
The Complete Mitochondrial Genome of a Neglected Breed, the Peruvian Creole Cattle (Bos taurus), and Its Phylogenetic Analysis

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
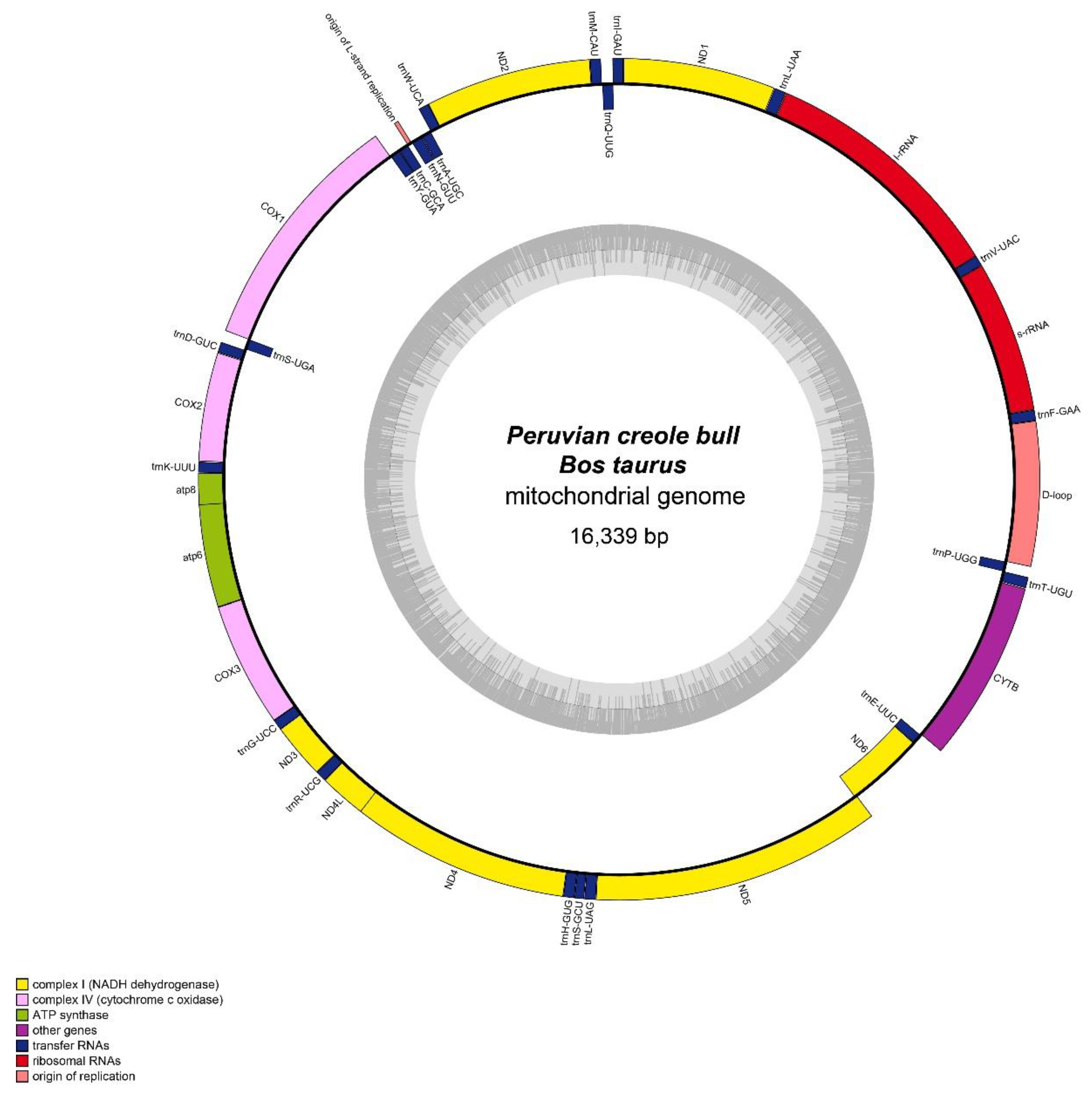
2.1. Mitochondrial Genome Organization
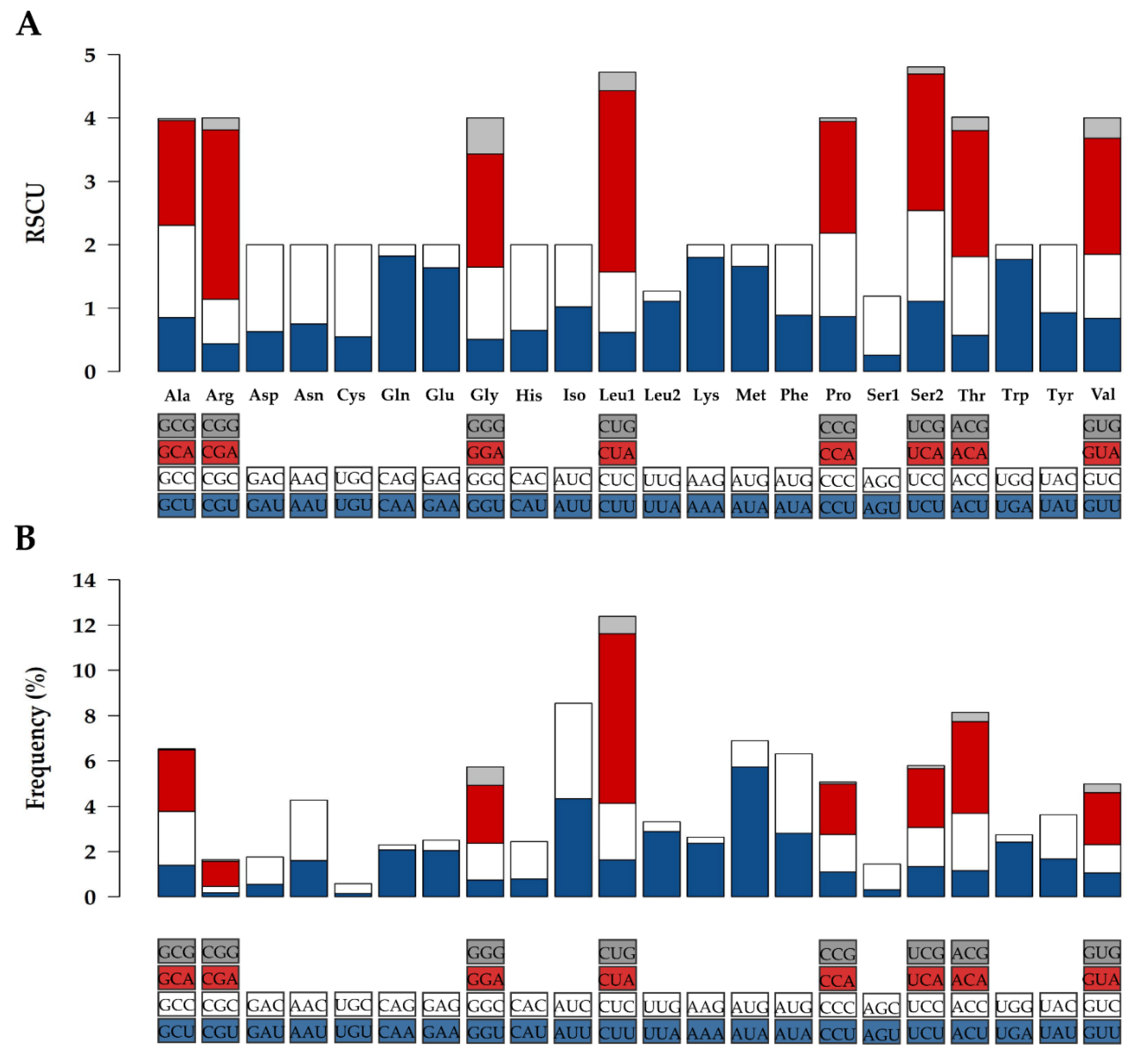
2.2. Protein Coding Genes (PCGs) and Codon Usage
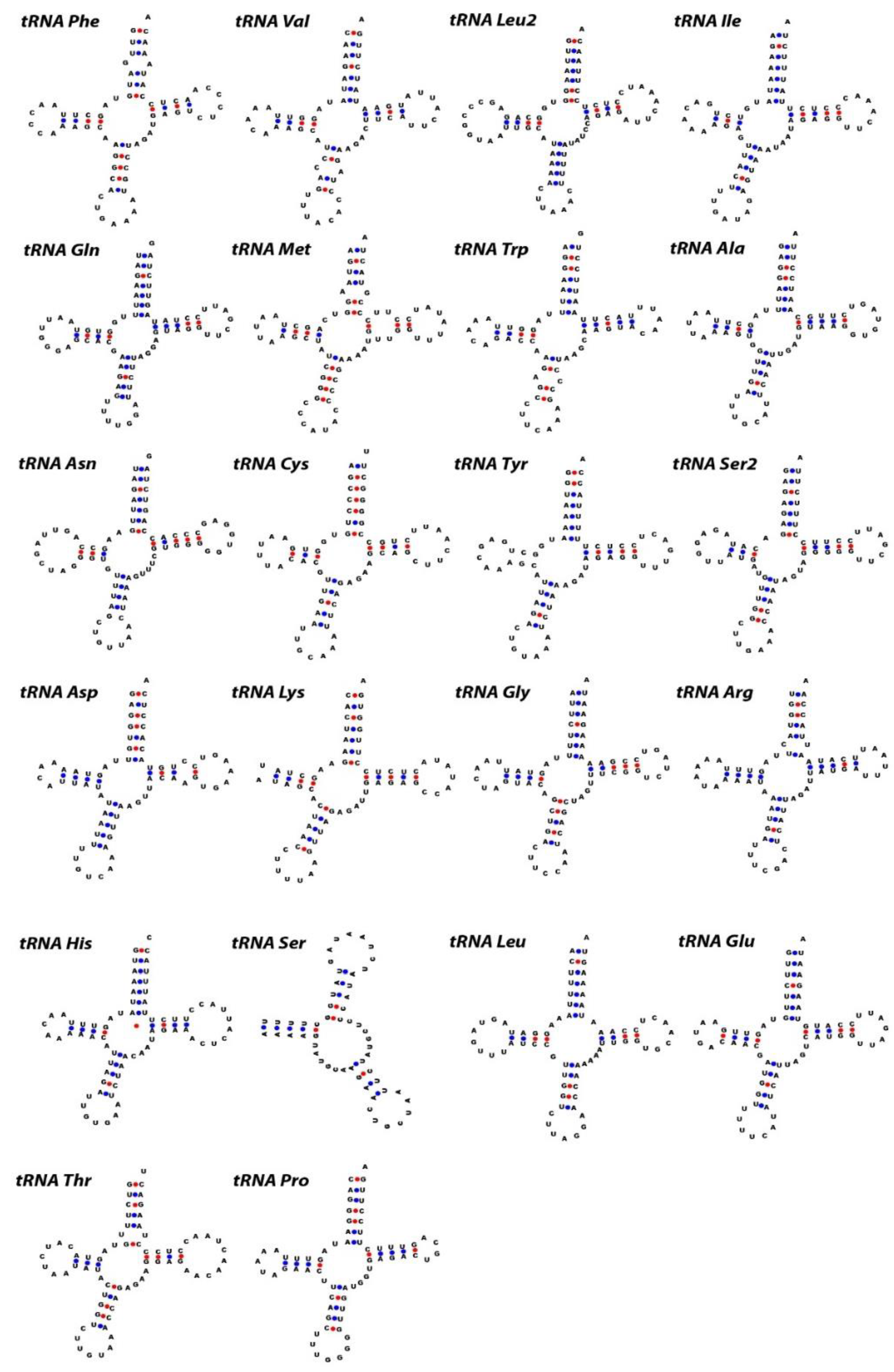
2.3. Ribosomal RNA, Transfer RNA, and Non-Coding Regions
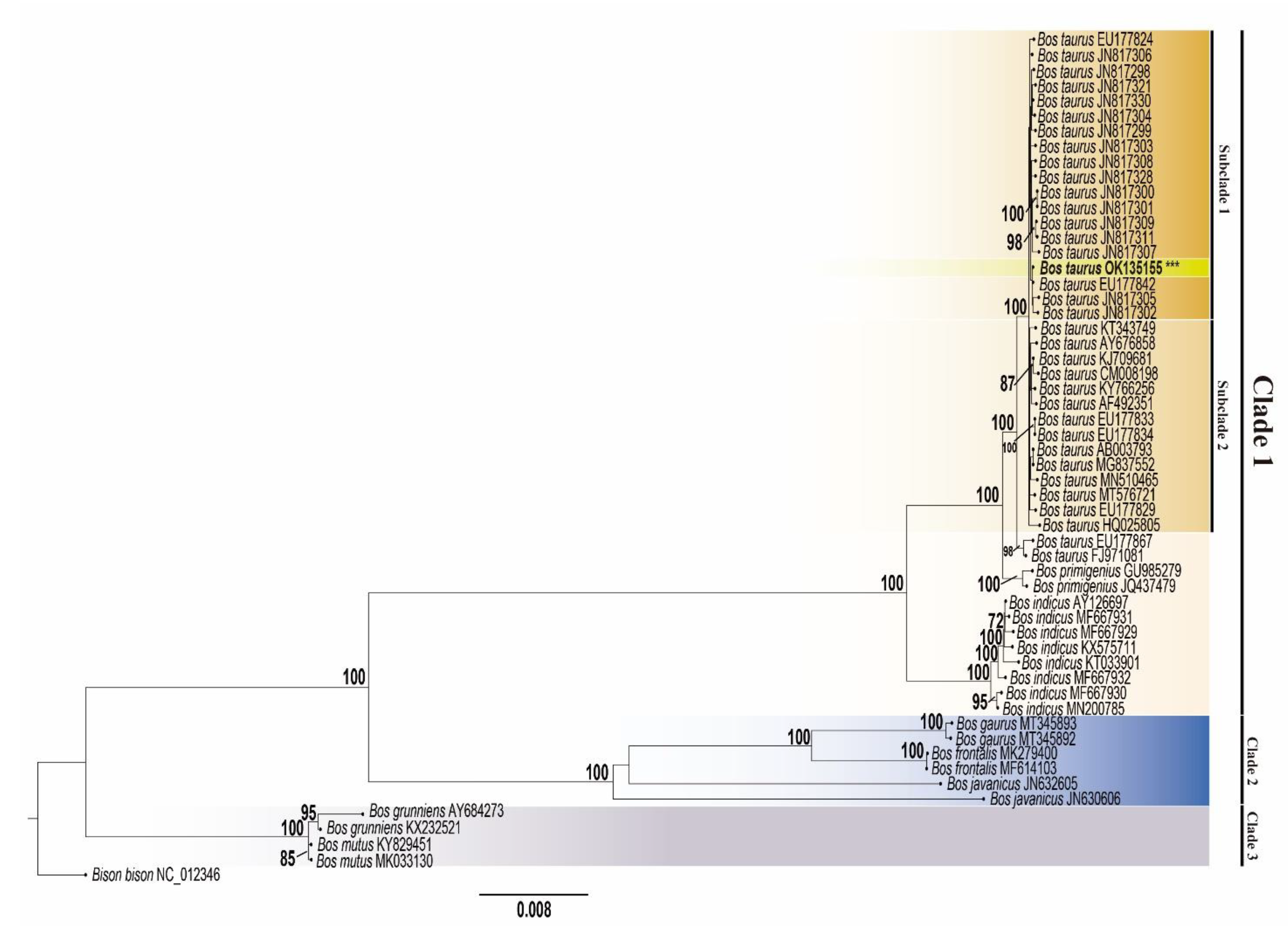
2.4. Phylogenetic Inference
3. Discussion
4. Materials and Methods
4.1. Sample Collection, DNA Extraction, and Sequencing
4.2. Assembly, Annotation, and Sequence Analysis
4.3. Codon Usage and tRNA Analysis
4.4. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herrero, M.; Grace, D.; Njuki, J.; Johnson, N.; Enahoro, D.; Silvestri, S.; Rufino, M.C. The Roles of Livestock in Developing Countries. Animal 2013, 7, 3–18. [Google Scholar] [CrossRef] [Green Version]
- McLeod, A. World Livestock 2011-Livestock in Food Security. In World Livestock 2011-Livestock in Food Security; Food and Agriculture Organization of the United Nations (FAO): Rome, Italy, 2011. [Google Scholar]
- Martínez, A.M.; Gama, L.T.; Cañón, J.; Ginja, C.; Delgado, J.V.; Dunner, S.; Landi, V.; Martín-Burriel, I.; Penedo, M.C.T.; Rodellar, C.; et al. Genetic Footprints of Iberian Cattle in America 500 Years after the Arrival of Columbus. PLoS ONE 2012, 7, e49066. [Google Scholar] [CrossRef] [Green Version]
- Yalta-Macedo, C.E.; Veli, E.A.; Díaz, G.R.; Vallejo-Trujillo, A. Paternal Ancestry of Peruvian Creole Cattle Inferred from Y-Chromosome Analysis. Livest. Sci. 2021, 244, 104376. [Google Scholar] [CrossRef]
- Ginja, C.; Gama, L.T.; Cortés, O.; Burriel, I.M.; Vega-Pla, J.L.; Penedo, C.; Sponenberg, P.; Cañón, J.; Sanz, A.; do Egito, A.A.; et al. The Genetic Ancestry of American Creole Cattle Inferred from Uniparental and Autosomal Genetic Markers. Sci. Rep. 2019, 9, 11486. [Google Scholar] [CrossRef]
- Villalobos-Cortés, A.; Martínez, A.; Vega-Pla, J.L.; Landi, V.; Quiroz, J.; Martínez, R.; López, R.M.; Sponenberg, P.; Armstrong, E.; Zambrano, D.; et al. Relationships between Panamanians and Some Creole Cattle Landraces in Latin America. Pesqui. Agropecu. Bras. 2012, 47, 1637–1646. [Google Scholar] [CrossRef] [Green Version]
- Instituto Nacional de Estadística e Informática IV Censo Nacional Agropecuario. 2012. Available online: http://censos.inei.gob.pe/Cenagro/redatam/# (accessed on 14 March 2022).
- Motta Zamalloa, E. El Astero de Plata. Anthropologica 1988, 6, 259–283. [Google Scholar]
- Espinoza, R.; Urviola, G. Biometría y Constantes Clínicas Del Bovino Criollo En El Centro de Investigación y Producción Chuquibambilla de Puno (Perú). Arch. Zootec. 2005, 54, 233–236. [Google Scholar]
- Dipas Vargas, E.S. Zoometría e Índices Corporales Del Vacuno Criollo En El Matadero de Quicapata de La Provincia de Huamanga, a 2720 m.s.n.m. Ayacucho-2014. Bachelor’s Thesis, Universidad Nacional San Cristóbal de Huamanga, Ayacucho, Perú, 2015. [Google Scholar]
- More Montoya, M.J. Caracterización Faneróptica y Morfométrica Del Vacuno Criollo En Ayacucho, Puno y Cajamarca. Master’s Thesis, Universidad Nacional Agraria la Molina, Lima, Perú, 2016. [Google Scholar]
- Ruiz, R.E.; Saucedo-uriarte, J.A.; Portocarrero-villegas, S.M.; Quispe-ccasa, H.A.; Cayo-colca, I.S. Zoometric Characterization of Creole Cows from the Southern Amazon Region of Peru. Diversity 2021, 13, 510. [Google Scholar] [CrossRef]
- Hiendleder, S.; Lewalski, H.; Janke, A. Complete Mitochondrial Genomes of Bos taurus and Bos indicus Provide New Insights into Intra-Species Variation, Taxonomy and Domestication. Cytogenet Genome Res 2008, 120, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Lirón, J.P.; Bravi, C.M.; Mirol, P.M.; Peral-García, P.; Giovambattista, G. African Matrilineages in American Creole Cattle: Evidence of Two Independent Continental Sources. Anim. Genet. 2006, 37, 379–382. [Google Scholar] [CrossRef]
- Bonfiglio, S.; Ginja, C.; De Gaetano, A.; Achilli, A.; Olivieri, A.; Colli, L.; Tesfaye, K.; Agha, S.H.; Gama, L.T.; Cattonaro, F.; et al. Origin and Spread of Bos taurus: New Clues from Mitochondrial Genomes Belonging to Haplogroup T1. PLoS ONE 2012, 7, e38601. [Google Scholar] [CrossRef] [Green Version]
- Veli, E.A.; Rivas Seoane, E.; Rivas Palma, V.; Aquino, Y.; Estrada, R. Variabilidad Genética Del Gen de Beta Lactoglobulina En Bovinos Criollos de Perú. Arch. Zootec. 2008, 57, 341–344. [Google Scholar]
- Almeyda, R.M.; Rosadio, A.R.; Maturrano, H.L. Genotypes of Kappa-Casein Gen in Creole Cattle of Bambamarca District (Cajamarca, Peru). Rev. Investig. Vet. Del Peru 2016, 27, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Ginja, C.; Penedo, M.C.T.; Melucci, L.; Quiroz, J.; Martínez López, O.R.; Revidatti, M.A.; Martínez-Martínez, A.; Delgado, J.V.; Gama, L.T. Origins and Genetic Diversity of New World Creole Cattle: Inferences from Mitochondrial and Y Chromosome Polymorphisms. Anim. Genet. 2010, 41, 128–141. [Google Scholar] [CrossRef]
- Georges, M.; Charlier, C.; Hayes, B. Harnessing Genomic Information for Livestock Improvement. Nat. Rev. Genet. 2019, 20, 135–156. [Google Scholar] [CrossRef]
- Prabhu, V.R.; Arjun, M.S.; Bhavana, K.; Kamalakkannan, R.; Nagarajan, M. Complete Mitochondrial Genome of Indian Mithun, Bos frontalis and Its Phylogenetic Implications. Mol. Biol. Rep. 2019, 46, 2561–2566. [Google Scholar] [CrossRef]
- Kamalakkannan, R.; Bhavana, K.; Prabhu, V.R.; Sureshgopi, D.; Singha, H.S.; Nagarajan, M. The Complete Mitochondrial Genome of Indian Gaur, Bos gaurus and Its Phylogenetic Implications. Sci. Rep. 2020, 10, 11936. [Google Scholar] [CrossRef]
- Chu, M.; Wu, X.; Liang, C.; Pei, J.; Ding, X.; Guo, X.; Bao, P.; Yan, P. The Complete Sequence of Mitochondrial Genome of Polled Yak (Bos grunniens). Mitochondrial DNA. Part A DNA Mapp. Seq. Anal. 2016, 27, 2032–2033. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.Q.; He, D.C.; Yang, X.M.; Li, B.; Wang, D.C.; Guang, J.; Xu, F.; Li, J.Y.; Gao, X.; et al. The Complete Mitochondrial Genome of Bos taurus coreanae (Korean Native Cattle). Mitochondrial DNA 2016, 27, 120–121. [Google Scholar] [CrossRef]
- Liu, S.-J.; Lv, J.-Z.; Tan, Z.-Y.; Ge, X.-Y. The Complete Mitochondrial Genome of Uruguayan Native Cattle (Bos taurus). Mitochondrial DNA Part B 2020, 5, 443–444. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, J.; Lamelas, L.; Aleix-Mata, G.; Arroyo, M.; Marchal, J.A.; Palomeque, T.; Lorite, P.; Sánchez, A. Complete Mitochondrial Genome of the Iberian Mole Talpa occidentalis (Talpidae, Insectivora) and Comparison with Talpa Europaea. Genetica 2018, 146, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Lamelas, L.; Aleix-Mata, G.; Rovatsos, M.; Marchal, J.A.; Palomeque, T.; Lorite, P.; Sánchez, A. Complete Mitochondrial Genome of Three Species of the Genus Microtus (Arvicolinae, Rodentia). Animals 2020, 10, 2130. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Malakar, A.K.; Chakraborty, S. Codon Usage Vis-a-Vis Start and Stop Codon Context Analysis of Three Dicot Species. J. Genet. 2018, 97, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Huang, X.; Hu, Y.; Tu, F. Description of the Mitogenome of Gansu Mole (Scapanulus oweni). Mitochondrial DNA. Part A DNA Mapp. Seq. Anal. 2016, 27, 2083–2084. [Google Scholar] [CrossRef]
- Kim, N.H.; Lim, S.J.; Chae, H.M.; Park, Y.C. Complete Mitochondrial Genome of the Amur Hedgehog Erinaceus amurensis (Erinaceidae) and Higher Phylogeny of the Family Erinaceidae. Genet. Mol. Res. 2017, 16, 1–8. [Google Scholar] [CrossRef]
- Hou, W.R.; Chen, Y.; Wu, X.; Hu, J.C.; Peng, Z.S.; Yang, J.; Tang, Z.X.; Zhou, C.Q.; Li, Y.M.; Yang, S.K.; et al. A Complete Mitochondrial Genome Sequence of Asian Black Bear Sichuan Subspecies (Ursus thibetanus mupinensis). Int. J. Biol. Sci. 2007, 3, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.J.; Kim, J.Y.; Lee, J.A.; Yoon, W.J.; Park, S.Y.; Jung, Y.H. Complete Mitochondrial Genome of the Rabbitfish Siganus fuscescens (Perciformes, Siganidae). DNA Seq.-J. DNA Seq. Mapp. 2007, 18, 295–301. [Google Scholar] [CrossRef]
- Prabhu, V.R.; Singha, H.S.; Kumar, R.G.; Gopalakrishnan, A.; Nagarajan, M. Characterization of the Complete Mitochondrial Genome of Barilius malabaricus and Its Phylogenetic Implications. Genomics 2020, 112, 2154–2163. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. TRNA Punctuation Model of RNA Processing in Human Mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Cha, S.Y.; Yoon, H.J.; Lee, E.M.; Yoon, M.H.; Hwang, J.S.; Jin, B.R.; Han, Y.S.; Kim, I. The Complete Nucleotide Sequence and Gene Organization of the Mitochondrial Genome of the Bumblebee, Bombus ignitus (Hymenoptera: Apidae). Gene 2007, 392, 206–220. [Google Scholar] [CrossRef]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon Usage in Yeast: Cluster Analysis Dearly Differentiates Highly and Lowly Expressed Genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, P.; Binti Othman, R.Y.; Mebus, K.; Ramakrishnan, N.; Ann Harikrishna, J. Codon Usage and Codon Pair Patterns in Non-Grass Monocot Genomes. Ann. Bot. 2017, 120, 893–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boore, J.L. Animal Mitochondrial Genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Silva, P.; Enriquez, J.A.; Montoya, J. Replication and Transcription of Mammalian Mitochondrial DNA. Exp. Physiol. 2003, 88, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Liu, S.; Zhang, X.; Chen, W.; Song, Z.; Peng, H.; Liu, Y.; Yue, B. Complete Mitochondrial Genome of a New Vole Proedromys liangshanensis (Rodentia: Cricetidae) and Phylogenetic Analysis with Related Species: Are There Implications for the Validity of the Genus Proedromys? Mitochondrial DNA 2011, 22, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, Y.C. Gene Organization and Characterization of the Complete Mitogenome of Hypsugo alaschanicus (Chiroptera: Vespertilionidae). Genet. Mol. Res. 2015, 14, 16325–16331. [Google Scholar] [CrossRef]
- Porter, V.; Alderson, L.; Hall, S.J.; Sponenberg, D.P. Mason’s World Encyclopedia of Livestock Breeds and Breeding, 2 Volume Pack; CABI: Worcester, MA, USA, 2016.
- Rubinoff, D.; Holland, B.S. Between Two Extremes: Mitochondrial DNA Is Neither the Panacea nor the Nemesis of Phylogenetic and Taxonomic Inference. Syst. Biol. 2005, 54, 952–961. [Google Scholar] [CrossRef]
- Cole, L.W.; Guo, W.; Mower, J.P.; Palmer, J.D. High and Variable Rates of Repeat-Mediated Mitochondrial Genome Rearrangement in a Genus of Plants. Mol. Biol. Evol. 2018, 35, 2773–2785. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.; Yang, J.B.; Song, Y.; Depamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A Fast and Versatile Toolkit for Accurate de Novo Assembly of Organelle Genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq-Versatile and Accurate Annotation of Organelle Genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Donath, A.; Jühling, F.; Al-Arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved Annotation of Protein-Coding Genes Boundaries in Metazoan Mitochondrial Genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. TRNAscan-SE 2.0: Improved Detection and Functional Classification of Transfer RNA Genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) Version 1.3.1: Expanded Toolkit for the Graphical Visualization of Organellar Genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinform. Appl. 2014, 30, 1312–1313. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Nucleotide Positions | Size (bp) | Strand 1 | Codon | Intergenic Spacer (bp) |
|---|---|---|---|---|---|
| tRNAPhe | 364–430 | 67 | H | TTC | |
| 12S rRNA | 431–1386 | 956 | H | ||
| tRNAVal | 1387–1453 | 67 | H | GTA | |
| 16S rRNA | 1454–3023 | 1570 | H | ||
| tRNALeu2 | 3025–3099 | 75 | H | TTA | 1 |
| Nd1 | 3102–4057 | 956 | H | 2 | |
| tRNAIle | 4058–4126 | 69 | H | ATC | |
| tRNAGln | 4124–4195 | 72 | L | CAA | −3 |
| tRNAMet | 4198–4266 | 69 | H | ATG | 2 |
| Nd2 | 4267–5309 | 1043 | H | ||
| tRNATrp | 5309–5375 | 67 | H | TGA | −1 |
| tRNAAla | 5377–5445 | 69 | L | GCA | 1 |
| tRNAAsn | 5447–5519 | 73 | L | AAC | 1 |
| Rep_origin | 5522–5552 | 31 | H | 2 | |
| tRNACys | 5552–5618 | 67 | L | TGC | −1 |
| tRNATyr | 5619–5686 | 68 | L | TAC | |
| Cox1 | 5688–7232 | 1545 | H | 1 | |
| tRNASer2 | 7230–7298 | 69 | L | TCA | −3 |
| tRNAAsp | 7306–7373 | 68 | H | GAC | 7 |
| Cox2 | 7375–8058 | 684 | H | 1 | |
| tRNALys | 8062–8128 | 67 | H | AAA | 3 |
| Atp8 | 8130–8330 | 201 | H | 1 | |
| Atp6 | 8291–8971 | 681 | H | −40 | |
| Cox3 | 8971–9755 | 785 | H | −1 | |
| tRNAGly | 9755–9823 | 69 | H | GGA | −1 |
| Nd3 | 9821–10170 | 350 | H | −3 | |
| tRNAArg | 10171–10239 | 69 | H | CGA | |
| Nd4L | 10240–10536 | 297 | H | ||
| Nd4 | 10530–11907 | 1378 | H | −7 | |
| tRNAHis | 11908–11977 | 70 | H | CAC | |
| tRNASer | 11978–12037 | 60 | H | AGC | |
| tRNALeu | 12039–12109 | 71 | H | CTA | 1 |
| Nd5 | 12110–13930 | 1821 | H | ||
| Nd6 | 13914–14441 | 528 | L | −17 | |
| tRNAGlu | 14442–14510 | 69 | L | GAA | |
| CytB | 14515–15654 | 1140 | H | 4 | |
| tRNAThr | 15658–15727 | 70 | H | 3 | |
| tRNAPro | 15727–15792 | 66 | L | CCA | −1 |
| D-loop | 15793–16339, 1–363 | 910 | H |
| Gene | Gene Length (bp) | A + T Content (%) | Start/Stop Codon | Protein Length (aa) |
|---|---|---|---|---|
| Nd1 | 956 | 59.4 | ATG/TA- | 318 |
| Nd2 | 1043 | 64.6 | ATA/TA- | 347 |
| Cox1 | 1545 | 58.3 | ATG/TAA | 514 |
| Cox2 | 684 | 61.7 | ATG/TAA | 227 |
| Atp8 | 201 | 68.2 | ATG/TAA | 66 |
| Atp6 | 681 | 61.5 | ATG/TAA | 226 |
| Cox3 | 785 | 55.9 | ATG/TA- | 261 |
| Nd3 | 350 | 58.0 | ATA/TA- | 116 |
| Nd4L | 297 | 63.9 | ATG/TAA | 98 |
| Nd4 | 1378 | 60.9 | ATG/T-- | 459 |
| Nd5 | 1821 | 60.3 | ATA/TAA | 606 |
| Nd6 | 528 | 63.1 | ATG/TAA | 175 |
| CytB | 1140 | 56.4 | ATG/AGA | 379 |
| Total | 11,409 | 3792 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arbizu, C.I.; Ferro-Mauricio, R.D.; Chávez-Galarza, J.C.; Vásquez, H.V.; Maicelo, J.L.; Poemape, C.; Gonzales, J.; Quilcate, C.; Corredor, F.-A. The Complete Mitochondrial Genome of a Neglected Breed, the Peruvian Creole Cattle (Bos taurus), and Its Phylogenetic Analysis. Data 2022, 7, 76. https://doi.org/10.3390/data7060076
Arbizu CI, Ferro-Mauricio RD, Chávez-Galarza JC, Vásquez HV, Maicelo JL, Poemape C, Gonzales J, Quilcate C, Corredor F-A. The Complete Mitochondrial Genome of a Neglected Breed, the Peruvian Creole Cattle (Bos taurus), and Its Phylogenetic Analysis. Data. 2022; 7(6):76. https://doi.org/10.3390/data7060076
Chicago/Turabian StyleArbizu, Carlos I., Rubén D. Ferro-Mauricio, Julio C. Chávez-Galarza, Héctor V. Vásquez, Jorge L. Maicelo, Carlos Poemape, Jhony Gonzales, Carlos Quilcate, and Flor-Anita Corredor. 2022. "The Complete Mitochondrial Genome of a Neglected Breed, the Peruvian Creole Cattle (Bos taurus), and Its Phylogenetic Analysis" Data 7, no. 6: 76. https://doi.org/10.3390/data7060076