Although it is well known that the main target of SNr is the thalamus [144], up to four neuron types have been identified based on the axonal projection targets. Type I cells project specifically to the thalamus, type II neurons target the thalamus, superior colliculus and pedundulopontine tegmental nucleus, type III cells project to the periaqueductal gray matter and thalamus, and type IV neurons send projections to the deep mesencephalic nucleus and the superior colliculus [145,146]. In addition to this long axonal arborization, SNr neurons are also highly collateralized [145,147], and most neurons receive robust inhibitory synaptic inputs even in the presence of strong activation [148,149].
In the SNr, GABAergic projection neurons are the largest cell population, but discrete clusters of dopaminergic neurons are also present in the caudomedial region of the nucleus [150]. The majority of GABAergic neurons in the SNr express PV [151,152] and a small subset of cells express calretinin [153,154], nitric oxide synthase, or acetylcholine transferase [150,155]. These cells differ in their neurochemical content and their topographical and morphological profiles. Thus, in the rostrolateral SNr, GABAergic cells are large and contain PV and nitric oxide synthase. In caudomedial positions, most of them are small and express only PV, and in rostromedial portions, they are predominantly small and contain either calretinin, nitric oxide synthase or PV [150]. As for receptor expression, the vast majority of PV-positive neurons also display GABAA receptor α1 subunits and at least one (GABABR2) of the heteromeric subunits of the GABAB receptor [156,157]. Moreover, cells with colocalized PV and calbindin also express purinergic receptor P2 [158].
Neurochemical analysis clearly distinguishes three different classes of cellular content, but electrophysiological characterization of GABAergic cells and correlation with the neurochemical profile has yet to be fully developed. PV-positive cells have been associated with high firing frequency [159], but this characteristic does not seem to be cell-specific. Indeed, calretinin-containing cells show similar projection targets, local arborization, morphological, and electrophysiological characteristics to PV-positive cells [160]. More recent studies describe four subtypes of GABAergic cells in the SNr, whose electrophysiological profile is complex and varies according to the posture and movement of the animal [161,162].
5.2. Ionic Conductances Underlying Pacemaking in Snr Neurons
Like other ESNs, SNr neurons are autonomously active. This regular spiking conveys an efficient tonic inhibitory drive onto motor thalamic nuclei [163,164,165]. Repetitive AP generation in SNr neurons is primarily supported by the subthreshold slowly inactivating NaV channels, while SK channels are critical to maintain the precision of autonomous pacemaking in these neurons [18]. HCN are also present in SNr cells but are not activated in the range of potential associated with pacemaking activity. It has also been shown that leak conductances are critical for the maintenance of SNr neurons at depolarized potential [166,167]. A molecular and electrophysiological study suggested that transient receptor potential channel family (TRPC3) channels are expressed in SNr neurons and contribute to their depolarized resting potential [167]. However, another study showed that spontaneous firing of SNr neurons is unaffected after genetic deletion of these channels [166], questioning the participation of these channels in the pacemaking of SNr neurons. This discrepancy can be explained by the lack of a specific blocker of non-specific cationic channels used by Zhou and colleagues to show the involvement of TRPC3 channels in the depolarized potential of SNr neurons. In addition, a more recent study has discovered that SNr neurons express the sodium leak channel, NALCN, and that genetic deletion of NALCN impairs spontaneous firing in these neurons [168]. Because the activity of NALCN is strongly dependent upon glycolysis, alterations in this metabolic pathway can significantly impair autonomous pacemaking in SNr neurons [166,168] and contributes to pathological activity of this nucleus. As with other ESNs, Kv3-channels are essential for SNr neurons to fire APs at high frequency, as they provide rapid repolarization of the membrane potential following AP depolarization [169]. An extensive description of the ionic mechanisms governing SNr neurons excitability can be found in the following review [170].
5.3. Dopamine Modulation of Intrinsic Excitability of Snr Neurons
Dopaminergic modulation of SNr is achieved through an unconventional release of DA by the dendrites of SNc neurons (for review, see [171]), which constitutes an ultra-short dopaminergic pathway. In situ hybridization and immunohistochemistry studies have shown that several DARs are expressed in the SNr. The most intense labeling is for D1Rs, especially in striato-nigral terminals. D4Rs and D5Rs are present in the SNr but mainly on perikarya [172,173,174,175,176], and D1/D5Rs have been found both on SNr neurons and astrocytes [177]. D1-like agonists excite SNr neurons, and this modulation is mediated by a PKA-dependent enhancement of the constitutively active TRPC3 channels, which depolarizes SNr neurons [178]. Interestingly, this effect was mimicked by artificially elevating ambient DA levels, which supports its physiological and functional relevance [178]. In addition, acute blockade of D1-like and D2-like receptors induces hyperpolarization of SNr neurons and a switch from tonic regular firing to irregular or burst firing [47]. This pharmacological manipulation resembles the activity of SNr neurons recorded in vivo in anesthetized [179,180,181,182] and awake [183] DD rodents. The cellular mechanisms underlying the changes in rate and pattern of SNr under DD have not been explored ex vivo. It will be of great interest to further investigate which intrinsic conductances of SNr neurons are directly impacted by the loss of DA.
5.4. Alteration of Gabaergic and Glutamatergic Transmission in Snr Neurons in Experimental Parkinson’s Disease Models
Anatomical studies have shown that single SNr neurons receive convergent afferents from dSPNs, the GP and the STN (for review [184]), supporting a strong integrating function of this output nucleus of the BG. Like in the GP, pallidal and striatal GABAergic inputs make symmetric synapses with the soma/proximal dendrites and distal dendrites of SNr neurons, respectively [185,186,187]. In addition to their subcellular locations, these two GABAergic synapses have also distinct properties. Striato-nigral (STR-SNr) IPSCs exhibit STF, while pallido-nigral (GP-SNr) synapses display STD [188]. These two synapses are also different regarding the pre-synaptic control exerted by DAR. GP-SNr synaptic transmission is reduced by D4R activation [189,190] while STR-SNr GABA release is augmented by D1Rs [189,191,192,193]. Because of its opposite action on STR-SNr and GP-SNr synapses, DA maintains a certain equilibrium between somatic and dendritic inhibitory inputs received by SNr neurons, whose precise function remains to be elucidated. The impact of DD on GP-SNr synaptic transmission is unknown but has been investigated for STR-SNr synapses [194]. In their study, the authors report that DD induces a strong increase in STR-SNr IPSC amplitude and show that dysfunctional GABAB receptors and loss of presynaptic reduction of GABA release probability was responsible for this augmented transmission [194].
The STN is the main provider of excitatory inputs to the SNr, and the properties of STN-SNr synapses have been well characterized. Electrical stimulation of STN axons triggers monosynaptic EPSCs [195,196], while the same type of stimulation delivered in the STN itself gives rise to complex EPSCs [197,198], which are believed to be generated by the activation of STN local axon collaterals [197,199]. Both D1Rs and D2Rs are present on STN-SNr synaptic terminals and activation of D1Rs enhances while activation of D2Rs decreases STN-SNr EPSC amplitude [196]. On the other hand, complex EPSCs which have been shown to promote burst-firing in SNr neurons [197,200] are reduced by the activation of D2-like receptors [200]. Only long-term depression (LTD) has been reported at STN-SNr. The induction of LTD requires post-synaptic D1R activation and is expressed through NMDAR-dependent endocytosis of AMPARs, which depresses EPSC amplitude by almost 50% [195]. This mechanism represents a specific feature of this synapse compared to cortico-striatal synapses [201], but like in the STR, DD completely abolishes STN-SNr LTD [195], supporting the hypothesis that increased synaptic transmission at STN-SNr synapses contributes to pathological activity of SNr neurons. DA-glutamate interplay dysfunctions at the post-synaptic density have been suggested as critical determinants of major psychiatric disorders [202,203] and could be emphasized as potential molecular targets at STN-SNr synapses to restore a normal level of activity of SNr neurons.
Overall, there is compelling evidence suggesting that both STR-SNr and STN-SNr synaptic transmission strength is regulated by DA and is pathologically-enhanced in experimental PD.
6. The Entopeduncular Nucleus
6.1. Anatomical Organization and Cellular Diversity in the EPN
The EPN is, in addition to the SNr, the other output nucleus of the BG. Early anatomical studies have suggested that the EPN is subdivided in rostral and caudal portions, with the former being enriched in somatostatin-positive (SOM+) neurons and the latter being composed of PV-expressing neurons [151,204,205]. More recently, a third neuronal population, negative for both SOM and PV, has been described [206]. The proportion of these three populations has been estimated as 28% (PV+/SOM-), 46% (PV-/SOM+), and 25% (PV-/SOM-). Additional molecular profiling has confirmed the existence of these three neuronal subtypes and has specified their molecular identity [207]. It has also been shown that the EPN is organized in a PV-rich core and a PV-poor shell fashion [206]. Retrograde tracing studies have demonstrated that SOM+ EPN neurons project to the lateral habenula (Lhb) [208,209], while PV+ EPN neurons innervate the motor thalamus [151,208]. Furthermore, Lhb- and thalamic-projecting EPN neurons are differentially innervated by subclasses of pallidal and striatal neurons [207,208]. EPN-thalamic neurons are innervated by PV+ GP neurons and matrix SPNs, while EPN-Lhb neurons receive inputs from PV- GP neurons and striosome SPNs, suggesting that these two EPN cell types are part of distinct functional networks. Indeed, it has been proposed that thalamic-projecting EPN neurons control motor program selection, while Lhb-projecting EPN neurons participate in the evaluation of the motor outcome [208].
6.2. Autonomous Pacemaking in EPN Neurons.
EPN neurons fire at a high rate in vivo both in anesthetized and awake rodents [210]. Like other ESN, it is likely that this elevated discharge relies on intrinsic properties of EPN neurons. Indeed, EPN neurons are spontaneously active ex vivo [211], but not much is known about the properties of the channels responsible for autonomous pacemaking in these neurons, and DA modulation of their excitability has never been investigated. Based on their firing pattern and properties ex vivo, two types of EPN neurons have been described [211,212]. Type I EPN neurons are spontaneously active, generate a sag upon membrane hyperpolarization, and generate a rebound-burst when hyperpolarizing current injection ends, suggesting that they express HCN and CaV3-type calcium channels, respectively. Type II EPN neurons are not autonomously active. They have low HCN channel expression and potassium A-type currents, which generate a slow depolarizing ramp at the end of hyperpolarizing pulses of currents [211]. Interestingly, most type I, but not type II, EPN neurons exhibit GABAergic IPSCs upon electrical stimulation of the striatum [212], which suggests differential innervation of these two cell types. Molecular and electrophysiological correlative studies would be required to determine if Lhb-projecting and thalamic-projecting EPN neurons possess specific electrophysiological signatures. To our knowledge, neither the modulation exerted by DA nor the direct consequences of DD on the excitability of EPN neurons have yet been investigated.
6.3. Dopamine Modulation of Gabaergic and Glutamatergic Transmission in the EPN
Like the SNr, the EPN receive convergent inputs from the striatal direct-pathway, the GP, and the STN [185,213]. Pallido-entopeduncular (GP-EPN) and striato-endopeduncular (STR-EPN) synapses display the same properties as STR-SNr and GP-SNr synapses, i.e., STF and STD, respectively [214].
All DAR subtypes have been found in the EPN [215,216]. Their presynaptic distribution appears to be very similar to that of the SNr: D2-like receptors are present on GP-EPN terminals, while D1Rs are found on STR-EPN synapses [215,216]. The modulation exerted by DA on these two pathways seems to also follow the principles described in the SNr, as D2-like receptors depress GP-EPN transmission whereas STR-EPN inputs are potentiated by D1Rs [215,217]. The impact of DD on these synapses has not been tested, but one may speculate that DD will facilitate GP-EPN synapses and depress the STR-EPN synapses, therefore generating an imbalance between direct and indirect pathway GABAergic inputs in the EPN.
7. Consequences of Cellular and Synaptic Dysfunctions for Abnormal Neural Dynamics in the Basal Ganglia during Parkinson’s Disease
One critical aspect that remains to be addressed is defining the link between the different molecular/synaptic alterations occurring after DA loss and the pathological BG activity recorded at the network level. This lack of knowledge raises important questions that need to be assessed to better understand how BG activity becomes dysfunctional in the DD state. In particular, it seems critical to define if the synaptic modifications are the cause of the neuronal network dysfunction present in PD or if it is the other way around. Also, being able to determine the respective contributions of the synaptic vs. the network alterations to Parkinson’s motor symptoms might help in designing new therapeutic strategies to target more precisely the pathophysiological process in PD. These are not trivial questions to tackle, especially considering the difficulty of determining if these changes are truly pathogenic, compensatory, or by-products of other (yet to be defined) neuronal changes. Another challenge facing the field is the fact that the network abnormalities underlying the motor disturbances of PD are still enigmatic. Indeed, while traditional views on the pathophysiological organization of BG circuits have highlighted the contribution of firing rate alterations to explain the motor impairments present in PD [8,220], other neuronal activities, such as changes in the firing pattern [182,221,222,223,224], or increases in neuronal synchronization [77,78,225,226], have also been associated with the Parkinson’s disease condition [227,228,229]. Whether these changes underlie the pathophysiology of PD is not known, but their presence is positively correlated with the PD bradykinesia and rigidity score [230] and can be used to distinguish the PD state [231]. Here, we will briefly review the principal neuronal network changes occurring after DD, the arguments in favor or against their causative contribution to the pathophysiology of PD, as well as how these network changes might be affected by the synaptic alterations previously described in this review.
First, considering the firing rate modification, it has been shown that the loss of DA creates an imbalance in activity in striatal projection neurons that leads to iSPN hyperactivity and dSPN hypoactivity [73,232,233,234]. This increase in iSPN activity induces a cascade of firing rate changes along the indirect pathway: notably decreased activity of GP neurons [14,71,235,236] and increased activity of STN neurons [221,237]. These modifications lead to increased firing of the BG output nuclei SNr/EPN [224,238], which translate into over-inhibition of the thalamo-cortical motor circuits [239]. The most compelling evidence in favor of a pathogenic contribution of the firing rate alterations comes from studies that have used cell-type-specific optogenetic [9,240,241] or pharmacogenetics [134,242] manipulations to reproduce the firing rate changes present in PD. In particular, specific excitation of iSPNs induced a reduction of locomotion mimicking the PD akinetic state [9,134,242], whereas activation of the dSPNs in PD model mice reduced the motor deficit and elicited locomotion [9]. In addition, a direct link between the increase in SNr output activity and motor suppression has been established consequently to iSPN opto-stimulation [240], thus supporting the view that BG outputs exert an inhibitory influence on movement control [146]. The timing at which the firing rate of striatal neurons is modified after DA loss is not known, but it is likely to be fast. Indeed, the expression of the messenger RNA for c-fos, a marker of neuronal reactivity often used as a correlate of neuronal activity [243], is increased in iSPNs 75 min after 6-OHDA injection. This timing parallels the DA loss [244]. Therefore, it seems reasonable to expect that the firing rate modifications along the indirect pathway are occurring before the synaptic reorganization (which might appear with longer time-scale after DA loss). This assumption favors the idea that some of the synaptic changes observed in the DD state might thus be corrective by nature, compensating for the abnormal firing rate activities established in the indirect pathway. With this in mind, the selective loss of glutamatergic synapses in iSPNs [245], the increased connectivity of striatal fast-spiking interneurons onto iSPNs [246] and the increased number [133] and strength of GP synapses onto STN neurons [133,134] might represent global homeostatic changes of the indirect pathway aiming to counterbalance the increased activity of iSPNs. Similarly, the strong synaptic depression of the cortico-STN excitatory inputs present both in rodents and monkey PD models [139,140] could also be an adaptive change to limit STN hyperactivity and, at the end-stage, the negative influence of BG output onto thalamo-cortical and brainstem circuits. The apparent discrepancy between the functional restoration of the Cx-STN pathway by motor cortex DBS (see Section 4.4) and an adaptive reduction of this pathway to counteract STN hyperactivity can be explain if motor cortex DBS acts by breaking pathological activity and promoting corticofugal [247] information transfer rather than simply increasing Cx-STN synaptic transmission. However, despite all the experimental evidence arguing for an important contribution of BG overall firing rate modification in experimental PD, it is still unknown if they represent the sole pathogenic cause of Parkinsonism [248,249,250]. Additionally, many experimental findings have directly challenged the predictions of the rate model. For example, while recordings in MPTP-treated monkeys have clearly established STN firing hyperactivity [221,235], changes in striatal firing activity as described by the rate model have been more conflicting. Indeed, while one study described a profound increase in striatal neuronal firing [251], another reported no change [235]. In addition, recordings in DD mice have shown a reduction in the autonomous properties of STN neurons which, when restored using chemogenetic excitation of STN, improves motor dysfunction in DD mice [125]. Considering the fact that motor recovery in PD is classically obtained using suppression [252] or reduction [238] of STN neuronal firing, these results were totally unexpected, and in contrast with the motor-suppressing effect predicted by the rate model when STN increase their activity. Another piece of experimental evidence that challenges the rate model is the fact that SNr firing output in DD mice is hypoactive and not hyperactive [183]. Taken together, the direction of the firing rate changes in various nodes of BG circuits might not be consistently present across different animal models of PD (i.e., rats vs. mice vs. monkeys), thus raising the possibility that other neuronal changes might more reliably contribute to the motor dysfunction of PD.
Abnormal expression of synchronized oscillatory activity in the beta frequency band (12-35 Hz) has been one of the most prominent neuronal changes consistently detected in PD patients and has been related to akinesia/bradykinesia syndromes in PD [253,254]. Accordingly, a correlation between the level of beta oscillation expression and the motor deficit has been described in the off-medicated state of PD [255] (but see [256]). In addition, imposing abnormal beta synchronization through external stimulation at beta frequency slows down movements both in healthy [257] and PD subjects [258], which suggests a pathogenic role (although this stimulation protocol likely altered the level of neuronal firing, which was not accounted for in the motor perturbation). What are the neuronal circuits generating this abnormal beta synchronization? In theory, any network with delayed negative feedback properties can generate oscillatory activity [259], and because the organization of the BG is principally composed of parallel feedback loops, many BG circuits could potentially generate beta oscillations [260,261,262]. It was first proposed that the abnormal BG synchronization is an emergent property of the GP-STN network [263], but this work was performed in organotypic culture and so far, there is no in vivo evidence that the GP-STN network could sustain synchronized rhythmic activity without the cortex [222,264]. Other proposed circuit generators of beta oscillations are the cortex [265], the striatum [266], the STR-GP network [267], and the hyperdirect loop [268]. One influential hypothesis suggests that beta oscillations are generated at the cortex at physiological levels, which propagate in BG circuits through the hyperdirect pathway where they are abnormally amplified by the reciprocally-connected GP-STN microcircuits [265,269,270]. There is indeed a strong rationale to suggest that the GP-STN network is involved in the maintenance and the propagation of oscillatory activity both in vivo and ex vivo [13,14,71,271,272]. Nonetheless, the specific contribution of these different circuit elements has never been tested with optogenetic tools that are reversible and offer high temporal resolution. One aspect to consider is the timing at which abnormal beta synchronization is generated after the DA loss, as it reveals important features regarding its contribution and interaction with the firing rate or synaptic modifications. Interestingly, beta oscillations are not detected in PD rat models until >4 days after the lesion and reach a plateau of expression after 10 days. This slow time-scale of generation contrasts with the fast motor deficits that appear within hours following the loss or the blockage of DA transmission [237,273,274]. Similarly, monkeys and rats present Parkinson’s associated motor symptoms following chronic MPTP or 6-OHDA treatment before the emergence of BG synchronous oscillatory activity [250,275]. Taken together, this suggests that beta synchronized oscillations might arise through long-term adaptive changes that are slower than the changes in firing rate, but which could parallel the timing of the synaptic alterations. This potentially highlights the relationship between these two kinds of activity, but whether one is the consequence of the other is currently not known. Similarly, the link between the firing rate modifications and the change in neuronal synchronization has never been investigated. We propose here the hypothesis that the GP firing rate alterations in PD trigger maladaptive modifications that favor GP neuronal synchronization. Indeed, in a normal animal, the firing of GP neurons is principally governed by their autonomous pacemaker properties, which are different across neurons due to variability in channel density [53,79]. Active decorrelation mechanisms caused by GP neurons’ intrinsic properties and mutual inhibition through axon collaterals prevent neuronal synchronization in normal conditions [226,276]. This indicates that the intra-GP GABAergic control in normal conditions is mainly achieved by GP neurons. In contrast, the loss of DA in PD, which causes the increase in iSPN firing, operates a switch in the GP GABAergic control from intrinsic (i.e., GP) to extrinsic (i.e., STR) sources. This change of GABA transmission is certainly at the origin of maladaptive changes at the molecular and synaptic level that favor the generation of synchronized oscillatory activity in GP neurons. Because of the critical dichotomous organization of GP neurons into prototypic and arkypallidal neurons [14], this abnormal oscillatory activity can then propagate to the whole BG circuit. In this scheme, the abnormal network synchronization is caused by maladaptive changes occurring at the cellular and synaptic levels and is a consequence of the firing rate perturbation in the indirect pathway. It is important to mention though that this abnormal beta synchronization has been detected in PD animal models, such as 6-OHDA-lesioned rats [237,247,277,278,279] and MPTP-treated monkeys [235,280], but not in 6-OHDA or α-synuclein mice [183,281], which raises the question of their contribution to the pathophysiology of PD. Indeed, future studies will have to precisely define the specific behavioural contributions of the different neuronal activities and how they impact the motor symptoms of PD.
STN or GPi DBS is highly effective to alleviate PD motor symptoms and it is widely thought that DBS achieves its therapeutic action by disrupting pathological oscillatory activity in the motor cortex/BG loops [282,283]. Considering the complex anatomical connectivity of the BG and the possible concomitant local and distant effects of STN-DBS [247,284,285,286], the mechanisms through which high frequency stimulation breaks down pathological activity and restores motor function remains highly debated. Because axons are known to be more responsive to electrical stimulation than cell bodies [287] and that globally ESN synapses are pathologically-enhanced in PD models one may speculate that STN-DBS has a stronger impact on synaptic transmission and neurotransmitter release rather than on neuronal excitability, which allow them to re-establish information transfer within the BG and execution of motor programs. In addition to what has been done already [142], the future development of optogenitically-based DBS protocols will certainly help to better understand where in the network and by which precise mechanism(s) STN-DBS restores motor function in PD.
8. Concluding Remarks
In this review of the literature, we have highlighted that every ESN of the BG receives a functional DA innervation that controls both the post-synaptic excitability as well as the strength of their afferent synaptic inputs. In PD, extrastriatal hypodopaminergy provoked by the loss of SNc neurons must participate, in addition to the alterations described in the striatum, in the pathophysiological activity of the BG network. According to all the evidence accumulated over the past decades, we propose that DA loss induces an imbalance between ESN neuron intrinsic excitability and ESN synapses, in favor of the latter. Autonomous pacemaking is reduced in all ESN while synaptic transmission is enhanced, leading to a switch from intrinsically-driven oscillatory activity in ESN to a synaptically-driven pattern of activity, which promotes hypersynchronous oscillatory activity in the BG network in PD (Figure 4B).
Funding
This research was funded by the French Agence Nationale de la Recherche (grants 08-JCJC-0087, 2014-CE13-0024-01 and 2015-CE37-0006), the Eurorégion Aquitaine-Euskadi (Neurosciences grant number 9), the University of the Basque Country (LD joint doctoral grant), the Association France Parkinson (SNR-PARK grant), the Spanish Government (SAF2016-77758-R [AEI/FEDER, UE]), and the LABEX BRAIN ANR-10-LABX-43 grant (MC post-doctoral grant). The University of Bordeaux and the Centre National de la Recherche Scientifique provided infrastructure support.
Acknowledgments
We are also grateful to Patricia Gongal for language assistance on the manuscript. The present review is based on a systematic review of the literature using Pubmed and BioRxiv. Our objective was to cite the latest research articles available in the literature as well as citing some older and pioneer experimental work when discussing key concept in the fields. When discussing specific points that have already been covered by review article(s), we have attempted to cite the most relevant and informative reviews.
Conflicts of Interest
The authors declare no competing financial interests.
References
- Graybiel, A.M. The basal ganglia: Learning new tricks and loving it. Curr. Opin. Neurobiol. 2005, 15, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Packard, M.G.; Knowlton, B.J. Learning and Memory Functions of the Basal Ganglia. Annu. Rev. Neurosci. 2002, 25, 563–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surmeier, D.J.; Graves, S.M.; Shen, W. Dopaminergic modulation of striatal networks in health and Parkinson’s disease. Curr. Opin. Neurobiol. 2014, 29, 109–117. [Google Scholar] [PubMed]
- Zhai, S.; Shen, W.; Graves, S.M.; Surmeier, D.J. Dopaminergic modulation of striatal function and Parkinson’s disease. J. Neural Transm. 2019, 126, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Smith, Y.; Kieval, J.Z. Anatomy of the dopamine system in the basal ganglia. Trends Neurosci. 2000, 23, S28–S33. [Google Scholar] [PubMed]
- Tepper, J.M.; Bolam, J.P. Functional diversity and specificity of neostriatal interneurons. Curr. Opin. Neurobiol. 2004, 14, 685–692. [Google Scholar] [PubMed]
- Tepper, J.M.; Koós, T.; Ibanez-Sandoval, O.; Tecuapetla, F.; Faust, T.W.; Assous, M. Heterogeneity and Diversity of Striatal GABAergic Interneurons: Update 2018. Front. Neuroanat. 2018, 12, 91. [Google Scholar] [CrossRef] [Green Version]
- Albin, R.L.; Young, A.B.; Penney, J.B. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989, 12, 366–375. [Google Scholar] [CrossRef]
- Kravitz, A.V.; Freeze, B.S.; Parker, P.R.L.; Kay, K.; Thwin, M.T.; Deisseroth, K.; Kreitzer, A.C. Regulation of parkinsonian motor behaviors by optogenetic control of basal ganglia circuitry. Nature 2010, 466, 622–626. [Google Scholar] [CrossRef]
- Cui, G.; Jun, S.B.; Jin, X.; Pham, M.D.; Vogel, S.S.; Lovinger, D.M.; Costa, R.M. Concurrent Activation of Striatal Direct and Indirect Pathways During Action Initiation. Nature 2013, 494, 238–242. [Google Scholar]
- Mink, J.W. The Basal Ganglia and involuntary movements: Impaired inhibition of competing motor patterns. Arch. Neurol. 2003, 60, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Nambu, A.; Tokuno, H.; Hamada, I.; Kita, H.; Imanishi, M.; Akazawa, T.; Ikeuchi, Y.; Hasegawa, N. Excitatory Cortical Inputs to Pallidal Neurons Via the Subthalamic Nucleus in the Monkey. J. Neurophysiol. 2000, 84, 289–300. [Google Scholar]
- Bevan, M. Move to the rhythm: Oscillations in the subthalamic nucleus–external globus pallidus network. Trends Neurosci. 2002, 25, 525–531. [Google Scholar] [CrossRef]
- Mallet, N.; Micklem, B.R.; Henny, P.; Brown, M.T.; Williams, C.; Bolam, J.P.; Nakamura, K.C.; Magill, P.J. Dichotomous Organization of the External Globus Pallidus. Neuron 2012, 74, 1075–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallet, N.; Schmidt, R.; Leventhal, D.; Chen, F.; Amer, N.; Boraud, T.; Berke, J.D. Arkypallidal cells send a Stop signal to Striatum. Neuron 2016, 89, 308–316. [Google Scholar] [PubMed]
- Cazorla, M.; De Carvalho, F.D.; Chohan, M.O.; Shegda, M.; Chuhma, N.; Rayport, S.; Ahmari, S.E.; Moore, H.; Kellendonk, C. Dopamine D2 Receptors Regulate the Anatomical and Functional Balance of Basal Ganglia Circuitry. Neuron 2014, 81, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, Y.; Wilson, C.; Emson, P. Projection subtypes of rat neostriatal matrix cells revealed by intracellular injection of biocytin. J. Neurosci. 1990, 10, 3421–3438. [Google Scholar] [CrossRef] [Green Version]
- Atherton, J.F.; Bevan, M.D. Ionic Mechanisms Underlying Autonomous Action Potential Generation in the Somata and Dendrites of GABAergic Substantia Nigra Pars Reticulata Neurons In Vitro. J. Neurosci. 2005, 25, 8272–8281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurrier, C.; Bioulac, B.; Hammond, C. Slowly Inactivating Sodium Current (I NaP) Underlies Single-Spike Activity in Rat Subthalamic Neurons. J. Neurophysiol. 2000, 83, 1951–1957. [Google Scholar]
- Bevan, M.D.; Wilson, C.J. Mechanisms Underlying Spontaneous Oscillation and Rhythmic Firing in Rat Subthalamic Neurons. J. Neurosci. 1999, 19, 7617–7628. [Google Scholar]
- Chan, C.S.; Shigemoto, R.; Mercer, J.N.; Surmeier, D.J. HCN2 and HCN1 Channels Govern the Regularity of Autonomous Pacemaking and Synaptic Resetting in Globus Pallidus Neurons. J. Neurosci. 2004, 24, 9921–9932. [Google Scholar] [CrossRef] [PubMed]
- Baranauskas, G.; Tkatch, T.; Nagata, K.; Yeh, J.Z.; Surmeier, D.J. Kv3.4 subunits enhance the repolarizing efficiency of Kv3.1 channels in fast-spiking neurons. Nat. Neurosci. 2003, 6, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Wigmore, M.A.; Lacey, M.G. A Kv3-like persistent, outwardly rectifying, Cs+-permeable, K+ current in rat subthalamic nucleus neurones. J. Physiol. 2000, 527, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Nisenbaum, E.; Wilson, C. Potassium currents responsible for inward and outward rectification in rat neostriatal spiny projection neurons. J. Neurosci. 1995, 15, 4449–4463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nisenbaum, E.S.; Xu, Z.C.; Wilson, C.J. Contribution of a slowly inactivating potassium current to the transition to firing of neostriatal spiny projection neurons. J. Neurophysiol. 1994, 71, 1174–1189. [Google Scholar] [CrossRef] [PubMed]
- Pidoux, M.; Mahon, S.; Deniau, J.M.; Charpier, S. Integration and propagation of somatosensory responses in the corticostriatal pathway: An intracellular study in vivo. J. Physiol. 2011, 589, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.L.; Day, M.; Surmeier, D.J. Synaptically driven state transitions in distal dendrites of striatal spiny neurons. Nat. Neurosci. 2011, 14, 881–888. [Google Scholar] [PubMed]
- Reig, R.; Silberberg, G. Multisensory Integration in the Mouse Striatum. Neuron 2014, 83, 1200–1212. [Google Scholar] [CrossRef] [Green Version]
- Sippy, T.; Lapray, D.; Crochet, S.; Petersen, C.C. Cell-Type-Specific Sensorimotor Processing in Striatal Projection Neurons during Goal-Directed Behavior. Neuron 2015, 88, 298–305. [Google Scholar] [Green Version]
- Surmeier, D.J.; Mercer, J.N.; Chan, C.S. Autonomous pacemakers in the basal ganglia: Who needs excitatory synapses anyway? Curr. Opin. Neurobiol. 2005, 15, 312–318. [Google Scholar] [CrossRef]
- Bevan, M.D.; Magill, P.J.; Hallworth, N.E.; Bolam, J.P.; Wilson, C.J. Regulation of the Timing and Pattern of Action Potential Generation in Rat Subthalamic Neurons In Vitro by GABA-A IPSPs. J. Neurophysiol. 2002, 87, 1348–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farries, M.A.; Wilson, C.J. Phase response curves of subthalamic neurons measured with synaptic input and current injection. J. Neurophysiol. 2012, 108, 1822–1837. [Google Scholar] [PubMed]
- Wilson, C.J. Active Decorrelation in the Basal Ganglia. Neuroscience 2013, 250, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Bevan, M.D.; Hallworth, N.E.; Baufreton, J. GABAergic control of the subthalamic nucleus. Prog. Brain Res. 2007, 160, 173–188. [Google Scholar] [PubMed]
- Chan, C.S.; Surmeier, D.J.; Yung, W.-H. Striatal Information Signaling and Integration in Globus Pallidus: Timing Matters. Neurosignals 2005, 14, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Bevan, M.D.; Wilson, C.J.; Bolam, J.P.; Magill, P.J. Equilibrium potential of GABA(A) current and implications for rebound burst firing in rat subthalamic neurons in vitro. J. Neurophysiol. 2000, 83, 3169–3172. [Google Scholar] [CrossRef] [PubMed]
- Hallworth, N.E.; Bevan, M.D. Globus Pallidus Neurons Dynamically Regulate the Activity Pattern of Subthalamic Nucleus Neurons through the Frequency-Dependent Activation of Postsynaptic GABAA and GABAB Receptors. J. Neurosci. 2005, 25, 6304–6315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kita, H. Short-term plasticity shapes activity pattern-dependent striato-pallidal synaptic transmission. J. Neurophysiol. 2013, 109, 932–939. [Google Scholar] [CrossRef]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single Nigrostriatal Dopaminergic Neurons Form Widely Spread and Highly Dense Axonal Arborizations in the Neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Surmeier, D.J.; Carrillo-Reid, L.; Bargas, J. Dopaminergic modulation of striatal neurons, circuits and assemblies. Neuroscience 2011, 198, 3–18. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Plotkin, J.; Shen, W. Dopamine and synaptic plasticity in dorsal striatal circuits controlling action selection. Curr. Opin. Neurobiol. 2009, 19, 621–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossette, M.; Levesque, M.; Parent, A. Extrastriatal dopaminergic innervation of human basal ganglia. Neurosci. Res. 1999, 34, 51–54. [Google Scholar] [CrossRef]
- Cragg, S.J.; Baufreton, J.; Xue, Y.; Bolam, J.P.; Bevan, M.D. Synaptic release of dopamine in the subthalamic nucleus. Eur. J. Neurosci. 2004, 20, 1788–1802. [Google Scholar] [CrossRef] [PubMed]
- Debeir, T.; Ginestet, L.; François, C.; Laurens, S.; Martel, J.-C.; Chopin, P.; Marien, M.; Colpaert, F.; Raisman-Vozari, R. Effect of intrastriatal 6-OHDA lesion on dopaminergic innervation of the rat cortex and globus pallidus. Exp. Neurol. 2005, 193, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Hassani, O.-K.; François, C.; Yelnik, J.; Féger, J. Evidence for a dopaminergic innervation of the subthalamic nucleus in the rat. Brain Res. 1997, 749, 88–94. [Google Scholar] [CrossRef]
- Rommelfanger, K.S.; Wichmann, T. Extrastriatal Dopaminergic Circuits of the Basal Ganglia. Front. Neuroanat. 2010, 4, 139. [Google Scholar] [CrossRef]
- Cáceres-Chávez, V.A.; Hernández-Martinez, R.; Pérez-Ortega, J.; Herrera-Valdez, M.A.; Aceves, J.J.; Galarraga, E.; Bargas, J. Acute dopamine receptor blockade in substantia nigra pars reticulata: A possible model for drug-induced Parkinsonism. J. Neurophysiol. 2018, 120, 2922–2938. [Google Scholar] [CrossRef]
- Hassani, O.-K.; Féger, J. Effects of intrasubthalamic injection of dopamine receptor agonists on subthalamic neurons in normal and 6-hydroxydopamine-lesioned rats: An electrophysiological and c-Fos study. Neuroscience 1999, 92, 533–543. [Google Scholar] [CrossRef]
- Mamad, O.; Delaville, C.; Benjelloun, W.; Benazzouz, A. Dopaminergic Control of the Globus Pallidus through Activation of D2 Receptors and Its Impact on the Electrical Activity of Subthalamic Nucleus and Substantia Nigra Reticulata Neurons. PLOS ONE 2015, 10, e0119152. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-C.; Xue, Y.; Diao, H.-L.; Chen, H.; Liu, H.-Y.; Han, X.-H.; Chen, L. Direct modulation of firing activity by dopamine D2 like receptors in the globus pallidus of both normal and parkinsonian rats. Sheng li xue bao [Acta Physiol. Sin.] 2016, 68, 699–707. [Google Scholar]
- Cooper, A.; Stanford, I. Calbindin D-28k positive projection neurones and calretinin positive interneurones of the rat globus pallidus. Brain Res. 2002, 929, 243–251. [Google Scholar] [CrossRef]
- Cooper, A.J.; Stanford, I.M. Electrophysiological and morphological characteristics of three subtypes of rat globus pallidus neurone in vitro. J. Physiol. 2000, 527, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Günay, C.; Edgerton, J.R.; Jaeger, D. Channel Density Distributions Explain Spiking Variability in the Globus Pallidus: A Combined Physiology and Computer Simulation Database Approach. J. Neurosci. 2008, 28, 7476–7491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoover, B.; Marshall, J. Further characterization of preproenkephalin mRNA-containing cells in the rodent globus pallidus. Neuroscience 2002, 111, 111–125. [Google Scholar] [CrossRef]
- Hoover, B.R.; Marshall, J.F. Molecular, chemical, and anatomical characterization of globus pallidus dopamine D2 receptor mRNA-containing neurons. Synapse 2004, 52, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Kita, H.; Kitai, S. Intracellular study of rat globus pallidus neurons: Membrane properties and responses to neostriatal, subthalamic and nigral stimulation. Brain Res. 1991, 564, 296–305. [Google Scholar] [CrossRef]
- Bugaysen, J.; Bronfeld, M.; Tischler, H.; Bar-Gad, I.; Korngreen, A. Electrophysiological Characteristics of Globus Pallidus Neurons. PLOS ONE 2010, 5, e12001. [Google Scholar] [CrossRef] [PubMed]
- Nóbrega-Pereira, S.; Gelman, D.; Bartolini, G.; Pla, R.; Pierani, A.; Marín, O. Origin and Molecular Specification of Globus Pallidus Neurons. J. Neurosci. 2010, 30, 2824–2834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; De Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030. [Google Scholar] [CrossRef]
- Assaf, F.; Schiller, Y. A chemogenetic approach for treating experimental Parkinson’s disease. Mov. Disord. 2019, 34, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Mastro, K.J.; Zitelli, K.T.; Willard, A.M.; Leblanc, K.H.; Kravitz, A.V.; Gittis, A.H. Cell-Specific Pallidal Intervention Induces Long-Lasting Motor Recovery in Dopamine Depleted Mice. Nat. Neurosci. 2017, 20, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Yan, Y.; Xi, W.; Zhou, R.; Lou, H.; Duan, S.; Chen, J.F.; Zhang, B. Optogenetic Stimulation of GABAergic Neurons in the Globus Pallidus Produces Hyperkinesia. Front. Behav. Neurosci. 2018, 12, 185. [Google Scholar] [CrossRef] [PubMed]
- Abdi, A.; Mallet, N.; Mohamed, F.Y.; Sharott, A.; Dodson, P.D.; Nakamura, K.C.; Suri, S.; Avery, S.V.; Larvin, J.T.; Garas, F.N.; et al. Prototypic and Arkypallidal Neurons in the Dopamine-Intact External Globus Pallidus. J. Neurosci. 2015, 35, 6667–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrahao, K.P.; Lovinger, D.M. Classification of GABAergic neuron subtypes from the globus pallidus using wild-type and transgenic mice. J. Physiol. 2018, 596, 4219–4235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, V.M.; Hegeman, D.J.; Cui, Q.; Kelver, D.A.; Fiske, M.P.; Glajch, K.E.; Pitt, J.E.; Huang, T.Y.; Justice, N.J.; Chan, C.S. Parvalbumin+ Neurons and Npas1+ Neurons Are Distinct Neuron Classes in the Mouse External Globus Pallidus. J. Neurosci. 2015, 35, 11830–11847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastro, K.J.; Bouchard, R.S.; Holt, H.A.K.; Gittis, A.H. Transgenic Mouse Lines Subdivide External Segment of the Globus Pallidus (GPe) Neurons and Reveal Distinct GPe Output Pathways. J. Neurosci. 2014, 34, 2087–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegeman, D.J.; Hong, E.S.; Hernández, V.M.; Chan, C.S. The External Globus Pallidus: Progress and Perspectives. Eur. J. Neurosci. 2016, 43, 1239–1265. [Google Scholar] [PubMed]
- Fujiyama, F.; Nakano, T.; Matsuda, W.; Furuta, T.; Udagawa, J.; Kaneko, T. A single-neuron tracing study of arkypallidal and prototypic neurons in healthy rats. Brain Struct. Funct. 2016, 221, 4733–4740. [Google Scholar] [PubMed]
- Saunders, A.; Huang, K.W.; Sabatini, B.L. Globus Pallidus Externus Neurons Expressing parvalbumin Interconnect the Subthalamic Nucleus and Striatal Interneurons. PLOS ONE 2016, 11, e0149798. [Google Scholar] [CrossRef] [PubMed]
- Dodson, P.D.; Larvin, J.T.; Duffell, J.M.; Garas, F.N.; Doig, N.M.; Kessaris, N.; Duguid, I.C.; Bogacz, R.; Butt, S.J.; Magill, P.J. Distinct Developmental Origins Manifest in the Specialized Encoding of Movement by Adult Neurons of the External Globus Pallidus. Neuron 2015, 86, 501–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallet, N.; Pogosyan, A.; Márton, L.F.; Bolam, J.P.; Brown, P.; Magill, P.J. Parkinsonian Beta Oscillations in the External Globus Pallidus and Their Relationship with Subthalamic Nucleus Activity. J. Neurosci. 2008, 28, 14245–14258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kita, H.; Kita, T. Role of Striatum in the Pause and Burst Generation in the Globus Pallidus of 6-OHDA-Treated Rats. Front. Syst. Neurosci. 2011, 5, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharott, A.; Vinciati, F.; Nakamura, K.C.; Magill, P.J. A Population of Indirect Pathway Striatal Projection Neurons Is Selectively Entrained to Parkinsonian Beta Oscillations. J. Neurosci. 2017, 37, 9977–9998. [Google Scholar] [Green Version]
- Zold, C.L.; Ballion, B.; Riquelme, L.A.; Gonon, F.; Murer, M.G. Nigrostriatal lesion induces D2-modulated phase-locked activity in the basal ganglia of rats. Eur. J. Neurosci. 2007, 25, 2131–2144. [Google Scholar] [PubMed]
- Yuan, X.S.; Wang, L.; Dong, H.; Qu, W.M.; Yang, S.R.; Cherasse, Y.; Lazarus, M.; Schiffmann, S.N.; d’Exaerde, A.K.; Li, R.X.; et al. Striatal adenosine A2A receptor neurons control active-period sleep via parvalbumin neurons in external globus pallidus. ELife 2017, 6, e29055. [Google Scholar] [CrossRef] [PubMed]
- Filion, M.; Tremblay, L. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res. 1991, 547, 140–144. [Google Scholar]
- Raz, A.; Frechter-Mazar, V.; Feingold, A.; Abeles, M.; Vaadia, E.; Bergman, H. Activity of Pallidal and Striatal Tonically Active Neurons Is Correlated in MPTP-Treated Monkeys But Not in Normal Monkeys. J. Neurosci. 2001, 21, RC128. [Google Scholar] [CrossRef]
- Raz, A.; Vaadia, E.; Bergman, H. Firing Patterns and Correlations of Spontaneous Discharge of Pallidal Neurons in the Normal and the Tremulous 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Vervet Model of Parkinsonism. J. Neurosci. 2000, 20, 8559–8571. [Google Scholar]
- Deister, C.A.; Dodla, R.; Barraza, D.; Kita, H.; Wilson, C.J. Firing rate and pattern heterogeneity in the globus pallidus arise from a single neuronal population. J. Neurophysiol. 2013, 109, 497–506. [Google Scholar] [CrossRef]
- Mercer, J.N.; Chan, C.S.; Tkatch, T.; Held, J.; Surmeier, D.J. Nav1.6 Sodium Channels Are Critical to Pacemaking and Fast Spiking in Globus Pallidus Neurons. J. Neurosci. 2007, 27, 13552–13566. [Google Scholar] [CrossRef]
- Deister, C.A.; Chan, C.S.; Surmeier, D.J.; Wilson, C.J. Calcium-activated SK channels influence voltage-gated ion channels to determine the precision of firing in globus pallidus neurons. J. Neurosci. 2009, 29, 8452–8461. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.Y.; Sims, J.R.; Bhide, P.G. Dopamine receptor mRNA and protein expression in the mouse corpus striatum and cerebral cortex during pre- and post-natal development. Brain Res. 2007, 1156, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, H.; Hauber, W. Reverse microdialysis of ionotropic glutamate receptor agonists in the rat globus pallidus increased extracellular dopamine. Neurosci. Lett. 2003, 343, 37–40. [Google Scholar] [CrossRef]
- Hauber, W.; Fuchs, H. Dopamine release in the rat globus pallidus characterised by in vivo microdialysis. Behav. Brain Res. 2000, 111, 39–44. [Google Scholar] [CrossRef]
- Meszaros, J.; Cheung, T.; Erler, M.M.; Kang, U.J.; Sames, D.; Kellendonk, C.; Sulzer, D. Evoked transients of pH-sensitive fluorescent false neurotransmitter reveal dopamine hot spots in the globus pallidus. eLife 2018, 7, e42383. [Google Scholar] [CrossRef] [PubMed]
- Stefani, A.; Spadoni, F.; Martorana, A.; Lavaroni, F.; Martella, G.; Sancesario, G.; Bernardi, G. D2-mediated modulation of N-type calcium currents in rat globus pallidus neurons following dopamine denervation. Eur. J. Neurosci. 2002, 15, 815–825. [Google Scholar]
- Napier, T.C.; Simson, P.E.; Givens, B.S. Dopamine electrophysiology of ventral pallidal/substantia innominata neurons: Comparison with the dorsal globus pallidus. J. Pharmacol. Exp. Ther. 1991, 258, 249–262. [Google Scholar] [PubMed]
- Chan, C.S.; Glajch, K.E.; Gertler, T.S.; Guzman, J.N.; Mercer, J.N.; Lewis, A.S.; Goldberg, A.B.; Tkatch, T.; Shigemoto, R.; Fleming, S.M.; et al. HCN channelopathy in external globus pallidus neurons in models of Parkinson’s disease. Nat. Neurosci. 2011, 14, 85–92. [Google Scholar] [CrossRef]
- Glajch, K.E.; Kelver, D.A.; Hegeman, D.J.; Cui, Q.; Xenias, H.S.; Augustine, E.C.; Hernandez, V.M.; Verma, N.; Huang, T.Y.; Luo, M.; et al. Npas1+ Pallidal Neurons Target Striatal Projection Neurons. J. Neurosci. 2016, 36, 5472–5488. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, K.; Takahashi, S.; Okamoto, S.; Karube, F.; Fujiyama, F. Substance P effects exclusively on prototypic neurons in mouse globus pallidus. Brain Struct. Funct. 2017, 222, 4089–4110. [Google Scholar] [CrossRef]
- Sadek, A.R.; Magill, P.J.; Bolam, J.P. A Single-Cell Analysis of Intrinsic Connectivity in the Rat Globus Pallidus. J. Neurosci. 2007, 27, 6352–6362. [Google Scholar] [CrossRef] [PubMed]
- Ingham, C.A.; Hood, S.H.; Mijnster, M.J.; Baldock, R.A.; Arbuthnott, G.W.; Baldock, R. Plasticity of striatopallidal terminals following unilateral lesion of the dopaminergic nigrostriatal pathway: A morphological study. Exp. Brain Res. 1997, 116, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Miguelez, C.; Morin, S.; Martinez, A.; Goillandeau, M.; Bezard, E.; Bioulac, B.; Baufreton, J. Altered pallido-pallidal synaptic transmission leads to aberrant firing of globus pallidus neurons in a rat model of Parkinson’s disease. J. Physiol. 2012, 590, 5861–5875. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.E.; Woodhall, G.L.; Wilson, C.L.; Stanford, I.M. Functional characterization of GABAergic pallidopallidal and striatopallidal synapses in the rat globus pallidus in vitro. Eur. J. Neurosci. 2008, 28, 2401–2408. [Google Scholar] [CrossRef] [PubMed]
- Charara, A.; Smith, Y.; Parent, A. Glutamatergic inputs from the pedunculopontine nucleus to midbrain dopaminergic neurons in primates: Phaseolus vulgaris-leucoagglutinin anterograde labeling combined with postembedding glutamate and GABA immunohistochemistry. J. Comp. Neurol. 1996, 364, 254–266. [Google Scholar] [CrossRef]
- Chen, L.; Chan, S.C.Y.; Yung, W.H. Rotational behavior and electrophysiological effects induced by GABA(B) receptor activation in rat globus pallidus. Neuroscience 2002, 114, 417–425. [Google Scholar] [CrossRef]
- Engler, B.; Freiman, I.; Urbanski, M.; Szabo, B. Effects of exogenous and endogenous cannabinoids on GABAergic neurotransmission between the caudate-putamen and the globus pallidus in the mouse. J. Pharmacol. Exp. Ther. 2006, 316, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, K.; Kita, H. Synaptically Released GABA Activates Both Pre- and Postsynaptic GABABReceptors in the Rat Globus Pallidus. J. Neurophysiol. 2005, 94, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Stanford, I.M.; Cooper, A.J. Presynaptic mu and delta opioid receptor modulation of GABAA IPSCs in the rat globus pallidus in vitro. J. Neurosci. 1999, 19, 4796–4803. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Kita, H. Activation of group III metabotropic glutamate receptors presynaptically reduces both GABAergic and glutamatergic transmission in the rat globus pallidus. Neuroscience 2003, 122, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Kita, H. Dynorphin Exerts Both Postsynaptic and Presynaptic Effects in the Globus Pallidus of the Rat. J. Neurophysiol. 2000, 83, 3366–3376. [Google Scholar] [CrossRef] [PubMed]
- Valenti, O.; Marino, M.J.; Wittmann, M.; Lis, E.; DiLella, A.G.; Kinney, G.G.; Conn, P.J. Group III Metabotropic Glutamate Receptor-Mediated Modulation of the Striatopallidal Synapse. J. Neurosci. 2003, 23, 7218–7226. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; Stanford, I.M. Dopamine D2 receptor mediated presynaptic inhibition of striatopallidal GABA(A) IPSCs in vitro. Neuropharmacology 2001, 41, 62–71. [Google Scholar] [CrossRef]
- Bugaysen, J.; Bar-Gad, I.; Korngreen, A. Continuous Modulation of Action Potential Firing by a Unitary GABAergic Connection in the Globus Pallidus In Vitro. J. Neurosci. 2013, 33, 12805–12809. [Google Scholar] [CrossRef] [PubMed]
- Shin, R.-M.; Masuda, M.; Miura, M.; Sano, H.; Shirasawa, T.; Song, W.-J.; Kobayashi, K.; Aosaki, T. Dopamine D4 receptor-induced postsynaptic inhibition of GABAergic currents in mouse globus pallidus neurons. J. Neurosci. 2003, 23, 11662–11672. [Google Scholar] [CrossRef] [PubMed]
- Charron, G.; Doudnikoff, E.; Canron, M.H.; Li, Q.; Vega, C.; Marais, S.; Baufreton, J.; Vital, A.; Oliet, S.H.; Bezard, E. Astrocytosis in parkinsonism: Considering tripartite striatal synapses in physiopathology? Front. Aging Neurosci. 2014, 6, 258. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, K.; Okuda, H.; Morita-Takemura, S.; Tanaka, T.; Isonishi, A.; Shinjo, T.; Terada, Y.; Wanaka, A. Voluntary Exercise Induces Astrocytic Structural Plasticity in the Globus Pallidus. Front. Cell. Neurosci. 2016, 10, 366. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Barres, B.A. Neuroscience: Glia—More than just brain glue. Nature 2009, 457, 675–677. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Fellin, T.; Takano, H.; Dong, J.-H.; Haydon, P.G. Synaptic Islands Defined by the Territory of a Single Astrocyte. J. Neurosci. 2007, 27, 6473–6477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volterra, A.; Meldolesi, J. Astrocytes, from brain glue to communication elements: The revolution continues. Nat. Rev. Neurosci. 2005, 6, 626–640. [Google Scholar] [CrossRef]
- Galvan, A.; Hu, X.; Smith, Y.; Wichmann, T. Localization and function of GABA transporters in the globus pallidus of parkinsonian monkeys. Exp. Neurol. 2010, 223, 505–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.-T.; Pare, J.-F.; Smith, Y. Differential localization and function of GABA transporters, GAT-1 and GAT-3, in the rat globus pallidus. Eur. J. Neurosci. 2011, 33, 1504–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Q.; Pitt, J.E.; Pamukcu, A.; Poulin, J.-F.; Mabrouk, O.S.; Fiske, M.P.; Fan, I.B.; Augustine, E.C.; Young, K.A.; Kennedy, R.T.; et al. Blunted mGluR Activation Disinhibits Striatopallidal Transmission in Parkinsonian Mice. Cell Rep. 2016, 17, 2431–2444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chazalon, M.; Paredes-Rodriguez, E.; Morin, S.; Martinez, A.; Cristóvão-Ferreira, S.; Vaz, S.; Sebastião, A.; Panatier, A.; Boué-Grabot, E.; Miguelez, C.; et al. GAT-3 Dysfunction Generates Tonic Inhibition in External Globus Pallidus Neurons in Parkinsonian Rodents. Cell Rep. 2018, 23, 1678–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochi, M.; Koga, K.; Kurokawa, M.; Kase, H.; Nakamura, J.; Kuwana, Y. Systemic administration of adenosine A2A receptor antagonist reverses increased GABA release in the globus pallidus of unilateral 6-hydroxydopamine-lesioned rats: a microdialysis study. Neuroscience 2000, 100, 53–62. [Google Scholar] [CrossRef]
- Ramanathan, S.; Tkatch, T.; Atherton, J.F.; Wilson, C.J.; Bevan, M.D. D2-Like Dopamine Receptors Modulate SKCaChannel Function in Subthalamic Nucleus Neurons Through Inhibition of Cav2.2 Channels. J. Neurophysiol. 2008, 99, 442–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svenningsson, P.; Le Moine, C. Dopamine D1/5 receptor stimulation induces c- fos expression in the subthalamic nucleus: Possible involvement of local D5 receptors. Eur. J. Neurosci. 2002, 15, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Baufreton, J.; Zhu, Z.-T.; Garret, M.; Bioulac, B.; Johnson, S.W.; Taupignon, A.I. Dopamine receptors set the pattern of activity generated in subthalamic neurons. FASEB J. 2005, 19, 1771–1777. [Google Scholar] [CrossRef] [Green Version]
- Beurrier, C.; Congar, P.; Bioulac, B.; Hammond, C. Subthalamic Nucleus Neurons Switch from Single-Spike Activity to Burst-Firing Mode. J. Neurosci. 1999, 19, 599–609. [Google Scholar] [CrossRef]
- Baufreton, J.; Garret, M.; Rivera, A.; De La Calle, A.; Gonon, F.; Dufy, B.; Bioulac, B.; Taupignon, A. D5 (Not D1) Dopamine Receptors Potentiate Burst-Firing in Neurons of the Subthalamic Nucleus by Modulating an L-Type Calcium Conductance. J. Neurosci. 2003, 23, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Loucif, K.C.; Wilson, C.L.; Baig, R.; Lacey, M.G.; Stanford, I.M. Functional interconnectivity between the globus pallidus and the subthalamic nucleus in the mouse brain slice. J. Physiol. 2005, 567, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Hutchison, W.D.; Lozano, A.M.; Dostrovsky, J.O. High-frequency Synchronization of Neuronal Activity in the Subthalamic Nucleus of Parkinsonian Patients with Limb Tremor. J. Neurosci. 2000, 20, 7766–7775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.; Cash, D.; Galley, K.; Chapman, H.; Lacey, M.; Stanford, I.M. Subthalamic nucleus neurones in slices from 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mice show irregular, dopamine-reversible firing pattern changes, but without synchronous activity. Neuroscience 2006, 143, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Bartol, M.; Shen, K.; Johnson, S.W. Excitatory effects of dopamine on subthalamic nucleus neurons: In vitro study of rats pretreated with 6-hydroxydopamine and levodopa. Brain Res. 2002, 945, 31–40. [Google Scholar] [CrossRef]
- McIver, E.L.; Atherton, J.F.; Chu, H.-Y.; Cosgrove, K.E.; Kondapalli, J.; Wokosin, D.; Surmeier, D.J.; Bevan, M.D. Maladaptive Downregulation of Autonomous Subthalamic Nucleus Activity following the Loss of Midbrain Dopamine Neurons. Cell Rep. 2019, 28, 992–1002.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, K.-Z.; Johnson, S.W. Chronic dopamine depletion augments the functional expression of K-ATP channels in the rat subthalamic nucleus. Neurosci. Lett. 2012, 531, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Zhang, J.-R.; Chen, L.; Ge, S.-N.; Wang, J.-L.; Yan, Z.-Q.; Jia, D.; Zhu, J.-L.; Gao, G.-D. Decreased HCN2 expression in STN contributes to abnormal high-voltage spindles in the cortex and globus pallidus of freely moving rats. Brain Res. 2015, 1618, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Baufreton, J.; Kirkham, E.; Atherton, J.F.; Menard, A.; Magill, P.J.; Bolam, J.P.; Bevan, M.D. Sparse but Selective and Potent Synaptic Transmission From the Globus Pallidus to the Subthalamic Nucleus. J. Neurophysiol. 2009, 102, 532–545. [Google Scholar] [CrossRef]
- Urbain, N.; Gervasoni, D.; Soulière, F.; Lobo, L.; Rentero, N.; Windels, F.; Astier, B.; Savasta, M.; Fort, P.; Renaud, B.; et al. Unrelated course of subthalamic nucleus and globus pallidus neuronal activities across vigilance states in the rat. Eur. J. Neurosci. 2000, 12, 3361–3374. [Google Scholar] [CrossRef] [PubMed]
- Atherton, J.F.; Menard, A.; Urbain, N.; Bevan, M.D. Short-term depression of external globus pallidus-subthalamic nucleus synaptic transmission and implications for patterning subthalamic activity. J. Neurosci. 2013, 33, 7130–7144. [Google Scholar] [CrossRef] [PubMed]
- Baufreton, J.; Bevan, M.D. D2-like dopamine receptor-mediated modulation of activity-dependent plasticity at GABAergic synapses in the subthalamic nucleus. J. Physiol. 2008, 586, 2121–2142. [Google Scholar] [PubMed]
- Abbott, L.F.; Regehr, W.G. Synaptic computation. Nature 2004, 431, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.Y.; Baufreton, J.; Surmeier, D.J.; Chan, C.S.; Bevan, M.D. Proliferation of external globus pallidus-subthalamic nucleus synapses following degeneration of midbrain dopamine neurons. J. Neurosci. 2012, 32, 13718–13728. [Google Scholar] [PubMed]
- Chu, H.-Y.; Atherton, J.F.; Wokosin, D.; Surmeier, D.J.; Bevan, M.D. Heterosynaptic Regulation of External Globus Pallidus Inputs to the Subthalamic Nucleus by the Motor Cortex. Neuron 2015, 85, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Bevan, M.D.; Francis, C.M.; Bolam, J.P. The glutamate-enriched cortical and thalamic input to neurons in the subthalamic nucleus of the rat: Convergence with GABA-positive terminals. J. Comp. Neurol. 1995, 361, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Mathai, A.; Smith, Y. The Corticostriatal and Corticosubthalamic Pathways: Two Entries, One Target. So What? Front. Syst. Neurosci. 2011, 5, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froux, L.; Le Bon-Jego, M.; Miguelez, C.; Normand, E.; Morin, S.; Fioramonti, S.; Barresi, M.; Frick, A.; Baufreton, J.; Taupignon, A. D5 dopamine receptors control glutamatergic AMPA transmission between the motor cortex and subthalamic nucleus. Sci. Rep. 2018, 8, 8858. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.-Z.; Johnson, S.W. Presynaptic dopamine D2 and muscarine M3 receptors inhibit excitatory and inhibitory transmission to rat subthalamic neurones in vitro. J. Physiol. 2000, 525, 331–341. [Google Scholar]
- Chu, H.-Y.; McIver, E.L.; Kovaleski, R.F.; Atherton, J.F.; Bevan, M.D. Loss Of Hyperdirect Pathway Cortico-Subthalamic Inputs Following Degeneration Of Midbrain Dopamine Neurons. Neuron 2017, 95, 1306–1318.e5. [Google Scholar] [CrossRef]
- Mathai, A.; Ma, Y.; Paré, J.-F.; Villalba, R.M.; Wichmann, T.; Smith, Y. Reduced cortical innervation of the subthalamic nucleus in MPTP-treated parkinsonian monkeys. Brain 2015, 138, 946–962. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-Y.; Wang, Y.; Jiang, H.-F.; Liu, J.-H.; Jia, J.; Wang, K.; Zhao, F.; Luo, M.-H.; Luo, M.-M.; Wang, X.-M. Impaired glutamatergic projection from the motor cortex to the subthalamic nucleus in 6-hydroxydopamine-lesioned hemi-parkinsonian rats. Exp. Neurol. 2018, 300, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Gradinaru, V.; Mogri, M.; Thompson, K.R.; Henderson, J.M.; Deisseroth, K. Optical Deconstruction of Parkinsonian Neural Circuitry. Science 2009, 324, 354–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, T.H.; Jaeger, D. Optogenetic Stimulation Of Cortico-Subthalamic Projections Is Sufficient To Ameliorate Bradykinesia In 6-Ohda Lesioned Mice. Neurobiol. Dis. 2016, 95, 225–237. [Google Scholar] [PubMed]
- Kha, H.T.; Finkelstein, D.I.; Tomas, D.; Drago, J.; Pow, D.V.; Horne, M.K. Projections from the substantia nigra pars reticulata to the motor thalamus of the rat: Single axon reconstructions and immunohistochemical study. J. Comp. Neurol. 2001, 440, 20–30. [Google Scholar] [PubMed]
- Cebrián, C.; Parent, A.; Prensa, L. Patterns of axonal branching of neurons of the substantia nigra pars reticulata and pars lateralis in the rat. J. Comp. Neurol. 2005, 492, 349–369. [Google Scholar] [CrossRef] [PubMed]
- Hikosaka, O. GABAergic output of the basal ganglia. Prog. Brain Res. 2007, 160, 209–226. [Google Scholar] [PubMed] [Green Version]
- Deniau, J.; Mailly, P.; Maurice, N.; Charpier, S. The pars reticulata of the substantia nigra: A window to basal ganglia output. Prog. Brain Res. 2007, 160, 151–172. [Google Scholar]
- Brown, J.; Pan, W.-X.; Dudman, J.T. The inhibitory microcircuit of the substantia nigra provides feedback gain control of the basal ganglia output. eLife 2014, 3, e02397. [Google Scholar] [CrossRef]
- Higgs, M.H.; Wilson, C.J. Unitary synaptic connections among substantia nigra pars reticulata neurons. J. Neurophysiol. 2016, 115, 2814–2829. [Google Scholar]
- González-Hernández, T.; Rodríguez, M. Compartmental organization and chemical profile of dopaminergic and GABAergic neurons in the substantia nigra of the rat. J. Comp. Neurol. 2000, 421, 107–135. [Google Scholar] [CrossRef]
- Rajakumar, N.; Elisevich, K.; Flumerfelt, B.A. Parvalbumin-containing GABAergic neurons in the basal ganglia output system of the rat. J. Comp. Neurol. 1994, 350, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Anderson, K.D. Co-occurrence of gamma-aminobutyric acid, parvalbumin and the neurotensin-related neuropeptide LANT6 in pallidal, nigral and striatal neurons in pigeons and monkeys. Brain Res. 1993, 624, 317–325. [Google Scholar] [CrossRef]
- Liang, C.-L.; Sinton, C.; German, D. Midbrain dopaminergic neurons in the mouse: Co-localization with Calbindin-D28k and calretinin. Neuroscience 1996, 75, 523–533. [Google Scholar] [CrossRef]
- McRitchie, D.; Hardman, C.; Halliday, G.; Halliday, G. Cytoarchitectural distribution of calcium binding proteins in midbrain dopaminergic regions of rats and humans. J. Comp. Neurol. 1996, 364, 121–150. [Google Scholar] [CrossRef]
- Martïanez-Murillo, R.; Villalba, R.; Montero-Caballero, M.I.; Rodrigo, J. Cholinergic somata and terminals in the rat substantia nigra: An immunocytochemical study with optical and electron microscopic techniques. J. Comp. Neurol. 1989, 281, 397–415. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.K.; Yung, K.K. Distinct cellular distribution of GABA(B)R1 and GABA(A)alpha1 receptor immunoreactivity in the rat substantia nigra. Neuroscience 2000, 99, 65–76. [Google Scholar] [CrossRef]
- Ng, T.K.; Yung, K.K. Subpopulations of neurons in rat substantia nigra display GABA(B)R2 receptor immunoreactivity. Brain Res. 2001, 920, 210–216. [Google Scholar] [CrossRef]
- Amadio, S.; Montilli, C.; Picconi, B.; Calabresi, P.; Volonté, C. Mapping P2X and P2Y receptor proteins in striatum and substantia nigra: An immunohistological study. Purinergic Signal. 2007, 3, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, M.; González-Hernández, T. Electrophysiological and Morphological Evidence for a GABAergic Nigrostriatal Pathway. J. Neurosci. 1999, 19, 4682–4694. [Google Scholar] [Green Version]
- Lee, C.R.; Tepper, J.M. Morphological and physiological properties of parvalbumin- and calretinin-containing gamma-aminobutyric acidergic neurons in the substantia nigra. J. Comp. Neurol. 2007, 500, 958–972. [Google Scholar] [CrossRef]
- Barter, J.W.; Castro, S.; Sukharnikova, T.; Rossi, M.A.; Yin, H.H. The role of the substantia nigra in posture control. Eur. J. Neurosci. 2014, 39, 1465–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barter, J.W.; Li, S.; Sukharnikova, T.; Rossi, M.A.; Bartholomew, R.A.; Yin, H.H. Basal Ganglia Outputs Map Instantaneous Position Coordinates during Behavior. J. Neurosci. 2015, 35, 2703–2716. [Google Scholar] [CrossRef] [PubMed]
- Bodor, Á.L.; Giber, K.; Rovó, Z.; Ulbert, I.; Acsády, L. Structural Correlates of Efficient GABAergic Transmission in the Basal Ganglia-Thalamus Pathway. J. Neurosci. 2008, 28, 3090–3102. [Google Scholar] [CrossRef] [PubMed]
- Kase, D.; Uta, D.; Ishihara, H.; Imoto, K. Inhibitory synaptic transmission from the substantia nigra pars reticulata to the ventral medial thalamus in mice. Neurosci. Res. 2015, 97, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, E.; Fujiyama, F.; Nakamura, K.C.; Tanaka, Y.; Hioki, H.; Kaneko, T. Complementary distribution of glutamatergic cerebellar and GABAergic basal ganglia afferents to the rat motor thalamic nuclei. Eur. J. Neurosci. 2011, 33, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Lutas, A.; Birnbaumer, L.; Yellen, G. Metabolism Regulates the Spontaneous Firing of Substantia Nigra Pars Reticulata Neurons via KATP and Nonselective Cation Channels. J. Neurosci. 2014, 34, 16336–16347. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.-W.; Matta, S.G.; Zhou, F.-M. Constitutively Active TRPC3 Channels Regulate Basal Ganglia Output Neurons. J. Neurosci. 2008, 28, 473–482. [Google Scholar] [CrossRef]
- Lutas, A.; Lahmann, C.; Soumillon, M.; Yellen, G. The leak channel NALCN controls tonic firing and glycolytic sensitivity of substantia nigra pars reticulata neurons. eLife 2016, 5, e15271. [Google Scholar] [CrossRef]
- Ding, S.; Matta, S.G.; Zhou, F.M. Kv3-like potassium channels are required for sustained high-frequency firing in basal ganglia output neurons. J. Neurophysiol. 2011, 105, 554–570. [Google Scholar] [CrossRef]
- Zhou, F.-M.; Lee, C.R. Intrinsic and integrative properties of substantia nigra pars reticulata neurons. Neuroscience 2011, 198, 69–94. [Google Scholar] [CrossRef] [Green Version]
- Rice, M.E.; Patel, J.C. Somatodendritic dopamine release: Recent mechanistic insights. Philos. Trans. R. Soc. B: Boil. Sci. 2015, 370, 20140185. [Google Scholar] [CrossRef] [PubMed]
- Ciliax, B.J.; Nash, N.; Heilman, C.; Sunahara, R.; Hartney, A.; Tiberi, M.; Rye, D.B.; Caron, M.G.; Niznik, H.B.; Levey, A.I. Dopamine D5 receptor immunolocalization in rat and monkey brain. Synapse 2000, 37, 125–145. [Google Scholar] [CrossRef]
- Khan, Z.; Gutierrez, A.; Martín, R.; Penafiel, A.; Rivera, A.; De La Calle, A.; Khan, Z. Dopamine D5 receptors of rat and human brain. Neuroscience 2000, 100, 689–699. [Google Scholar] [CrossRef]
- Kliem, M.A.; Pare, J.-F.; Khan, Z.U.; Wichmann, T.; Smith, Y. Ultrastructural localization and function of dopamine D1-like receptors in the substantia nigra pars reticulata and the internal segment of the globus pallidus of parkinsonian monkeys. Eur. J. Neurosci. 2010, 31, 836–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrzljak, L.; Bergson, C.; Pappy, M.; Huff, R.; Levenson, R.; Goldman-Rakic, P.S. Localization of dopamine D4 receptors in GABAergic neurons of the primate brain. Nature 1996, 381, 245–248. [Google Scholar] [CrossRef]
- Rivera, A.; Trías, S.; Peñafiel, A.; Narváez, J.A.; Díaz-Cabiale, Z.; Moratalla, R.; De La Calle, A. Expression of D4 dopamine receptors in striatonigral and striatopallidal neurons in the rat striatum. Brain Res. 2003, 989, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Nagatomo, K.; Suga, S.; Saitoh, M.; Kogawa, M.; Kobayashi, K.; Yamamoto, Y.; Yamada, K. Dopamine D1 Receptor Immunoreactivity on Fine Processes of GFAP-Positive Astrocytes in the Substantia Nigra Pars Reticulata of Adult Mouse. Front. Neuroanat. 2017, 11, 182. [Google Scholar] [CrossRef]
- Zhou, F.-W.; Jin, Y.; Matta, S.G.; Xu, M.; Zhou, F.-M. An ultra-short dopamine pathway regulates basal ganglia output. J. Neurosci. 2009, 29, 10424–10435. [Google Scholar] [CrossRef]
- Lobb, C.; Jaeger, D. Bursting activity of substantia nigra pars reticulata neurons in mouse parkinsonism in awake and anesthetized states. Neurobiol. Dis. 2015, 75, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Seeger-Armbruster, S.; von Ameln-Mayerhofer, A. Short- and long-term unilateral 6-hydroxydopamine lesions in rats show different changes in characteristics of spontaneous firing of substantia nigra pars reticulata neurons. Exp. Brain Res. 2013, 224, 15–24. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Q.J.; Liu, J.; Ali, U.; Gui, Z.H.; Hui, Y.P.; Chen, L.; Wang, T. Changes in firing rate and pattern of GABAergic neurons in subregions of the substantia nigra pars reticulata in rat models of Parkinson’s disease. Brain Res. 2010, 1324, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, T.; Soares, J. Neuronal Firing Before and After Burst Discharges in the Monkey Basal Ganglia Is Predictably Patterned in the Normal State and Altered in Parkinsonism. J. Neurophysiol. 2006, 95, 2120–2133. [Google Scholar] [CrossRef] [Green Version]
- Willard, A.M.; Isett, B.R.; Whalen, T.C.; Mastro, K.J.; Ki, C.S.; Mao, X.; Gittis, A.H. State transitions in the substantia nigra reticulata predict the onset of motor deficits in models of progressive dopamine depletion in mice. eLife 2019, 8, e42746. [Google Scholar] [CrossRef] [PubMed]
- Smith, Y.; Bevan, M.D.; Shink, E.; Bolam, J.P. Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience 1998, 86, 353–387. [Google Scholar] [PubMed]
- Bolam, J.P.; Smith, Y. The striatum and the globus pallidus send convergent synaptic inputs onto single cells in the entopeduncular nucleus of the rat: A double anterograde labelling study combined with postembedding immunocytochemistry for GABA. J. Comp. Neurol. 1992, 321, 456–476. [Google Scholar] [CrossRef] [PubMed]
- Von Krosigk, M.; Smith, Y.; Bolam, J.P.; Smith, A. Synaptic organization of gabaergic inputs from the striatum and the globus pallidus onto neurons in the substantia nigra and retrorubral field which project to the medullary reticular formation. Neuroscience 1992, 50, 531–549. [Google Scholar] [CrossRef]
- Bolam, J.P.; Smith, Y.; Ingham, C.A.; von Krosigk, M.; Smith, A.D. Convergence of synaptic terminals from the striatum and the globus pallidus onto single neurones in the substantia nigra and the entopeduncular nucleus. Prog. Brain. Res. 1993, 99, 73–88. [Google Scholar]
- Connelly, W.M.; Schulz, J.M.; Lees, G.; Reynolds, J.N.J. Differential Short-Term Plasticity at Convergent Inhibitory Synapses to the Substantia Nigra Pars Reticulata. J. Neurosci. 2010, 30, 14854–14861. [Google Scholar] [CrossRef]
- Galarraga, E.; Aceves, J.J.; Rueda-Orozco, P.E.; Hernandez-Martinez, R.; Bargas, J. Bidirectional plasticity in striatonigral synapses: A switch to balance direct and indirect basal ganglia pathways. Learn. Mem. 2011, 18, 764–773. [Google Scholar]
- Erlij, D.; Acosta-García, J.; Rojas-Márquez, M.; González-Hernández, B.; Escartín-Perez, E.; Aceves, J.; Florán, B. Dopamine D4 receptor stimulation in GABAergic projections of the globus pallidus to the reticular thalamic nucleus and the substantia nigra reticulata of the rat decreases locomotor activity. Neuropharmacology 2012, 62, 1111–1118. [Google Scholar] [CrossRef]
- Acosta-García, J.; Hernández-Chan, N.; Paz-Bermúdez, F.; Sierra, A.; Erlij, D.; Aceves, J.; Florán, B. D4 and D1 dopamine receptors modulate [3H]GABA release in the substantia nigra pars reticulata of the rat. Neuropharmacology 2009, 57, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Chuhma, N.; Tanaka, K.F.; Hen, R.; Rayport, S. Functional Connectome of the Striatal Medium-Spiny Neuron. J. Neurosci. 2011, 31, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Radnikow, G.; Misgeld, U. Dopamine D1 receptors facilitate GABAA synaptic currents in the rat substantia nigra pars reticulata. J. Neurosci. 1998, 18, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Borgkvist, A.; Avegno, E.M.; Wong, M.Y.; Kheirbek, M.A.; Sonders, M.S.; Hen, R.; Sulzer, D. Loss of Striatonigral GABAergic Presynaptic Inhibition Enables Motor Sensitization in Parkinsonian Mice. Neuron 2015, 87, 976–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, J.P.; Feyder, M.; Miguelez, C.; Garcia, L.; Morin, S.; Choquet, D.; Hosy, E.; Bezard, E.; Fisone, G.; Bioulac, B.H.; et al. Dopamine-Dependent Long-Term Depression at Subthalamo-Nigral Synapses Is Lost in Experimental Parkinsonism. J. Neurosci. 2013, 33, 14331–14341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibanez-Sandoval, O.; Hernández, A.; Florán, B.; Galarraga, E.; Tapia, D.; Valdiosera, R.; Erlij, D.; Aceves, J.; Bargas, J.; Hernandez-Cortes, A. Control of the Subthalamic Innervation of Substantia Nigra Pars Reticulata by D1and D2Dopamine Receptors. J. Neurophysiol. 2006, 95, 1800–1811. [Google Scholar] [CrossRef] [PubMed]
- Ammari, R.; Lopez, C.; Bioulac, B.; Garcia, L.; Hammond, C. Subthalamic nucleus evokes similar long lasting glutamatergic excitations in pallidal, entopeduncular and nigral neurons in the basal ganglia slice. Neuroscience 2010, 166, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.-Z.; Johnson, S.W. Subthalamic stimulation evokes complex EPSCs in the rat substantia nigra pars reticulata in vitro. J. Physiol. 2006, 573, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Gouty-Colomer, L.A.; Michel, F.J.; Baude, A.; Lopez-Pauchet, C.; Dufour, A.; Cossart, R.; Hammond, C. Mouse subthalamic nucleus neurons with local axon collaterals. J. Comp. Neurol. 2018, 526, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.-Z.; Johnson, S.W. Regulation of polysynaptic subthalamonigral transmission by D2, D3 and D4 dopamine receptors in rat brain slices. J. Physiol. 2012, 590, 2273–2284. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, J.P.; Bioulac, B.H.; Baufreton, J. Long-term depression at distinct glutamatergic synapses in the basal ganglia. Rev. Neurosci. 2014, 25, 741–754. [Google Scholar] [CrossRef] [PubMed]
- De Berardis, D.; Fornaro, M.; Valchera, A.; Cavuto, M.; Perna, G.; Di Nicola, M.; Serafini, G.; Carano, A.; Pompili, M.; Vellante, F.; et al. Eradicating Suicide at Its Roots: Preclinical Bases and Clinical Evidence of the Efficacy of Ketamine in the Treatment of Suicidal Behaviors. Int. J. Mol. Sci. 2018, 19, 2888. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Iasevoli, F.; Buonaguro, E.F.; De Berardis, D.; Fornaro, M.; Fiengo, A.L.C.; Martinotti, G.; Orsolini, L.; Valchera, A.; Di Giannantonio, M.; et al. Treating the Synapse in Major Psychiatric Disorders: The Role of Postsynaptic Density Network in Dopamine-Glutamate Interplay and Psychopharmacologic Drugs Molecular Actions. Int. J. Mol. Sci. 2017, 18, 135. [Google Scholar] [CrossRef] [PubMed]
- Hontanilla, B.; Parent, A.; Heras, S.D.L.; Giménez-Amaya, J.M. Distribution of calbindin D-28k and parvalbumin neurons and fibers in the rat basal ganglia. Brain Res. Bull. 1998, 47, 107–116. [Google Scholar] [CrossRef]
- Hontanilla, B.; Parent, A.; Giménez-Amaya, J.M. Parvalbumin and calbindin D-28k in the entopeduncular nucleus, subthalamic nucleus, and substantia nigra of the rat as revealed by double-immunohistochemical methods. Synapse 1997, 25, 359–367. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Fukuda, T. Immunohistochemical study on the neuronal diversity and three-dimensional organization of the mouse entopeduncular nucleus. Neurosci. Res. 2015, 94, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.L.; Saunders, A.; Huang, K.W.; Philson, A.C.; Goldman, M.; Macosko, E.Z.; McCarroll, S.A.; Sabatini, B.L. Genetically distinct parallel pathways in the entopeduncular nucleus for limbic and sensorimotor output of the basal ganglia. Neuron 2017, 94, 138–152.e5. [Google Scholar] [CrossRef]
- Stephenson-Jones, M.; Yu, K.; Ahrens, S.; Tucciarone, J.M.; Van Huijstee, A.N.; Mejia, L.A.; Penzo, M.A.; Tai, L.-H.; Wilbrecht, L.; Li, B. A basal ganglia circuit for evaluating action outcomes. Nature 2016, 539, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Vincent, S.R.; Brown, J.C. Somatostatin immunoreactivity in the entopeduncular projection to the lateral habenula in the rat. Neurosci. Lett. 1986, 68, 160–164. [Google Scholar] [CrossRef]
- Benhamou, L.; Cohen, D. Electrophysiological characterization of entopeduncular nucleus neurons in anesthetized and freely moving rats. Front. Syst. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [Green Version]
- Nakahishi, H.; Kita, H.; Kitai, S. Intracellular study of rat entopeduncular nucleus neurons in an in vitro slice preparation: electrical membrane properties. Brain Res. 1990, 527, 81–88. [Google Scholar] [CrossRef]
- Kita, H. Neostriatal and globus pallidus stimulation induced inhibitory postsynaptic potentials in entopeduncular neurons in rat brain slice preparations. Neuroscience 2001, 105, 871–879. [Google Scholar] [CrossRef]
- Bevan, M.D.; Clarke, N.P.; Bolam, J.P. Synaptic Integration of Functionally Diverse Pallidal Information in the Entopeduncular Nucleus and Subthalamic Nucleus in the Rat. J. Neurosci. 1997, 17, 308–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavian, H.; Korngreen, A. Inhibitory short-term plasticity modulates neuronal activity in the rat entopeduncular nucleus in vitro. Eur. J. Neurosci. 2016, 43, 870–884. [Google Scholar] [CrossRef] [PubMed]
- Lavian, H.; Almog, M.; Madar, R.; Loewenstern, Y.; Bar-Gad, I.; Okun, E.; Korngreen, A. Dopaminergic Modulation of Synaptic Integration and Firing Patterns in the Rat Entopeduncular Nucleus. J. Neurosci. 2017, 37, 7177–7187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavian, H.; Loewenstern, Y.; Madar, R.; Almog, M.; Bar-Gad, I.; Okun, E.; Korngreen, A. Dopamine receptors in the rat entopeduncular nucleus. Brain Struct. Funct. 2018, 223, 2673–2684. [Google Scholar] [CrossRef] [PubMed]
- Ferre, S.; O’Connor, W.T.; Svenningsson, P.; Bjorklund, L.; Lindberg, J.; Tinner, B.; Stromberg, I.; Goldstein, M.; Ogren, S.O.; Ungerstedt, U.; et al. Dopamine D1 receptor-mediated facilitation of GABAergic neurotransmission in the rat strioentopenduncular pathway and its modulation by adenosine A1 receptor-mediated mechanisms. Eur. J. Neurosci. 1996, 8, 1545–1553. [Google Scholar] [CrossRef]
- Nauta, H.J.W.; Cole, M. Efferent projections of the subthalamic nucleus: An autoradiographic study in monkey and cat. J. Comp. Neurol. 1978, 180, 1–16. [Google Scholar] [CrossRef]
- Gorodetski, L.; Zeira, R.; Lavian, H.; Korngreen, A. Long-term plasticity of glutamatergic input from the subthalamic nucleus to the entopeduncular nucleus. Eur. J. Neurosci. 2018, 48, 2139–2151. [Google Scholar] [CrossRef]
- Delong, M.R. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990, 13, 281–285. [Google Scholar] [CrossRef]
- Bergman, H.; Wichmann, T.; Karmon, B.; DeLong, M.R. The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J. Neurophysiol. 1994, 72, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Magill, P.; Bolam, J.P.; Bevan, M.; Magill, P. Dopamine regulates the impact of the cerebral cortex on the subthalamic nucleus–globus pallidus network. Neuroscience 2001, 106, 313–330. [Google Scholar] [CrossRef]
- Tseng, K.Y.; Kasanetz, F.; Kargieman, L.; Pazo, J.H.; Murer, M.; A Riquelme, L. Subthalamic nucleus lesions reduce low frequency oscillatory firing of substantia nigra pars reticulata neurons in a rat model of Parkinson’s disease. Brain Res. 2001, 904, 93–103. [Google Scholar] [CrossRef]
- Wichmann, T.; Bergman, H.; Starr, P.A.; Delong, M.R.; Watts, R.L.; Subramanian, T. Comparison of MPTP-induced changes in spontaneous neuronal discharge in the internal pallidal segment and in the substantia nigra pars reticulata in primates. Exp. Brain Res. 1999, 125, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.A.; Boraud, T.; Maraton, S.; Haber, S.N.; Vaadia, E.; Bergman, H. Enhanced synchrony among primary motor cortex neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine primate model of Parkinson’s disease. J. Neurosci. 2002, 22, 4639–4653. [Google Scholar] [CrossRef] [PubMed]
- Nini, A.; Feingold, A.; Slovin, H.; Bergman, H. Neurons in the globus pallidus do not show correlated activity in the normal monkey, but phase-locked oscillations appear in the MPTP model of parkinsonism. J. Neurophysiol. 1995, 74, 1800–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellens, D.J.; Leventhal, D.K. Electrophysiology of Basal Ganglia and Cortex in Models of Parkinson Disease. J. Park. Dis. 2013, 3, 241–254. [Google Scholar]
- Nelson, A.B.; Kreitzer, A.C. Reassessing Models of Basal Ganglia Function and Dysfunction. Annu. Rev. Neurosci. 2014, 37, 117–135. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, J.L.; Goldberg, J.A. Thinking Outside the Box (and Arrow): Current Themes in Striatal Dysfunction in Movement Disorders. Neuroscientist 2018, 1073858418807887. [Google Scholar] [CrossRef]
- Sharott, A.; Gulberti, A.; Zittel, S.; Jones, A.A.T.; Fickel, U.; Münchau, A.; Köppen, J.A.; Gerloff, C.; Westphal, M.; Buhmann, C.; et al. Activity Parameters of Subthalamic Nucleus Neurons Selectively Predict Motor Symptom Severity in Parkinson’s Disease. J. Neurosci. 2014, 34, 6273–6285. [Google Scholar] [CrossRef]
- Sanders, T.H.; Clements, M.A.; Wichmann, T. Parkinsonism-related features of neuronal discharge in primates. J. Neurophysiol. 2013, 110, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Ketzef, M.; Spigolon, G.; Johansson, Y.; Bonito-Oliva, A.; Fisone, G.; Silberberg, G. Dopamine Depletion Impairs Bilateral Sensory Processing in the Striatum in a Pathway-Dependent Manner. Neuron 2017, 94, 855–865.e5. [Google Scholar] [CrossRef] [PubMed]
- Mallet, N.; Ballion, B.; Le Moine, C.; Gonon, F. Cortical Inputs and GABA Interneurons Imbalance Projection Neurons in the Striatum of Parkinsonian Rats. J. Neurosci. 2006, 26, 3875–3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.G.; Marshall, J.D.; Ahanonu, B.; Wu, Y.-W.; Kim, T.H.; Grewe, B.F.; Zhang, Y.; Li, J.Z.; Ding, J.B.; Ehlers, M.D.; et al. Diametric neural ensemble dynamics in parkinsonian and dyskinetic states. Nature 2018, 557, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Deffains, M.; Iskhakova, L.; Katabi, S.; Haber, S.N.; Israel, Z.; Bergman, H. Subthalamic, not striatal, activity correlates with basal ganglia downstream activity in normal and parkinsonian monkeys. eLife 2016, 5, e16443. [Google Scholar] [CrossRef] [PubMed]
- Heimer, G.; Bar-Gad, I.; Goldberg, J.A.; Bergman, H. Dopamine Replacement Therapy Reverses Abnormal Synchronization of Pallidal Neurons in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Primate Model of Parkinsonism. J. Neurosci. 2002, 22, 7850–7855. [Google Scholar] [CrossRef] [Green Version]
- Mallet, N.; Pogosyan, A.; Sharott, A.; Csicsvari, J.; Bolam, J.P.; Brown, P.; Magill, P.J. Disrupted Dopamine Transmission and the Emergence of Exaggerated Beta Oscillations in Subthalamic Nucleus and Cerebral Cortex. J. Neurosci. 2008, 28, 4795–4806. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, T.; Bergman, H.; Delong, M.R. The primate subthalamic nucleus. III. Changes in motor behavior and neuronal activity in the internal pallidum induced by subthalamic inactivation in the MPTP model of parkinsonism. J. Neurophysiol. 1994, 72, 521–530. [Google Scholar] [CrossRef]
- Pasquereau, B.; DeLong, M.R.; Turner, R.S. Primary motor cortex of the parkinsonian monkey: Altered encoding of active movement. Brain 2016, 139, 127–143. [Google Scholar] [CrossRef]
- Freeze, B.S.; Kravitz, A.V.; Hammack, N.; Berke, J.D.; Kreitzer, A.C. Control of Basal Ganglia Output by Direct and Indirect Pathway Projection Neurons. J. Neurosci. 2013, 33, 18531–18539. [Google Scholar] [CrossRef]
- Roseberry, T.K.; Lee, A.M.; Lalive, A.L.; Wilbrecht, L.; Bonci, A.; Kreitzer, A.C. Cell-Type-Specific Control of Brainstem Locomotor Circuits by Basal Ganglia. Cell 2016, 164, 526–537. [Google Scholar] [CrossRef] [Green Version]
- Lemos, J.C.; Friend, D.M.; Kaplan, A.R.; Shin, J.H.; Rubinstein, M.; Kravitz, A.V.; Alvarez, V.A. Enhanced GABA transmission drives bradykinesia following loss of dopamine D2 receptor signaling. Neuron 2016, 90, 824–838. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, T.; Okuno, H.; Bito, H. A new era for functional labeling of neurons: Activity-dependent promoters have come of age. Front. Neural Circuits 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Svenningsson, P.; Fourreau, L.; Bloch, B.; Fredholm, B.; Gonon, F.; Le Moine, C. Opposite tonic modulation of dopamine and adenosine on c-fos gene expression in striatopallidal neurons. Neuroscience 1999, 89, 827–837. [Google Scholar] [CrossRef]
- Day, M.; Wang, Z.; Ding, J.; An, X.; A Ingham, C.; Shering, A.F.; Wokosin, D.; Ilijic, E.; Sun, Z.; Sampson, A.R.; et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci. 2006, 9, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Gittis, A.H.; Hang, G.B.; LaDow, E.S.; Shoenfeld, L.R.; Atallah, B.V.; Finkbeiner, S.; Kreitzer, A.C. Rapid target-specific remodeling of fast-spiking inhibitory circuits after loss of dopamine. Neuron 2011, 71, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ke, Y.; Chan, D.C.; Qian, Z.-M.; Yung, K.K.; Ko, H.; Arbuthnott, G.W.; Yung, W.-H. Therapeutic Deep Brain Stimulation in Parkinsonian Rats Directly Influences Motor Cortex. Neuron 2012, 76, 1030–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGregor, M.M.; Nelson, A.B. Circuit Mechanisms of Parkinson’s Disease. Neuron 2019, 101, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Nambu, A.; Tachibana, Y.; Chiken, S. Cause of parkinsonian symptoms: Firing rate, firing pattern or dynamic activity changes? Basal Ganglia 2015, 5, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Quiroga-Varela, A.; Walters, J.; Brazhnik, E.; Marin, C.; Obeso, J. What basal ganglia changes underlie the parkinsonian state? The significance of neuronal oscillatory activity. Neurobiol. Dis. 2013, 58, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Liang, L.; Kaneoke, Y.; Cao, X.; Papa, S.M. Dopamine regulates distinctively the activity patterns of striatal output neurons in advanced parkinsonian primates. J. Neurophysiol. 2015, 113, 1533–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergman, H.; Wichmann, T.; Delong, M. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science 1990, 249, 1436–1438. [Google Scholar] [CrossRef] [PubMed]
- Brown, P. Abnormal oscillatory synchronisation in the motor system leads to impaired movement. Curr. Opin. Neurobiol. 2007, 17, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Dostrovsky, J.; Bergman, H. Oscillatory activity in the basal ganglia--relationship to normal physiology and pathophysiology. Brain 2004, 127, 721–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, W.-J.; Degen, K.; Schneider, G.-H.; Brücke, C.; Huebl, J.; Brown, P.; Kühn, A.A. Subthalamic Synchronized Oscillatory Activity Correlates With Motor Impairment in Patients With Parkinson’s Disease. Mov. Disord. 2016, 31, 1748–1751. [Google Scholar] [CrossRef] [PubMed]
- Beudel, M.; Oswal, A.; Jha, A.; Foltynie, T.; Zrinzo, L.; Hariz, M.; Limousin, P.; Litvak, V. Oscillatory Beta Power Correlates With Akinesia-Rigidity in the Parkinsonian Subthalamic Nucleus. Mov. Disord. 2017, 32, 174–175. [Google Scholar] [CrossRef]
- Pogosyan, A.; Gaynor, L.D.; Eusebio, A.; Brown, P. Boosting Cortical Activity at Beta-Band Frequencies Slows Movement in Humans. Curr. Boil. 2009, 19, 1637–1641. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.; Litvak, V.; Gilbertson, T.; Kühn, A.; Lu, C.S.; Lee, S.T.; Tsai, C.H.; Tisch, S.; Limousin, P.; Hariz, M. Excessive synchronization of basal ganglia neurons at 20 Hz slows movement in Parkinson’s disease. Exp. Neurol. 2007, 205, 214–221. [Google Scholar] [CrossRef]
- Ermentrout, B.; Pascal, M.; Gutkin, B. The Effects of Spike Frequency Adaptation and Negative Feedback on the Synchronization of Neural Oscillators. Neural Comput. 2001, 13, 1285–1310. [Google Scholar] [CrossRef]
- Pavlides, A.; Hogan, S.J.; Bogacz, R. Computational Models Describing Possible Mechanisms for Generation of Excessive Beta Oscillations in Parkinson’s Disease. PLoS Comput. Boil. 2015, 11, e1004609. [Google Scholar] [CrossRef]
- Rubin, J.E. Computational models of basal ganglia dysfunction: The dynamics is in the details. Curr. Opin. Neurobiol. 2017, 46, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Shouno, O.; Tachibana, Y.; Nambu, A.; Doya, K. Computational Model of Recurrent Subthalamo-Pallidal Circuit for Generation of Parkinsonian Oscillations. Front. Neuroanat. 2017, 11, 366. [Google Scholar] [CrossRef] [PubMed]
- Plenz, D.; Kital, S.T. A basal ganglia pacemaker formed by the subthalamic nucleus and external globus pallidus. Nature 1999, 400, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Magill, P.J.; Bolam, J.P.; Bevan, M.D. Relationship of Activity in the Subthalamic Nucleus–Globus Pallidus Network to Cortical Electroencephalogram. J. Neurosci. 2000, 20, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Brittain, J.S.; Brown, P. Oscillations and the basal ganglia: Motor control and beyond. NeuroImage 2014, 85, 637–647. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.M.; Moore-Kochlacs, C.; Gu, X.; Boyden, E.S.; Han, X.; Kopell, N. Striatal origin of the pathologic beta oscillations in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2011, 108, 11620–11625. [Google Scholar] [CrossRef] [PubMed]
- Corbit, V.L.; Whalen, T.C.; Zitelli, K.T.; Crilly, S.Y.; Rubin, J.E.; Gittis, A.H. Pallidostriatal Projections Promote β Oscillations in a Dopamine-Depleted Biophysical Network Model. J. Neurosci. 2016, 36, 5556–5571. [Google Scholar] [CrossRef]
- Leblois, A.; Boraud, T.; Meissner, W.; Bergman, H.; Hansel, D. Competition between Feedback Loops Underlies Normal and Pathological Dynamics in the Basal Ganglia. J. Neurosci. 2006, 26, 3567–3583. [Google Scholar] [CrossRef] [Green Version]
- Nambu, A.; Tachibana, Y. Mechanism of parkinsonian neuronal oscillations in the primate basal ganglia: Some considerations based on our recent work. Front. Syst. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Tachibana, Y.; Iwamuro, H.; Kita, H.; Takada, M.; Nambu, A. Subthalamo-pallidal interactions underlying parkinsonian neuronal oscillations in the primate basal ganglia. Eur. J. Neurosci. 2011, 34, 1470–1484. [Google Scholar] [CrossRef]
- Baufreton, J.; Atherton, J.F.; Surmeier, D.J.; Bevan, M.D. Enhancement of Excitatory Synaptic Integration by GABAergic Inhibition in the Subthalamic Nucleus. J. Neurosci. 2005, 25, 8505–8517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terman, D.; Rubin, J.E.; Yew, A.C.; Wilson, C.J. Activity Patterns in a Model for the Subthalamopallidal Network of the Basal Ganglia. J. Neurosci. 2002, 22, 2963–2976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degos, B.; Deniau, J.M.; Chavez, M.; Maurice, N. Chronic but not acute dopaminergic transmission interruption promotes a progressive increase in cortical beta frequency synchronization: Relationships to vigilance state and akinesia. Cereb. Cortex 2009, 19, 1616–1630. [Google Scholar] [CrossRef] [PubMed]
- Galati, S.; Stanzione, P.; D’Angelo, V.; Fedele, E.; Marzetti, F.; Sancesario, G.; Procopio, T.; Stefani, A. The pharmacological blockade of medial forebrain bundle induces an acute pathological synchronization of the cortico–subthalamic nucleus–globus pallidus pathway. J. Physiol. 2009, 587, 4405–4423. [Google Scholar] [CrossRef] [PubMed]
- Leblois, A.; Meissner, W.; Bioulac, B.; Gross, C.E.; Hansel, D.; Boraud, T. Late emergence of synchronized oscillatory activity in the pallidum during progressive parkinsonism. Eur. J. Neurosci. 2007, 26, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Bar-Gad, I.; Heimer, G.; Ritov, Y.; Bergman, H. Functional Correlations between Neighboring Neurons in the Primate Globus Pallidus Are Weak or Nonexistent. J. Neurosci. 2003, 23, 4012–4016. [Google Scholar] [CrossRef] [Green Version]
- Avila, I.; Parr-Brownlie, L.C.; Brazhnik, E.; Castaneda, E.; Bergstrom, D.A.; Walters, J.R. Beta frequency synchronization in basal ganglia output during rest and walk in a hemiparkinsonian rat. Exp. Neurol. 2010, 221, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Brazhnik, E.; McCoy, A.J.; Novikov, N.; Hatch, C.E.; Walters, J.R. Ventral Medial Thalamic Nucleus Promotes Synchronization of Increased High Beta Oscillatory Activity in the Basal Ganglia–Thalamocortical Network of the Hemiparkinsonian Rat. J. Neurosci. 2016, 36, 4196–4208. [Google Scholar] [CrossRef]
- Sharott, A.; Magill, P.J.; Harnack, D.; Kupsch, A.; Meissner, W.; Brown, P. Dopamine depletion increases the power and coherence of beta-oscillations in the cerebral cortex and subthalamic nucleus of the awake rat. Eur. J. Neurosci. 2005, 21, 1413–1422. [Google Scholar] [CrossRef]
- Deffains, M.; Iskhakova, L.; Katabi, S.; Israel, Z.; Bergman, H. Longer β oscillatory episodes reliably identify pathological subthalamic activity in Parkinsonism. Mov. Disord. 2018, 33, 1609–1618. [Google Scholar] [CrossRef]
- Lobb, C.J.; Zaheer, A.K.; Smith, Y.; Jaeger, D. In vivo electrophysiology of nigral and thalamic neurons in alpha-synuclein-overexpressing mice highlights differences from toxin-based models of parkinsonism. J. Neurophysiol. 2013, 110, 2792–2805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guridi, J.; Alegre, M. Oscillatory activity in the basal ganglia and deep brain stimulation. Mov. Disord. 2017, 32, 64–69. [Google Scholar] [CrossRef] [PubMed]
- McConnell, G.C.; So, R.Q.; Hilliard, J.D.; Lopomo, P.; Grill, W.M. Effective deep brain stimulation suppresses low frequency network oscillations in the basal ganglia by regularizing neural firing patterns. J. Neurosci. 2012, 32, 15657–15668. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.; Ammari, R.; Bioulac, B.; García, L. Latest view on the mechanism of action of deep brain stimulation. Mov. Disord. 2008, 23, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.; Bergman, H.; Brown, P. Pathological synchronization in Parkinson’s disease: networks, models and treatments. Trends Neurosci. 2007, 30, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, T.; Delong, M.R. Deep Brain Stimulation for Movement Disorders of Basal Ganglia Origin: Restoring Function or Functionality? Neurotherapeutics 2016, 13, 264–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranck, J.B. Which elements are excited in electrical stimulation of mammalian central nervous system: A review. Brain Res. 1975, 98, 417–440. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Basal ganglia circuitry in rodents. (A) Schematic representing the main connections of the basal ganglia network in a sagittal section of the rodent brain. (B) Corresponding anatomical-functional diagram adapted from the Albin model of the basal ganglia [8]. Dopaminergic, GABAergic, and glutamatergic projections and nuclei are depicted in blue, purple and red, respectively. A: arkypallidal neuron; dSPN: direct-pathway spiny projection neuron; EPN: entopedoncular nucleus; GP: globus pallidus; iSPN: indirect-pathway spiny projection neuron; P: prototypic neuron; SNc: substantia nigra pars compacta; SNr: Substantia nigra pars reticulata; SIN: Striatal interneurons; STN: subthalamic nucleus.
Figure 1.
Basal ganglia circuitry in rodents. (A) Schematic representing the main connections of the basal ganglia network in a sagittal section of the rodent brain. (B) Corresponding anatomical-functional diagram adapted from the Albin model of the basal ganglia [8]. Dopaminergic, GABAergic, and glutamatergic projections and nuclei are depicted in blue, purple and red, respectively. A: arkypallidal neuron; dSPN: direct-pathway spiny projection neuron; EPN: entopedoncular nucleus; GP: globus pallidus; iSPN: indirect-pathway spiny projection neuron; P: prototypic neuron; SNc: substantia nigra pars compacta; SNr: Substantia nigra pars reticulata; SIN: Striatal interneurons; STN: subthalamic nucleus.

Figure 2.
Mechanisms underlying GABAergic synaptic alterations in the GP in experimental PD. (A) In DA-intact conditions, D2 receptors present on astrocyte membranes elevate intracellular calcium levels and promote glutamate gliotransmission. Glutamate activates group III mGluR (GluRIII), which reduces GABA release probability at striato-pallidal synapses. D2Rs also regulate GAT-3 activity by an unknown mechanism, preventing GABA spillover from GABAergic synapses and hence activation of extrasynaptic GABAA receptors. (B) Under DA-depleted conditions, D2Rs are no longer activated, glutamate gliotransmission is reduced and mGluRIII-dependent reduction of GABA release is lost, leading to an increase in striato-pallidal transmission. The loss of D2R modulation of GAT-3 triggers a downregulation of the expression of the transporters, and the elevation of extracellular GABA concentrations leads to the activation of extrasynaptic GABAA receptors, which favors tonic inhibition.
Figure 2.
Mechanisms underlying GABAergic synaptic alterations in the GP in experimental PD. (A) In DA-intact conditions, D2 receptors present on astrocyte membranes elevate intracellular calcium levels and promote glutamate gliotransmission. Glutamate activates group III mGluR (GluRIII), which reduces GABA release probability at striato-pallidal synapses. D2Rs also regulate GAT-3 activity by an unknown mechanism, preventing GABA spillover from GABAergic synapses and hence activation of extrasynaptic GABAA receptors. (B) Under DA-depleted conditions, D2Rs are no longer activated, glutamate gliotransmission is reduced and mGluRIII-dependent reduction of GABA release is lost, leading to an increase in striato-pallidal transmission. The loss of D2R modulation of GAT-3 triggers a downregulation of the expression of the transporters, and the elevation of extracellular GABA concentrations leads to the activation of extrasynaptic GABAA receptors, which favors tonic inhibition.

Figure 3.
Cellular and synaptic alterations in the STN in experimental PD. (A) Schematic of the main post-synaptic and pre-synaptic molecular pathways involved in DA-dependent cellular excitability and synaptic plasticity. (B) Under DD conditions, post-synaptic and pre-synaptic DA modulation is lost. This triggers NMDA-dependent M1 cortex synapse pruning, NMDA-dependent and NO-dependent heterosynaptic LTP (synapse proliferation) at pallido-subthalamic GABAergic synapses and NMDA-dependent activation of KATP channels, which reduces STN autonomous pacemaking.
Figure 3.
Cellular and synaptic alterations in the STN in experimental PD. (A) Schematic of the main post-synaptic and pre-synaptic molecular pathways involved in DA-dependent cellular excitability and synaptic plasticity. (B) Under DD conditions, post-synaptic and pre-synaptic DA modulation is lost. This triggers NMDA-dependent M1 cortex synapse pruning, NMDA-dependent and NO-dependent heterosynaptic LTP (synapse proliferation) at pallido-subthalamic GABAergic synapses and NMDA-dependent activation of KATP channels, which reduces STN autonomous pacemaking.

Figure 4.
Cellular and synaptic alterations in the ESN in experimental PD. (A) Summary diagram of the main modifications in cellular excitability and synaptic transmission observed in the BG network under DA-depleted conditions. Black arrows indicate increase or decrease in synaptic transmission or intrinsic excitability (represented by black APs). Astr.: astrocyte involvement in GABAergic dysfunction in GP. GABAergic and glutamatergic projections and nuclei are depicted in blue and red, respectively. Loss of SNc DA neurons is represented by the dashed purple circle. (B) Schematic illustrating the modification in the balance between intrinsic neuronal excitability and synaptic strength in ESN as a function of the levels of DA.
Figure 4.
Cellular and synaptic alterations in the ESN in experimental PD. (A) Summary diagram of the main modifications in cellular excitability and synaptic transmission observed in the BG network under DA-depleted conditions. Black arrows indicate increase or decrease in synaptic transmission or intrinsic excitability (represented by black APs). Astr.: astrocyte involvement in GABAergic dysfunction in GP. GABAergic and glutamatergic projections and nuclei are depicted in blue and red, respectively. Loss of SNc DA neurons is represented by the dashed purple circle. (B) Schematic illustrating the modification in the balance between intrinsic neuronal excitability and synaptic strength in ESN as a function of the levels of DA.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mallet, N.; Delgado, L.; Chazalon, M.; Miguelez, C.; Baufreton, J. Cellular and Synaptic Dysfunctions in Parkinson’s Disease: Stepping Out of the Striatum. Cells 2019, 8, 1005. https://doi.org/10.3390/cells8091005
AMA Style
Mallet N, Delgado L, Chazalon M, Miguelez C, Baufreton J. Cellular and Synaptic Dysfunctions in Parkinson’s Disease: Stepping Out of the Striatum. Cells. 2019; 8(9):1005. https://doi.org/10.3390/cells8091005
Chicago/Turabian StyleMallet, Nicolas, Lorena Delgado, Marine Chazalon, Cristina Miguelez, and Jérôme Baufreton. 2019. "Cellular and Synaptic Dysfunctions in Parkinson’s Disease: Stepping Out of the Striatum" Cells 8, no. 9: 1005. https://doi.org/10.3390/cells8091005
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.