Customized Massive Parallel Sequencing Panel for Diagnosis of Pulmonary Arterial Hypertension

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Cohort Description
2.2. Statistical Analysis
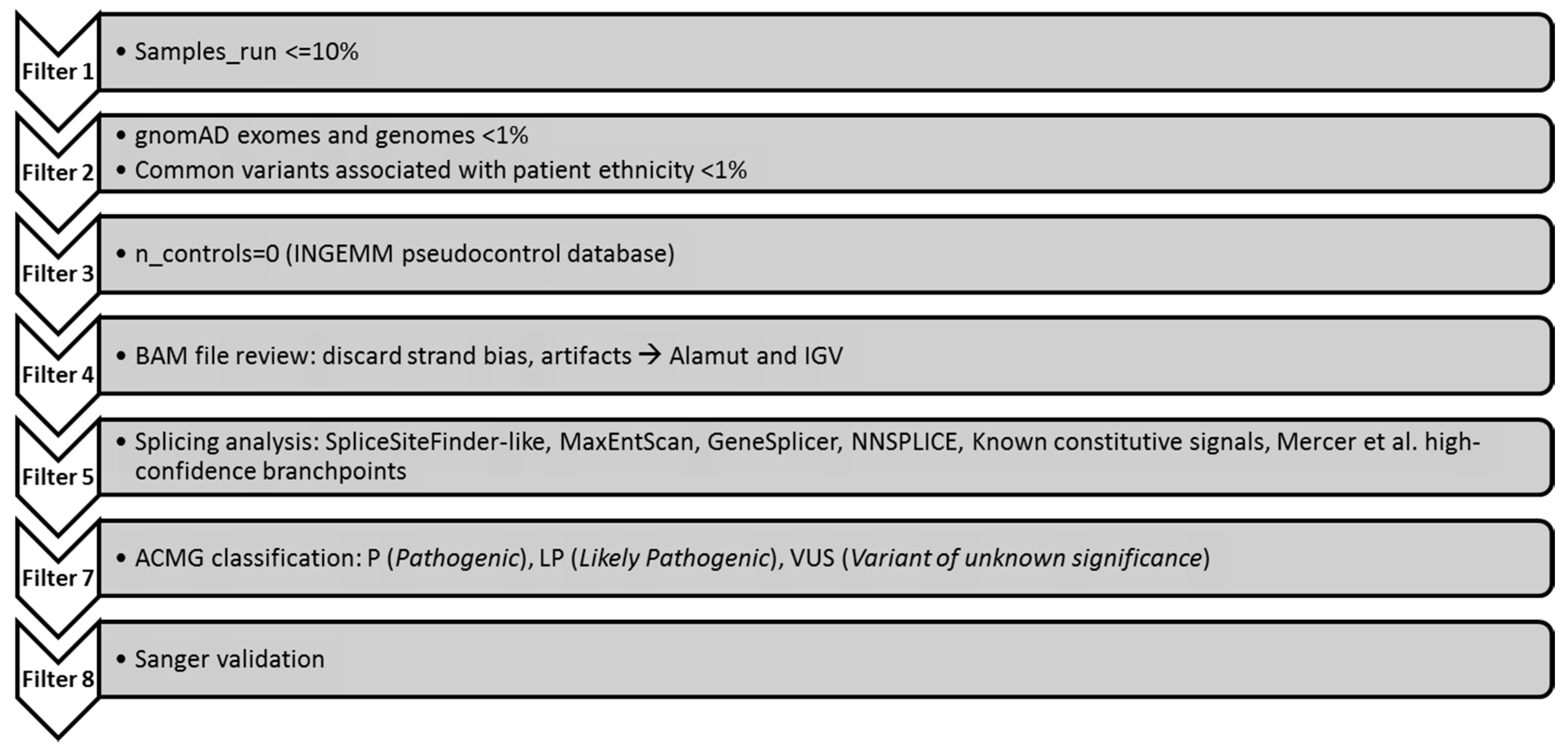
2.3. Genetic Analysis
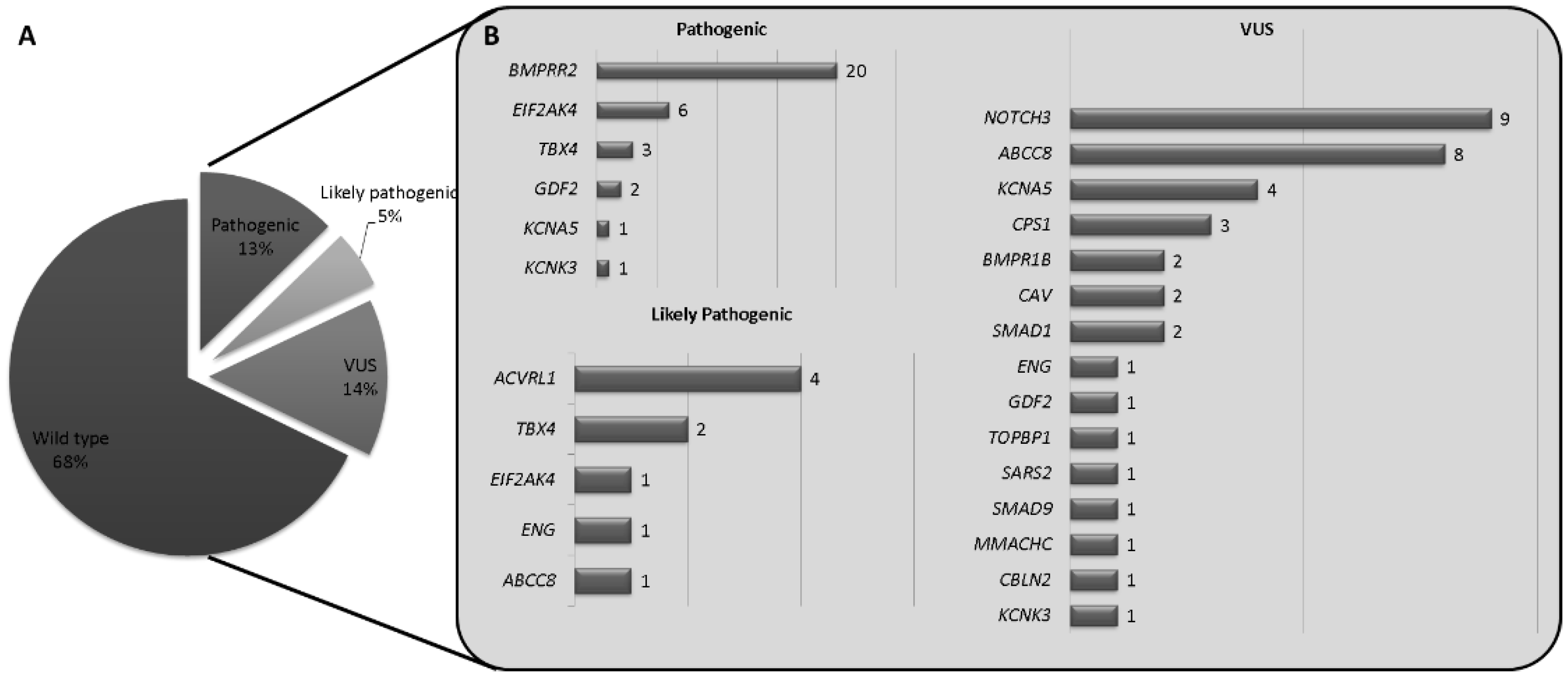
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Escribano-Subias, P.; Blanco, I.; Lopez-Meseguer, M.; Lopez-Guarch, C.J.; Roman, A.; Morales, P.; Castillo-Palma, M.J.; Segovia, J.; Gomez-Sanchez, M.A.; Barbera, J.A.; et al. Survival in pulmonary hypertension in Spain: Insights from the Spanish registry. Eur. Respir. J. 2012, 40, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Rev. Esp. Cardiol. 2016, 69, 177. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.R.; Machado, R.D.; Pauciulo, M.W.; Morgan, N.V.; Humbert, M.; Elliott, G.C.; Ward, K.; Yacoub, M.; Mikhail, G.; Rogers, P.; et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J. Med. Genet. 2000, 37, 741–745. [Google Scholar] [CrossRef]
- Navas, P.; Tenorio, J.; Quezada, C.A.; Barrios, E.; Gordo, G.; Arias, P.; Lopez Meseguer, M.; Santos-Lozano, A.; Palomino Doza, J.; Lapunzina, P.; et al. Molecular analysis of BMPR2, TBX4, and KCNK3 and genotype-phenotype correlations in Spanish patients and families with idiopathic and hereditary pulmonary arterial hypertension. Rev. Esp. Cardiol. 2016, 69, 1011–1019. [Google Scholar] [CrossRef]
- Machado, R.D.; Eickelberg, O.; Elliott, C.G.; Geraci, M.W.; Hanaoka, M.; Loyd, J.E.; Newman, J.H.; Phillips, J.A., 3rd; Soubrier, F.; Trembath, R.C.; et al. Genetics and genomics of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54 (Suppl. S1), S32–S42. [Google Scholar] [CrossRef] [Green Version]
- Antigny, F.; Hautefort, A.; Meloche, J.; Belacel-Ouari, M.; Manoury, B.; Rucker-Martin, C.; Pechoux, C.; Potus, F.; Nadeau, V.; Tremblay, E.; et al. Potassium channel subfamily K member 3 (KCNK3) contributes to the development of pulmonary arterial hypertension. Circulation 2016, 133, 1371–1385. [Google Scholar] [CrossRef]
- Austin, E.D.; Ma, L.; LeDuc, C.; Berman Rosenzweig, E.; Borczuk, A.; Phillips, J.A., 3rd; Palomero, T.; Sumazin, P.; Kim, H.R.; Talati, M.H.; et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ. Cardiovasc. Genet. 2012, 5, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Chida, A.; Shintani, M.; Matsushita, Y.; Sato, H.; Eitoku, T.; Nakayama, T.; Furutani, Y.; Hayama, E.; Kawamura, Y.; Inai, K.; et al. Mutations of NOTCH3 in childhood pulmonary arterial hypertension. Mol. Genet. Genom. Med. 2014, 2, 229–239. [Google Scholar] [CrossRef]
- De Jesus Perez, V.A.; Yuan, K.; Lyuksyutova, M.A.; Dewey, F.; Orcholski, M.E.; Shuffle, E.M.; Mathur, M.; Yancy, L., Jr.; Rojas, V.; Li, C.G.; et al. Whole-exome sequencing reveals TopBP1 as a novel gene in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 1260–1272. [Google Scholar] [CrossRef] [Green Version]
- Eyries, M.; Montani, D.; Girerd, B.; Perret, C.; Leroy, A.; Lonjou, C.; Chelghoum, N.; Coulet, F.; Bonnet, D.; Dorfmuller, P.; et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat. Genet. 2014, 46, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Kerstjens-Frederikse, W.S.; Bongers, E.M.; Roofthooft, M.T.; Leter, E.M.; Douwes, J.M.; Van Dijk, A.; Vonk-Noordegraaf, A.; Dijk-Bos, K.K.; Hoefsloot, L.H.; Hoendermis, E.S.; et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J. Med. Genet. 2013, 50, 500–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2018, 1801899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Pauciulo, M.W.; Welch, C.L.; Lutz, K.A.; Coleman, A.W.; Gonzaga-Jauregui, C.; Wang, J.; Grimes, J.M.; Martin, L.J.; He, H.; et al. Novel risk genes and mechanisms implicated by exome sequencing of 2572 individuals with pulmonary arterial hypertension. Genome Med. 2019, 11, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenorio, J.; Navas, P.; Barrios, E.; Fernandez, L.; Nevado, J.; Quezada, C.A.; Lopez-Meseguer, M.; Arias, P.; Mena, R.; Lobo, J.L.; et al. A founder EIF2AK4 mutation causes an aggressive form of pulmonary arterial hypertension in Iberian Gypsies. Clin. Genet. 2015, 88, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Best, D.H.; Sumner, K.L.; Austin, E.D.; Chung, W.K.; Brown, L.M.; Borczuk, A.C.; Rosenzweig, E.B.; Bayrak-Toydemir, P.; Mao, R.; Cahill, B.C.; et al. EIF2AK4 mutations in pulmonary capillary hemangiomatosis. Chest 2014, 145, 231–236. [Google Scholar] [CrossRef] [Green Version]
- Pousada, G.; Baloira, A.; Valverde, D. Complex inheritance in pulmonary arterial hypertension patients with several mutations. Sci. Rep. 2016, 6, 33570. [Google Scholar] [CrossRef] [Green Version]
- Eichstaedt, C.A.; Song, J.; Benjamin, N.; Harutyunova, S.; Fischer, C.; Grunig, E.; Hinderhofer, K. EIF2AK4 mutation as “second hit” in hereditary pulmonary arterial hypertension. Respir. Res. 2016, 17, 141. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Knight, L.; Ji, R.; Lawrence, P.; Kanaan, U.; Li, L.; Das, A.; Cui, B.; Zou, W.; Penny, D.J.; et al. Early onset severe pulmonary arterial hypertension with ’two-hit’ digenic mutations in both BMPR2 and KCNA5 genes. Int. J. Cardiolo. 2014, 177, e167. [Google Scholar] [CrossRef]
- Abou Hassan, O.K.; Haidar, W.; Nemer, G.; Skouri, H.; Haddad, F.; BouAkl, I. Clinical and genetic characteristics of pulmonary arterial hypertension in Lebanon. BMC Med. Genet. 2018, 19, 89. [Google Scholar] [CrossRef]
- Lazaro Salvador, M.; Quezada Loaiza, C.A.; Rodriguez Padial, L.; Barbera, J.A.; Lopez-Meseguer, M.; Lopez-Reyes, R.; Sala Llinas, E.; Alcolea, S.; Blanco, I.; Escribano Subias, P.; et al. Portopulmonary hypertension: Prognosis and management in the current treatment era. Results from the REHAP Registry. Intern. Med. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Gonzalez, R.; Lopez-Guarch, C.J.; Subirana-Domenech, M.T.; Ruiz, J.M.; Gonzalez, I.O.; Cubero, J.S.; del Cerro, M.J.; Salvador, M.L.; Dos Subira, L.; Gallego, P.; et al. Pulmonary hypertension and congenital heart disease: An insight from the REHAP national registry. Int. J. Cardiol. 2015, 184, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Hernandez-Gonzalez, I.; Tenorio, J.; Palomino-Doza, J.; Martinez Menaca, A.; Morales Ruiz, R.; Lago-Docampo, M.; Valverde Gomez, M.; Gomez Roman, J.; Enguita Valls, A.B.; Perez-Olivares, C.; et al. Clinical heterogeneity of pulmonary arterial hypertension associated with variants in TBX4. PLoS ONE 2020, 15, e0232216. [Google Scholar] [CrossRef]
- Mercer, T.R.; Clark, M.B.; Andersen, S.B.; Brunck, M.E.; Haerty, W.; Crawford, J.; Taft, R.J.; Nielsen, L.K.; Dinger, M.E.; Mattick, J.S. Genome-wide discovery of human splicing branchpoints. Genome Res. 2015, 25, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Eichstaedt, C.A.; Viales, R.R.; Benjamin, N.; Harutyunova, S.; Fischer, C.; Grunig, E.; Hinderhofer, K. Identification of genetic defects in pulmonary arterial hypertension by a new gene panel diagnostic tool. Clin. Sci. 2016, 130, 2043–2052. [Google Scholar] [CrossRef]
- Barozzi, C.; Galletti, M.; Tomasi, L.; De Fanti, S.; Palazzini, M.; Manes, A.; Sazzini, M.; Galie, N. A combined targeted and whole exome sequencing approach identified novel candidate genes involved in heritable pulmonary arterial hypertension. Sci. Rep. 2019, 9, 753. [Google Scholar] [CrossRef]
- Zhang, H.S.; Liu, Q.; Piao, C.M.; Zhu, Y.; Li, Q.Q.; Du, J.; Gu, H. Genotypes and phenotypes of Chinese pediatric patients with idiopathic and heritable pulmonary arterial hypertension—A single-center study. Can. J. Cardiol. 2019, 35, 1851–1856. [Google Scholar] [CrossRef]
- Hansmann, G.; Koestenberger, M.; Alastalo, T.P.; Apitz, C.; Austin, E.D.; Bonnet, D.; Budts, W.; D’Alto, M.; Gatzoulis, M.A.; Hasan, B.S.; et al. 2019 updated consensus statement on the diagnosis and treatment of pediatric pulmonary hypertension: The European pediatric pulmonary vascular disease network (EPPVDN), endorsed by AEPC, ESPR and ISHLT. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 2019, 38, 879–901. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, X.; Leathers, R.; Makino, A.; Huang, C.; Parsa, P.; Macias, J.; Yuan, J.X.; Jamieson, S.W.; Thistlethwaite, P.A. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat. Med. 2009, 15, 1289–1297. [Google Scholar] [CrossRef] [Green Version]
- Bohnen, M.S.; Ma, L.; Zhu, N.; Qi, H.; McClenaghan, C.; Gonzaga-Jauregui, C.; Dewey, F.E.; Overton, J.D.; Reid, J.G.; Shuldiner, A.R.; et al. Loss-of-function ABCC8 mutations in pulmonary arterial hypertension. Circ. Genom. Precis. Med. 2018, 11, e002087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lago-Docampo, M.; Tenorio, J.; Hernandez-Gonzalez, I.; Perez-Olivares, C.; Escribano-Subias, P.; Pousada, G.; Baloira, A.; Arenas, M.; Lapunzina, P.; Valverde, D. Characterization of rare ABCC8 variants identified in Spanish pulmonary arterial hypertension patients. Sci. Rep. 2020, 10, 15135. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.E.; McElroy, J.J.; Wong, W.P.; Yen, E.; Widlitz, A.; Barst, R.J.; Knowles, J.A.; Morse, J.H. BMPR2 mutations in pulmonary arterial hypertension with congenital heart disease. Eur. Respir. J. 2004, 24, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liu, Q.Q.; Guan, L.H.; Jiang, X.; Zhou, D.X.; Beghetti, M.; Qu, J.M.; Jing, Z.C. BMPR2 mutation is a potential predisposing genetic risk factor for congenital heart disease associated pulmonary vascular disease. Int. J. Cardiol. 2016, 211, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Stout, K.K.; Daniels, C.J.; Aboulhosn, J.A.; Bozkurt, B.; Broberg, C.S.; Colman, J.M.; Crumb, S.R.; Dearani, J.A.; Fuller, S.; Gurvitz, M.; et al. 2018 AHA/ACC guideline for the management of adults with congenital heart disease: Executive summary: A Report of the American college of cardiology/American heart association task force on clinical practice guidelines. Circulation 2019, 139, e637–e697. [Google Scholar] [CrossRef]
- Galambos, C.; Mullen, M.P.; Shieh, J.T.; Schwerk, N.; Kielt, M.J.; Ullmann, N.; Boldrini, R.; Stucin-Gantar, I.; Haass, C.; Bansal, M.; et al. Phenotype characterisation of TBX4 mutation and deletion carriers with neonatal and paediatric pulmonary hypertension. Eur. Respir. J. 2019, 54. [Google Scholar] [CrossRef] [PubMed]
- Canter, J.A.; Summar, M.L.; Smith, H.B.; Rice, G.D.; Hall, L.D.; Ritchie, M.D.; Motsinger, A.A.; Christian, K.G.; Drinkwater, D.C., Jr.; Scholl, F.G.; et al. Genetic variation in the mitochondrial enzyme carbamyl-phosphate synthetase I predisposes children to increased pulmonary artery pressure following surgical repair of congenital heart defects: A validated genetic association study. Mitochondrion 2007, 7, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Kaluarachchi, D.C.; Smith, C.J.; Klein, J.M.; Murray, J.C.; Dagle, J.M.; Ryckman, K.K. Polymorphisms in urea cycle enzyme genes are associated with persistent pulmonary hypertension of the newborn. Pediatr. Res. 2018, 83, 142–147. [Google Scholar] [CrossRef]
- McGoon, M.D.; Benza, R.L.; Escribano-Subias, P.; Jiang, X.; Miller, D.P.; Peacock, A.J.; Pepke-Zaba, J.; Pulido, T.; Rich, S.; Rosenkranz, S.; et al. Pulmonary arterial hypertension: Epidemiology and registries. Turk. Kardiyol. Dern. Ars. 2014, 42 (Suppl. S1), 67–77. [Google Scholar] [CrossRef] [Green Version]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, M.D. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef]
- Rosenkranz, S.; Dumitrescu, D. Pulmonary hypertension-back to the future. Rev. Esp. Cardiol. 2017, 70, 901–904. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
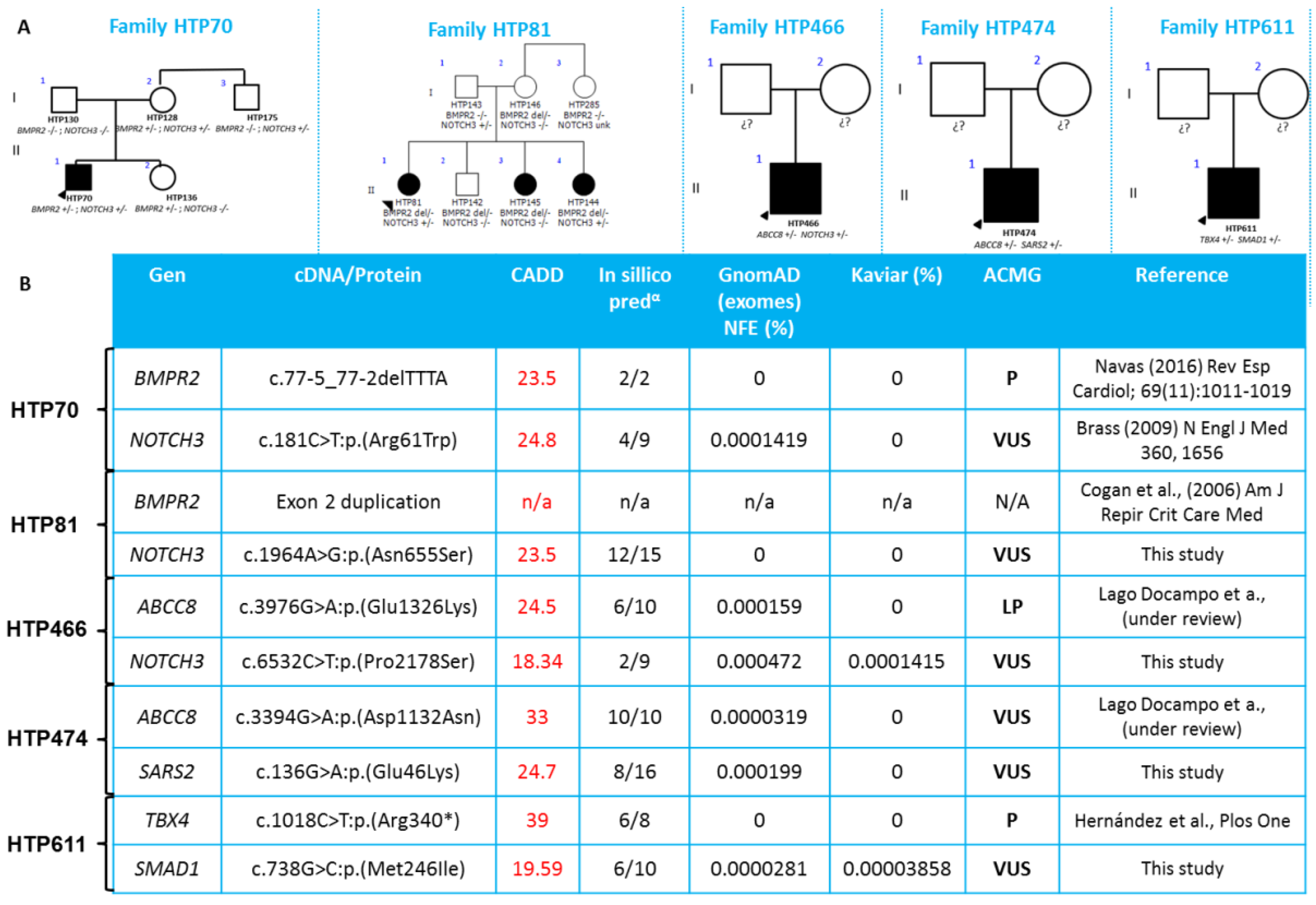
| Patient ID | PAH Etiology | Variant 1 | ACMG Classification | Variant 2 | ACMG Classification |
|---|---|---|---|---|---|
| HTP070 | IPAH | BMPR2:NM_001204.6:c.77-5_77-2delTTTA | P | NOTCH3:NM_000435.2:c.181C>T:p.(Arg61Trp) | VUS |
| HTP081 | IPAH | BMPR2:Exon2 duplication | N/A | NOTCH3:NM_000435.2:c.1964A>G:p.(Asn655Ser) | VUS |
| HTP466 | IPAH | ABCC8:NM_000352.4:c.3976G>A:p.(Glu1326Lys) | LP | NOTCH3:NM_000435.2:c.6532C>T:p.(Pro2178Ser) | VUS |
| HTP474 | CHD | ABCC8:NM_000352.4(ABCC8):c.3394G>A:p.(Asp1132Asn) | VUS | SARS2:NM_017827.3:c.136G>A:p.(Glu46Lys) | VUS |
| HTP611 | CHD | TBX4:NM_018488.3:c.1018C>T:p.(Arg340 *) | P | SMAD1:NM_005900.2:c.738G>C:p.(Met246Ile) | VUS |
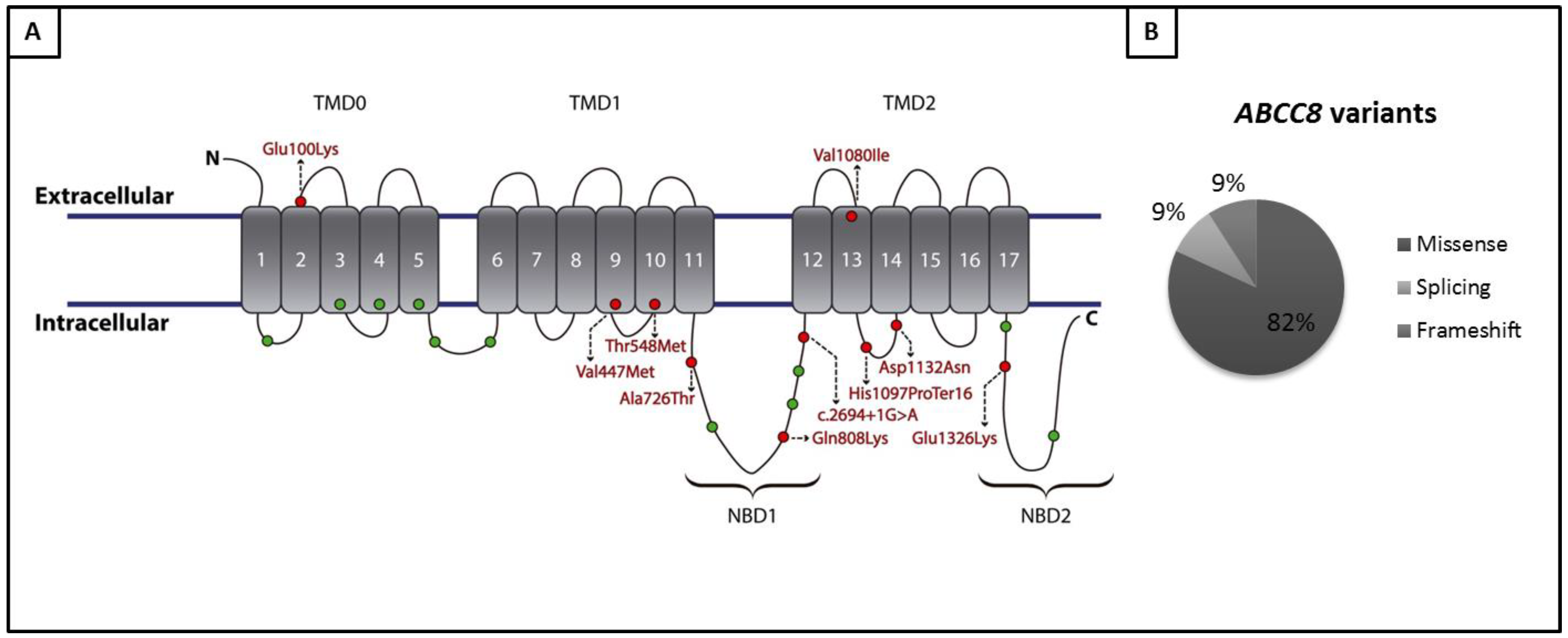
| Patient ID | PAH Etiology | Variant |
|---|---|---|
| HTP114 | IPAH | ABCC8:NM_000352.4:c.1643C>T:p.(Thr548Met) |
| HTP88 | IPAH | ABCC8:NM_000352:exon26:c.3288_3289del:p.(His1097Profs*16) |
| HTP151 | IPAH | ABCC8:NM_000352.4:c.3238G>A:p.(Val1080Ile) |
| HTP159 | IPAH | ABCC8:NM_000352.4:c.2422C>A:p.(Gln808Lys) |
| HTP162 | IPAH | ABCC8:NM_000352.4:c.1429G>A:p.(Val477Met) |
| HTP466 | IPAH | ABCC8:NM_000352.4:c.3976G>A:p.Glu1326Lys |
| HTP37 | IPAH | ABCC8:NM_000352.3:c.579+5G>A |
| HTP78 | CREST-PAH | ABCC8:NM_000352.3:c.2694+1G>A |
| HTP474 | CHD | ABCC8:NM_000352.4:c.3394G>A:p.Asp1132Asn |
| ID | PAH Etiology | cDNA and Protein Position | GT | ACMG Classification |
|---|---|---|---|---|
| HTP501 | CHD | CPS1:NM_001122633.2(CPS1):c.3047C>T:p.(Thr1016Met) | Hom | VUS |
| HTP536 | CHD | BMPR2:NM_001204.6:c.2674delG: p.(Glu892Asnfs*4) | Het | P |
| HTP541 | CHD | BMPR2:NM_001204.6:c.2674delG: p.(Glu892Asnfs*4) | Het | P |
| HTP474 | CHD | ABCC8:NM_000352.4(ABCC8):c.3394G>A:p.(Asp1132Asn) SARS2:NM_017827.3:c.136G>A:p.(Glu46Lys) | Het Het | VUS |
| HTP558 | CHD | SMAD5:NM_001001420.2:c.763A>G:p.(Ile255Val) | Het | VUS |
| HTP262 | CHD | NOTCH3:NM_000435.3:c.6097C>G:p.(Pro2033Ala) | Het | VUS |
| HTP472 | CHD | CPS1:NM_001122633.2:c.1036G>A:p.(Ala346Thr) | Het | VUS |
| HTP611 | CHD | TBX4:NM_018488.3:c.1018C>T:p.(Arg340*) SMAD1:NM_005900.2:c.738G>C:p.(Met246Ile) | Het Het | P VUS |
| HTP551 | CTD | GDF2:NM_016204:c.642G>A:p.(Trp214*) | Het | VUS |
| HTP355 | CTD | CPS1:NM_001875.4:c.4252C>T:p.(Pro1418Ser) | Het | VUS |
| HTP452 | CTD | NOTCH3:NM_000435:c.5203G>A:p.(Glu1735Lys) | Het | VUS |
| HTP564 | CTD | TBX4:NM_018488.2:c.1112dupC:p.(Pro372Serfs*14) | Het | P |
| HTP78 | CTD | ABCC8:NM_000352.3:c.2694+1G>A | Het | VUS |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castaño, J.A.T.; Hernández-Gonzalez, I.; Gallego, N.; Pérez-Olivares, C.; Ochoa Parra, N.; Arias, P.; Granda, E.; Acebo, G.G.; Lago-Docampo, M.; Palomino-Doza, J.; et al. Customized Massive Parallel Sequencing Panel for Diagnosis of Pulmonary Arterial Hypertension. Genes 2020, 11, 1158. https://doi.org/10.3390/genes11101158
Castaño JAT, Hernández-Gonzalez I, Gallego N, Pérez-Olivares C, Ochoa Parra N, Arias P, Granda E, Acebo GG, Lago-Docampo M, Palomino-Doza J, et al. Customized Massive Parallel Sequencing Panel for Diagnosis of Pulmonary Arterial Hypertension. Genes. 2020; 11(10):1158. https://doi.org/10.3390/genes11101158
Chicago/Turabian StyleCastaño, Jair Antonio Tenorio, Ignacio Hernández-Gonzalez, Natalia Gallego, Carmen Pérez-Olivares, Nuria Ochoa Parra, Pedro Arias, Elena Granda, Gonzalo Gómez Acebo, Mauro Lago-Docampo, Julian Palomino-Doza, and et al. 2020. "Customized Massive Parallel Sequencing Panel for Diagnosis of Pulmonary Arterial Hypertension" Genes 11, no. 10: 1158. https://doi.org/10.3390/genes11101158