
Prevalent ALMS1 Pathogenic Variants in Spanish Alström Patients
by
 , , and
, , and
Brais Bea-Mascato
1,2, 
Carlos Solarat
1,2 ,
,
Irene Perea-Romero
3,4,
Teresa Jaijo
3,5,
Fiona Blanco-Kelly
3,4,
José M. Millán
3,5,
Carmen Ayuso
3,4 and
Diana Valverde
1,2,* 1
CINBIO, Universidad de Vigo, 36310 Vigo, Spain
2
Grupo de Investigación en Enfermedades Raras y Medicina Pediátrica, Instituto de Investigación Sanitaria Galicia Sur (IIS Galicia Sur), SERGAS-UVIGO, 36310 Vigo, Spain
3
Centro de Investigación Biomédica en Red en Enfermedades Raras (CIBERER), ISCIII, 28029 Madrid, Spain
4
Departamento de Genética Clínica, Instituto de Investigación Sanitaria Hospital Universitario Fundación Jiménez Díaz, (IIS-FJD, UAM), 28040 Madrid, Spain
5
Unidad de Genética, Hospital Universitario y Politécnico La Fe. Biomedicina Molecular Celular y Genómica, Instituto Investigación Sanitaria La Fe, 46026 Valencia, Spain
*
Author to whom correspondence should be addressed.
Genes 2021, 12(2), 282; https://doi.org/10.3390/genes12020282
Submission received: 19 January 2021
/
Revised: 11 February 2021
/
Accepted: 12 February 2021
/
Published: 16 February 2021
(This article belongs to the Section Human Genomics and Genetic Diseases)
Abstract
:Alström syndrome (ALMS) is an ultrarare disease with an estimated prevalence lower than 1 in 1,000,000. It is associated with disease-causing mutations in the Alström syndrome 1 (ALMS1) gene, which codifies for a structural protein of the basal body and centrosomes. The symptomatology involves nystagmus, type 2 diabetes mellitus (T2D), obesity, dilated cardiomyopathy (DCM), neurodegenerative disorders and multiorgan fibrosis. We refined the clinical and genetic diagnosis data of 12 patients from 11 families, all of them from Spain. We also studied the allelic frequency of the different variants present in this cohort and performed a haplotype analysis for the most prevalent allele. The genetic analysis revealed 2 novel homozygous variants located in the exon 8, p.(Glu929Ter) and p.(His1808GlufsTer20) in 2 unrelated patients. These 2 novel variants were classified as pathogenic after an in silico experiment (computer analysis). On the other hand, 2 alleles were detected at a high frequency in our cohort: p.(Tyr1714Ter) (25%) and p.(Ser3872TyrfsTer19) (16.7%). The segregation analysis showed that the pathogenic variant p.(Tyr1714Ter) in 3 families is linked to a rare missense polymorphism, p.(Asn1787Asp). In conclusion, 2 novel pathological mutations have been discovered in homozygosis, as well as a probable founder effect in 3 unrelated families.
1. Introduction
Alström Syndrome (ALMS; OMIM #203800) is an ultrarare recessive disorder, with an estimated prevalence lower than 1 in 1,000,000 in European-descent populations. As in the case of other rare syndromes, consanguineous and/or geographically isolated populations have higher frequency values [1,2,3]. About 1000 cases have been described worldwide for this pathology, of which 13 have been diagnosed in Spain [4,5,6,7].
ALMS is a pleiotropic and multisystemic disorder characterized by a high inter- and intrafamilial variability, regarding the phenotype displayed, the age of onset and the severity of symptoms [4,5]. The cardinal features include childhood obesity, insulin resistance, cone-rod retinal dystrophy, sensorineural hearing loss, type 2 diabetes mellitus (T2D), hypertriglyceridemia and dilated cardiomyopathy (DCM) [8]. Other secondary features include seizures, hyporeflexia or multiorgan fibrosis that develops from adolescence onwards. This latter is very variable and can affect the liver, kidneys, lungs and gonads [8]. The first clinical feature, visual dysfunction (photophobia and nystagmus), usually develops between a few weeks after birth and the first year of life [5]. The remaining signs evolve slowly during childhood and adolescence, although the most severe features can be detected before the first decade [4].
The clinical diagnosis of ALMS is based on the presence of primary and secondary features, considering the age of onset throughout the development [9].
ALMS is a monogenic disorder caused by pathogenic variants in the ALMS1 gene (MIM #606844), which represents an unusual phenomenon among ciliopathies, normally described with high genetic heterogeneity. ALMS1 is located on chromosome 2 (region 2p13.1) and consists of 224 kilobases (kb) containing 23 coding exons [10,11]. Several splicing isoforms have been reported, which could produce different protein isoforms with specific functions [10,12,13,14].
To date, over 298 pathogenic variants have been involved in ALMS development, of which 96% are nonsense or frameshift changes (insertions and deletions) that could originate truncated, nonfunctional proteins [4,15]. Most of the deleterious variants are clustered in exons 8 (6.1 kb), 10 (1.9 kb) and 16 (1.2 kb), which are considered mutational hotspots as they comprise 85–97% of the total mutational load for ALMS1 in the different cohorts [4,15]. Hence, the direct sequencing of these 3 exons represents the standard strategy when ALMS is suspected. However, the progressive implementation of high-throughput sequencing (HTS) techniques, such as whole-exome sequencing (WES) and targeted gene panels, is replacing the classical approach to ALMS molecular diagnosis [16,17,18].
The vast majority of changes in ALMS1 have been described once, most cases worldwide are compound heterozygotes, and several groups of patients have shown a founder effect, like the Acadian, English or Turkish population [1,3,19]. The knowledge of the mutational load in this gene could be interesting for understanding the genotype-phenotype correlation and the molecular basis of this disorder.
2. Materials and Methods
2.1. Cohort Presentation
This study included 12 patients from 11 unrelated families clinically diagnosed with ALMS (Patients 4 and 5 are siblings). Here, we reported the genetic characterization of 5 males and 7 females (Table 1) of Caucasian ethnicity.
The clinical history for 11 of the 12 patients was obtained through collaboration with medical doctors and the National Association of Alström syndrome Spain. The main clinical characteristics are described in Table 2.
Most of the families were molecularly described elsewhere [4], and, as part of the Spanish ciliopathy cohort, they have been studied clinically and molecularly by our group [6,7]. Families GBB-28 and UG-26225 have been described for the first time in this study, and their molecular characterization was deposited in ClinVar.
2.2. DNA Extraction and Sanger Sequencing
DNA was extracted from peripheral blood from participants (Patients 1, 3, 6, 7 and 8) and available family members. We used the Flexigene DNA kit 250 (Qiagen, Hilden, Germany), following the manufacturer’s protocol.
After DNA extraction, we analyzed the exonic DNA of ALMS1. We amplified the DNA by polymerase chain reaction (PCR) in an MJ MiniTM Gradient Thermal Cycler (Bio-Rad, Hercules, CA, USA) with the primers described by Collin et al. [10] (Table S2). PCR reactions were performed using 100 ng of genomic DNA, 1 μL at 10 μM of each primer and 12.5 µL Supreme NZYTaq II 2x Green Master Mix (Nzytech, Lisbon, Portugal) in a final volume of 25 µL per sample. The amplification program applied to samples was as follows: initial denaturing at 95 °C for 5 min, followed by 35 cycles of 94 °C for 30 s, 52–66 °C for 30 s, and 72 °C for 30 s and a final extension step at 72 °C for 10 min [6]. Then, the PCR products were resolved and stained on a 2% agarose gel with 0.05% ethidium bromide.
After that, 4.5 µL of each PCR was purified using ExoSAP (Thermo-Fisher, Waltham, CA, USA) in a final volume of 6 µL, incubating the reaction for 15 min at 37 °C and 15 min at 80 °C. The products were sequenced directly using the BigDye® Terminator v1.3 Cycle Sequencing Kit (Life Technologies, Foster City, CA, USA) in a 10 μL reaction. The program was as follows: initial denaturing at 98 °C for 3 min, followed by 25 cycles of 96 °C for 10 s, 50 °C for 5 s and 60 °C for 4 min. The sequencing products were precipitated and dried using MgCl2, ethanol at 4 °C and Microfuge 18® (Beckman-Coulter™, Krefeld, Germany) [6]. The final product was resolved in an ABI PRISM 3130 (Life Technologies, Foster City, CA, USA) genetic analyzer.
Finally, all sequences were visualized with the BioEdit 7.2, and the reference sequences ENST00000613296.6/ENSP00000482968.1 were used for the nucleotide and amino acid numbering of pathogenic variants.
2.3. Relative Allele Frequency Calculation
The calculation of the relative allele frequency (P) was performed using the number of alleles with a specific change (i) divided by the total number of alleles in our cohort (N = 24). Then, the result was multiplied by 100 to obtain a percentage.
P = i/N * 100
2.4. In Silico Analysis of Variants
To predict the mutational effect, the novel pathogenic ALMS1 variants were analyzed with the following most used software: PolyPhen2 (p. (Glu929Ter) mutation was excluded from this analysis because the software does not analyze STOP mutations, considering them to always be pathogenic) [21], SIFT [22], MutPred-LOF [23] and PROVEAN [24]. The score provided by these software were used to classify the variants following the American College of Medical Genetics and Genomics (ACMG) guidelines [25].
3. Results
3.1. Patients Characteristics
All of the 12 patients, (5 males and 7 females) from the 11 families have a positive molecular diagnosis that is biallelic according to a recessive model.
In all cases, we established 2 pathogenic variants. Families carrying the same variant did not have any kindred relationship between them and came from different Spanish locations across the country.
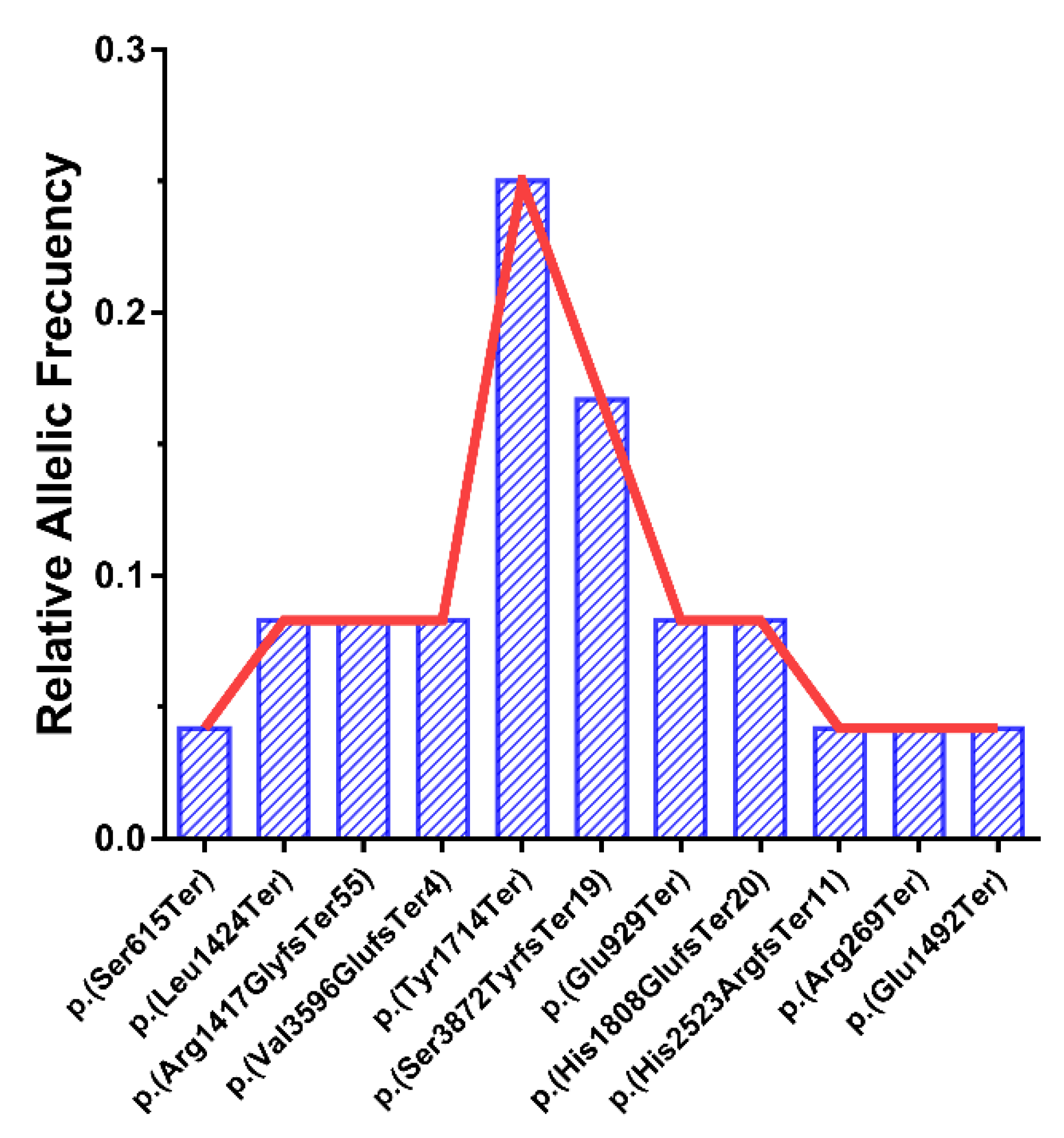
We detected 11 different pathogenic variants (Table 1), 6 patients from the families, GBB-45, GBB-46, RP-793, RP-1232, GBB-28 and UG-26225, being homozygous. Regarding pathogenic variants, 1 is located in exon 5 [20], 7 are located in exon 8 [4,6,7], 1 in exon 9 [4], 1 in exon 16 [19] and 1 in exon 17 [4] of the ALMS1 gene, and all of them lead to a stop codon. The variant p.(Tyr1714Ter) in exon 8 has a high frequency in our pool of patients, at 25%, appearing 6 times in 5 patients (1 homozygous and 4 compound heterozygous). The pathogenic variant p.(Ser3872TyrfsTer19) has been detected 4 times in 3 patients (1 homozygous and 2 compound heterozygous), rising to a 16.7% frequency. Most of the variants detected as being pathogenic were not shown in the gnomAD and ClinVar databases (Table S1), as no population information was available.
Some of the mutations have been uploaded into the LOVD database from the REWBA project by the labs where the molecular analysis was performed. For the data that was found, the pathogenic variant p.(Tyr1714Ter) has been described 5 times [4,15], including 3 of our samples. The pathogenic variants p.(Ser3872TyrfsTer19) and p.(Val3596GlufsTer4) have been described 3 times [4,9], a heterozygous sample for p.(Ser3872TyrfsTer19) that has been reported is 1 of our patients.
All patients included in this study present cone-rod dystrophy and/or nystagmus. Obesity (BMI > 95%), overweight (BMI > 85%), insulin resistance or T2D were present in 91% of the cases. DCM was found in 6 of 11 patients (54.5%). Hearing loss was reported in 8 of 11 patients (73%) (Table 2).
The second group of symptoms with a low incidence was reported too: hepatic dysfunction (55%), renal failure (18%), short stature (45%), thyroid disorders (55%) and hypogonadism/irregular menses (36%) (Table 2).
3.2. Novel ALMS1 pathogenic variants
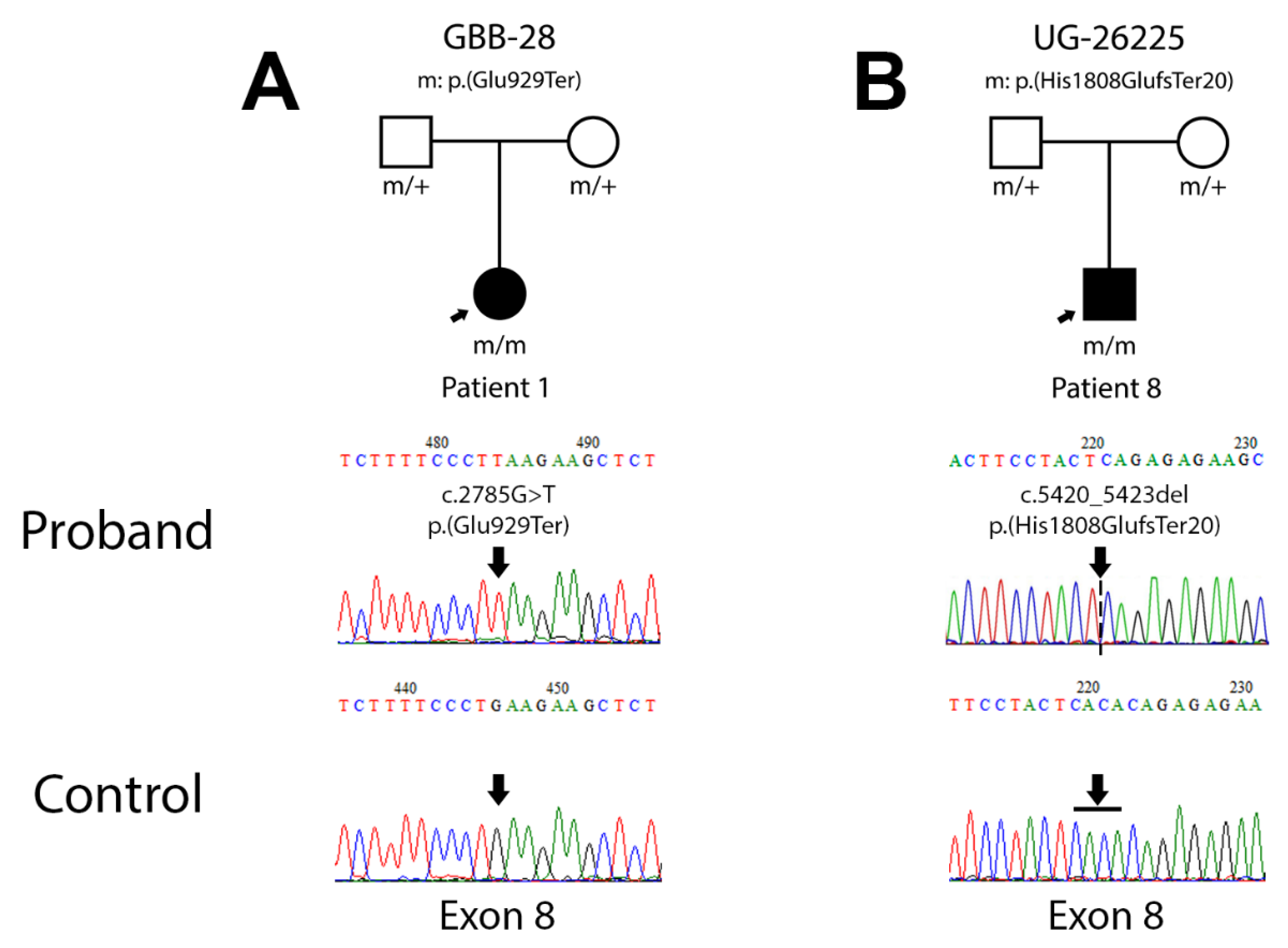
3.2.1. Patient 1 (Family GBB-28)
Patient 1 is a young girl (11 years old) from consanguineous parents. The main symptoms of this patient are nystagmus, photophobia, and rod and cone dystrophy with decreased visual acuity; morbid obesity, DCM and bronchospasm. No further Alström spectrum symptoms have been reported to date (Table 2).
A novel homozygous amino acid change on exon 8, c.2785G>T leading to p.(Glu929Ter), was detected by Sanger sequencing in this patient (Figure 1A). The in silico prediction of pathogenicity through different bioinformatic tools resulted in deleterious variant scores in MutPred-LOF (0.432), SIFT (0) and PROVEAN (−3.376). According to the ACMG, this variant should be classified as pathogenic (PVS1, very strong evidence of pathogenicity) (Table 3).
3.2.2. Patient 8 (Family UG-26225)
Patient 8 is a young boy (3 years old) from consanguineous parents. The main symptoms of this patient are horizontal nystagmus within a few months of birth, DCM of birth, obesity and hypothyroidism. He has a normal size and sexual development according to his age (Table 2).
A novel homozygous amino acid change on exon 8 of ALMS1, c.5420_5423del leading to p.(His1808GlufsTer20), was detected by Sanger sequencing in this patient (Figure 1B). In this case, the in silico analysis shows deleterious scores in PolyPhen2 (0.852), MutPred-LOF (0.422), SIFT (0.05) and PROVEAN (−2.720). Following the criteria of ACMG, this variant should also be classified as pathogenic (PVS1, very strong evidence of pathogenicity) (Table 3).
3.3. Relative Allele Frequencies
We detected 2 specific alleles with a high frequency in ALMS1: p.(Tyr1714Ter) and p.(Ser3872TyrfsTer19), with only 1 family for each of these pathogenic variants being homozygous. The relative frequencies of these alleles in the cohort were 0.25 (25%) and 0.167 (16.7%), respectively (Figure 2).
3.4. Segregation Study
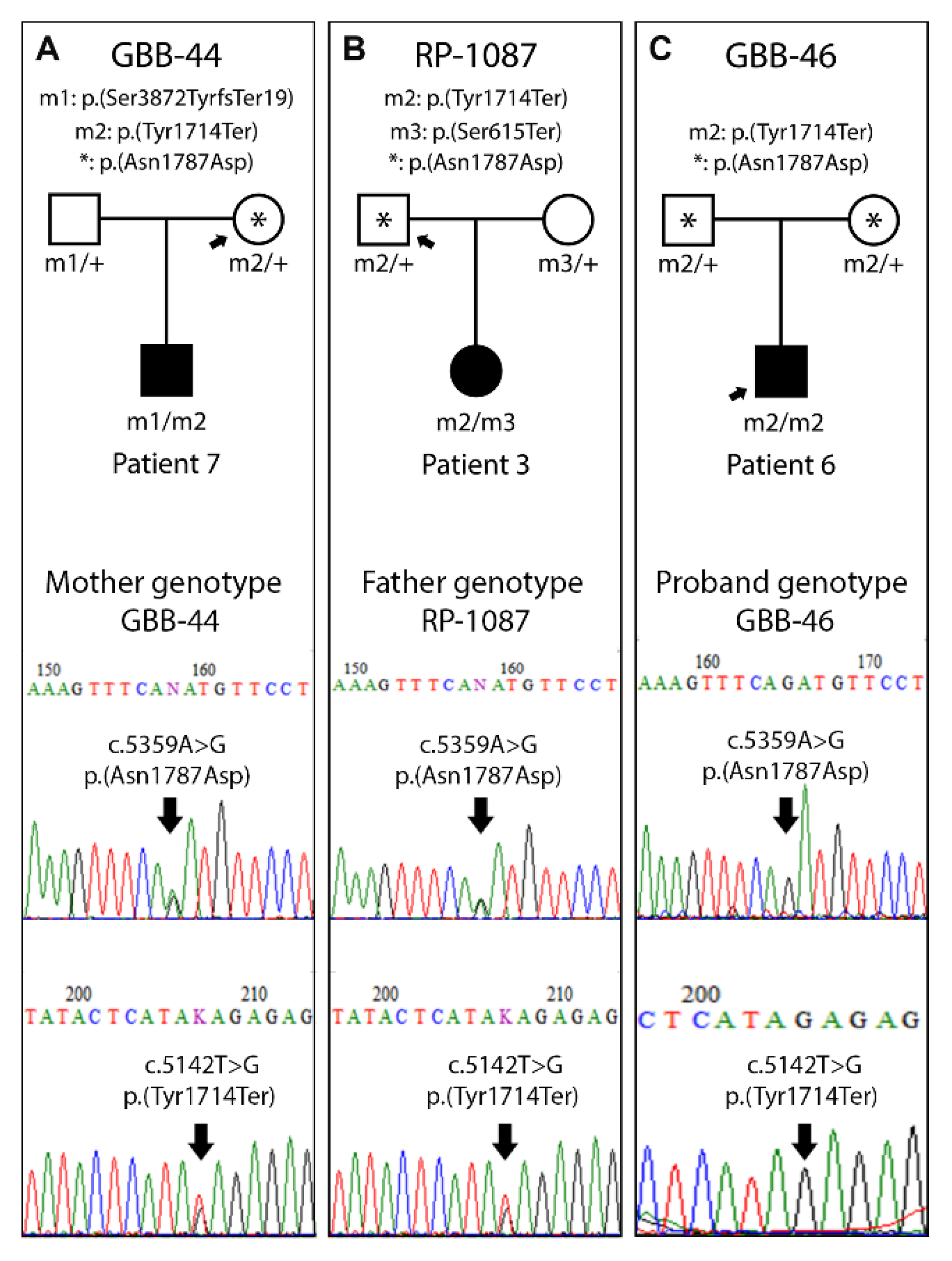
In patients carrying the p.(Tyr1714Ter) pathogenic variant, we detected a single nucleotide polymorphism (SNP) with a low frequency (0.017) in the European population. This SNP, p.(Asn1787Asp) (c.5359A>G; rs45608038), is located at exon 8 of ALMS1 (Table 4). Thus, we evaluated whether the allele p.(Asn1787Asp) segregated with the pathogenic variant p.(Tyr1714Ter) in 3 families, and we concluded that it was linked to the latter (Figure 3).
3.5. Haplogroup Classification
4. Discussion
Alström Syndrome is a complex disease that affects multiple organs and induces a metabolic disorder. Its huge heterogenic interpatient symptomatology and its low incidence in population worldwide makes it very difficult to perform any phenotype–genotype correlation.
In our cohort, we analyzed 12 patients from 11 families with ALMS pathogenic variants. Most of them have been previously clinically and molecularly characterized [4,6,7,19], but patient 1 (GBB-28) and patient 8 (UG-26225) have been described in this study for the first time. These 2 patients are carriers for novel ALMS1 pathogenic variants in homozygosity. The analysis of the open reading frame (ORF) sequence showed the generation of a premature stop codon, resulting in a truncated protein in both cases. For patient 1 (GBB-28), the amino acid change affects the glutamate located in position 929 generating a stop codon (TAA). Regarding patient 8 (UG-26225), the microdeletion of 4 pb (CACA) changes the histidine in position 1808 to glutamate and generates a frameshift mutation that leads to a stop codon (TGA), 20 amino acids downstream. Until now, approximately 298 pathogenic or likely pathogenic variants have been described in ALMS1. A great percentage of cases harbour private mutations. Here, we are expanding this mutational spectrum with 2 novel ALMS1 mutations.
Moreover, 2 highly prevalent pathogenic variants were detected within our cohort. Both pathogenic variants, p.(Ser3872TyrfsTer19) and p.(Tyr1714Ter), located in exons 17 and 8, respectively, generate a premature stop codon resulting in a truncated protein. In this point 1 of these pathogenic variants, p.(Tyr1714Ter), cosegregates with a low-frequency SNP, p.(Asn1787Asp) (c.5359A>G; rs45608038), in the 3 analyzed families (Figure 2), which allows us to establish a potential common origin of this allele in these Spanish patients. Furthermore, based on the haplogroups described for ALMS1, this haplotype is grouped with the ancestral [26], which has been detected in the south of Europe (France, Spain and Portugal) and has a high presence in the African continent. This fact could be explained as an introduction of this ALMSallele in the Iberian Peninsula from the African continent.
In this study, no genotype–phenotype correlation for the pathogenic variants p.(Tyr1714Ter) and p.(Ser3872TyrfsTer19) was detected. Even in siblings with the same genotype (p.(Tyr1714Ter)/p.(Leu1424Ter); patients 4 and 5, family GAS-37), the phenotype was not the same. In this case, patient 4 reported DCM, but his brother did not.
Due to the complexity of ALMS, the effect of only one gene does not seem to be enough to completely explain the heterogeneous symptoms seen in these patients. The prevalence of symptoms like DCM, hepatic dysfunction or hearing loss could be conditioned by other agents like common mutational load or epigenetic regulation, whose study would be interesting.
ALMS is a very rare disease that shares clinical features with other ciliopathic syndromes, so a clinical diagnosis is quite difficult in some cases due to this phenotypic overlap, highlighting the Bardet–Biedl syndrome (in presenting a multiorgan pathology) and Usher syndrome (in combining retinitis pigmentosa with hearing loss). The difficulty in achieving a diagnosis and the lack of a global point of view leads to the fact that some patients are still underdiagnosed and seek medical attendance when the symptoms are exacerbated. Given that the sample size is one of the main barriers to establishing a genotype–phenotype correlation in rare diseases, it would be interesting to establish an international registry of patients that reflects the causal mutations of each patient accompanied by their standardized symptoms. In this respect, an international effort is underway to enrol these patients into national and international associations that provide updated information to patients and put them in touch with clinicians and investigations [27] that facilitate future observational studies and clinical trials.
5. Conclusions
In all, we described 2 novel ALMS pathogenic variants in the exon 8 of the ALMS1 gene that leads to a truncated protein. These 2 variants were reported in homozygosity in nonrelated patients, which showed an Alström syndrome clinical spectrum according to age. Furthermore, we detected 2 prevalent ALMS1 pathogenic variants, p.(Tyr1714Ter) and p.(Ser3872TyrfsTer19), in our Spanish cohort. The pathogenic variant p.(Tyr1714Ter) cosegregates with a benign missense variant, p.(Asn1787Asp). Finally, 3 families with the p.(Tyr1714Ter) pathogenic variant shared the ancestral haplotype for ALMS1 that is predominant in the African continent, which could have arisen by a founder effect.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4425/12/2/282/s1, Table S1. Pathogenic variants register in gnomAD and clinVar from the total alleles in our cohort. Table S2. Oligonucleotides for genomic amplification and sequencing of ALMS1 described in Collin et al., 2002.
Author Contributions
B.B.-M., C.S. and D.V. designed the study. B.B.-M. and C.S. performed the experiments. I.P.-R., T.J., F.B.-K., J.M.M. and C.A. collected clinical and genetic data. B.B.-M., C.S., C.A. and D.V. wrote the manuscript. All authors have read the draft and provided approval for publication.
Funding
This work was funded by Instituto de Salud Carlos III de Madrid FIS project PI15/00049 and PI19/00332, Xunta de Galicia (Centro de Investigación de Galicia CINBIO 2019-2022) Ref. ED431G-2019/06, Consolidación e estructuración de unidades de investigación competitivas e outras accións de fomento (ED431C-2018/54). Brais Bea-Mascato (FPU17/01567) and Carlos Solarat (FPU19/00175) were supported by graduate studentship awards (FPU predoctoral fellowship) from the Spanish Ministry of Education, Culture and Sports.
Institutional Review Board Statement
This study adhered to the tenets of the Declaration of Helsinki and was approved by an ethics committee (Comité Ético de Investigaciones Clínicas de Galicia, Spain, 2006/08).
Informed Consent Statement
Informed consent was obtained from all study participants or their guardians after the nature of the procedures to be performed in this study were fully explained.
Data Availability Statement
The data presented in this study are openly available in ClinVar.
Acknowledgments
We would like to thank the Asociación Española del Síndrome de Alström for their help and ongoing cooperation. Furthermore, we appreciate the collaboration of all colleagues that provided us with the patients’ clinical information.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Marshall, J.D.; Ludman, M.D.; Shea, S.E.; Salisbury, S.R.; Willi, S.M.; LaRoche, R.G.; Nishina, P.M. Genealogy, natural history, and phenotype of Alström syndrome in a large Acadian kindred and three additional families. Am. J. Med. Genet. 1997, 73, 150–161. [Google Scholar] [CrossRef]
- Aldahmesh, M.A.; Safieh, L.A.; Alkuraya, H.; Al-Rajhi, A.; Shamseldin, H.; Hashem, M.; Alzahrani, F.; Khan, A.O.; Alqahtani, F.; Rahbeeni, Z.; et al. Molecular characterization of retinitis pigmentosa in Saudi Arabia. Mol. Vis. 2009, 15, 2464–2469. [Google Scholar] [PubMed]
- Ozantürk, A.; Marshall, J.D.; Collin, G.B.; Düzenli, S.; Marshall, R.P.; Candan, Ş.; Tos, T.; Esen, İ.; Taşkesen, M.; Çayır, A.; et al. The phenotypic and molecular genetic spectrum of Alström syndrome in 44 Turkish kindreds and a literature review of Alström syndrome in Turkey. J. Hum. Genet. 2014, 60, 1. [Google Scholar] [CrossRef]
- Marshall, J.D.; Muller, J.; Collin, G.B.; Milan, G.; Kingsmore, S.F.; Dinwiddie, D.; Farrow, E.G.; Miller, N.A.; Favaretto, F.; Maffei, P.; et al. Alström Syndrome: Mutation Spectrum of ALMS1. Hum. Mutat. 2015, 36, 660–668. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.D.; Maffei, P.; Collin, G.B.; Naggert, J.K. Alström syndrome: Genetics and clinical overview. Curr. Genomics 2011, 12, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Piñeiro-Gallego, T.; Cortón, M.; Ayuso, C.; Baiget, M.; Valverde, D. Molecular approach in the study of Alström syndrome: Analysis of ten Spanish families. Mol. Vis. 2012, 18, 1794–1802. [Google Scholar]
- Sanchez-Navarro, I.; da Silva, L.R.J.; Blanco-Kelly, F.; Zurita, O.; Sanchez-Bolivar, N.; Villaverde, C.; Lopez-Molina, M.I.; Garcia-Sandoval, B.; Tahsin-Swafiri, S.; Minguez, P.; et al. Combining targeted panel-based resequencing and copy-number variation analysis for the diagnosis of inherited syndromic retinopathies and associated ciliopathies. Sci. Rep. 2018, 8, 5285. [Google Scholar] [CrossRef] [PubMed]
- Tahani, N.; Maffei, P.; Dollfus, H.; Paisey, R.; Valverde, D.; Milan, G.; Han, J.C.; Favaretto, F.; Madathil, S.C.; Dawson, C.; et al. Consensus clinical management guidelines for Alström syndrome. Orphanet J. Rare Dis. 2020, 15, 253. [Google Scholar] [CrossRef]
- Marshall, J.D.; Beck, S.; Maffei, P.; Naggert, J.K. Alström Syndrome. Eur. J. Hum. Genet. 2007, 15, 1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, G.B.; Marshall, J.D.; Ikeda, A.; So, W.V.; Russell-Eggitt, I.; Maffei, P.; Beck, S.; Boerkoel, C.F.; Sicolo, N.; Martin, M.; et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat. Genet. 2002, 31, 74. [Google Scholar] [CrossRef]
- Hearn, T.; Renforth, G.L.; Spalluto, C.; Hanley, N.A.; Piper, K.; Brickwood, S.; White, C.; Connolly, V.; Taylor, J.F.N.; Russell-Eggitt, I.; et al. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alström syndrome. Nat. Genet. 2002, 31, 79. [Google Scholar] [CrossRef] [PubMed]
- Collin, G.B.; Cyr, E.; Bronson, R.; Marshall, J.D.; Gifford, E.J.; Hicks, W.; Murray, S.A.; Zheng, Q.Y.; Smith, R.S.; Nishina, P.M.; et al. Alms1-disrupted mice recapitulate human Alström syndrome. Hum. Mol. Genet. 2005, 14, 2323–2333. [Google Scholar] [CrossRef] [PubMed]
- Hearn, T.; Spalluto, C.; Phillips, V.J.; Renforth, G.L.; Copin, N.; Hanley, N.A.; Wilson, D.I. Subcellular Localization of ALMS1 Supports Involvement of Centrosome and Basal Body Dysfunction in the Pathogenesis of Obesity, Insulin Resistance, and Type 2 Diabetes. Diabetes 2005, 54, 1581–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braune, K.; Volkmer, I.; Staege, M.S. Characterization of Alstrom Syndrome 1 (ALMS1) Transcript Variants in Hodgkin Lymphoma Cells. PLoS ONE 2017, 12, e0170694. [Google Scholar] [CrossRef] [PubMed]
- Astuti, D.; Sabir, A.; Fulton, P.; Zatyka, M.; Williams, D.; Hardy, C.; Milan, G.; Favaretto, F.; Yu-Wai-Man, P.; Rohayem, J.; et al. Monogenic diabetes syndromes: Locus-specific databases for Alström, Wolfram, and Thiamine-responsive megaloblastic anemia. Hum. Mutat. 2017, 38, 764–777. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Cao, M.; Li, Z.; Chen, X.; Patenia, C.; Gore, A.; Abboud, E.B.; Al-Rajhi, A.A.; Lewis, R.A.; et al. Whole-exome sequencing identifies ALMS1, IQCB1, CNGA3, and MYO7A mutations in patients with Leber congenital amaurosis. Hum. Mutat. 2011, 32, 1450–1459. [Google Scholar] [CrossRef] [Green Version]
- Katagiri, S.; Yoshitake, K.; Akahori, M.; Hayashi, T.; Furuno, M.; Nishino, J.; Ikeo, K.; Tsuneoka, H.; Iwata, T. Whole-exome sequencing identifies a novel ALMS1 mutation (p.Q2051X) in two Japanese brothers with Alström syndrome. Mol. Vis. 2013, 19, 2393–2406. [Google Scholar]
- Long, P.A.; Evans, J.M.; Olson, T.M. Exome sequencing establishes diagnosis of Alström syndrome in an infant presenting with non-syndromic dilated cardiomyopathy. Am. J. Med. Genet. A 2015, 167A, 886–890. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.D.; Hinman, E.G.; Collin, G.B.; Beck, S.; Cerqueira, R.; Maffei, P.; Milan, G.; Zhang, W.; Wilson, D.I.; Hearn, T.; et al. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alström syndrome. Hum. Mutat. 2007, 28, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Lazar, C.H.; Kimchi, A.; Namburi, P.; Mutsuddi, M.; Zelinger, L.; Beryozkin, A.; Ben-Simhon, S.; Obolensky, A.; Ben-Neriah, Z.; Argov, Z.; et al. Nonsyndromic Early-Onset Cone-Rod Dystrophy and Limb-Girdle Muscular Dystrophy in a Consanguineous Israeli Family are Caused by Two Independent yet Linked Mutations in ALMS1 and DYSF. Hum. Mutat. 2015, 36, 836–841. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Scheinfeldt, L.B.; Biswas, S.; Madeoy, J.; Connelly, C.F.; Schadt, E.E.; Akey, J.M. Population genomic analysis of ALMS1 in humans reveals a surprisingly complex evolutionary history. Mol. Biol. Evol. 2009, 26, 1357–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Groenendael, S.; Giacovazzi, L.; Davison, F.; Holtkemper, O.; Huang, Z.; Wang, Q.; Parkinson, K.; Barrett, T.; Geberhiwot, T. High quality, patient centred and coordinated care for Alstrom syndrome: A model of care for an ultra-rare disease. Orphanet J. Rare Dis. 2015, 10, 149. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Pedigree chart and electropherogram for the patients carrying novel mutations. (A) Pedigree chart for family GBB-28, carrier of mutation c.2785C>T; p.(Glu929Ter) and electropherogram of the proband sequence compared to the control. (B) Pedigree chart for family UG-26225, carrier of mutation c.5420_5423del; p.(His1808GlufsTer20) and electropherogram of the proband sequence compared to the control.
Figure 1.
Pedigree chart and electropherogram for the patients carrying novel mutations. (A) Pedigree chart for family GBB-28, carrier of mutation c.2785C>T; p.(Glu929Ter) and electropherogram of the proband sequence compared to the control. (B) Pedigree chart for family UG-26225, carrier of mutation c.5420_5423del; p.(His1808GlufsTer20) and electropherogram of the proband sequence compared to the control.

Figure 2.
Different relative allele frequencies for the pathogenic variants detected in the cohort, expressed as a fraction of total alleles (N = 24).
Figure 2.
Different relative allele frequencies for the pathogenic variants detected in the cohort, expressed as a fraction of total alleles (N = 24).

Figure 3.
Single nucleotide polymorphism (SNP) segregation study in 3 families. (A) The mother´s genotype from family GBB-44, carrier of p.(Asn1787Asp) and p.(Tyr1714Ter) in heterozygosis. (B) The father´s genotype from the RP-1087 family, carrier of p.(Asn1787Asp) and p.(Tyr1714Ter) in heterozygosis. (C) The genotype of the GBB-46 proband with p.(Asn1787Asp) and p.(Tyr1714Ter) in homozygosis. *: p.(Asn1787Asp) (c.5359A>G; rs45608038).
Figure 3.
Single nucleotide polymorphism (SNP) segregation study in 3 families. (A) The mother´s genotype from family GBB-44, carrier of p.(Asn1787Asp) and p.(Tyr1714Ter) in heterozygosis. (B) The father´s genotype from the RP-1087 family, carrier of p.(Asn1787Asp) and p.(Tyr1714Ter) in heterozygosis. (C) The genotype of the GBB-46 proband with p.(Asn1787Asp) and p.(Tyr1714Ter) in homozygosis. *: p.(Asn1787Asp) (c.5359A>G; rs45608038).


{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of the genotype of patients, their author reference, and the family and patient codes. The reference sequence for ALMS1 (ENST00000613296.5/ ENSP00000482968.1) was used.
Table 1.
Summary of the genotype of patients, their author reference, and the family and patient codes. The reference sequence for ALMS1 (ENST00000613296.5/ ENSP00000482968.1) was used.
| Allele 1 | Allele 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient | Family | Reference | ALMS1 Pathogenic Variant 1 c.DNA | Exon | ALMS1 Pathogenic Variant 1 Protein | ALMS1 Pathogenic Variant 2 c.DNA | Exon | ALMS1 Pathogenic Variant 2 Protein | Genotype Status |
| 1 | GBB-28 | This study | c.2785G>T | 8 | p.(Glu929Ter) | c.2785G>T | 8 | p.(Glu929Ter) | Homozygous |
| 2 | RP-1232 | [7] | c.4249del | 8 | p.(Arg1417GlyfsTer55) | c.4249del | 8 | p.(Arg1417GlyfsTer55) | Homozygous |
| 3 | RP-1087 | [6] | c.5142T>G | 8 | p.(Tyr1714Ter) | c.1844C>G | 8 | p.(Ser615Ter) | Heterozygous |
| 4 | GAS-37 | Allele 1 [6]; Allele 2 [4] | c.5142T>G | 8 | p.(Tyr1714Ter) | c.4271T>G | 8 | p.(Leu1424Ter) | Heterozygous |
| 5 | GAS-37 | Allele 1 [6]; Allele 2 [4] | c.5142T>G | 8 | p.(Tyr1714Ter) | c.4271T>G | 8 | p.(Leu1424Ter) | Heterozygous |
| 6 | GBB-46 | Allele 1 [6]; Allele 2 [4] | c.5142T>G | 8 | p.(Tyr1714Ter) | c.5142T>G | 8 | p.(Tyr1714Ter) | Homozygous |
| 7 | GBB-44 | Allele 1 [6]; Allele 2 [4] | c.5142T>G | 8 | p.(Tyr1714Ter) | c.11615_11616del | 17 | p.(Ser3872TyrfsTer19) | Heterozygous |
| 8 | UG-26225 | This study | c.5420_5423del | 8 | p.(His1808GlufsTer20) | c.5420_5423del | 8 | p.(His1808GlufsTer20) | Homozygous |
| 9 | RP-2186 | [4] | c.7568_7569del | 9 | p.(His2523ArgfsTer11) | c.4474G>T | 8 | p.(Glu1492Ter) | Heterozygous |
| 10 | RP-793 | [19] | c.10787_10788del | 16 | p.(Val3596GlufsTer4) | c.10787_10788del | 16 | p.(Val3596GlufsTer4) | Homozygous |
| 11 | GBB-45 | [4] | c.11615_11616del | 17 | p.(Ser3872TyrfsTer19) | c.11615_11616del | 17 | p.(Ser3872TyrfsTer19) | Homozygous |
| 12 | RP-2177 | Allele1 [4]; Allele 2 [20] | c.11615_11616del | 17 | p.(Ser3872TyrfsTer19) | c.805C>T | 5 | p.(Arg269Ter) | Heterozygous |
Table 2.
Phenotype summary based on the diagnostic criteria for Alström syndrome according to Marshall et al. (2007) [9] for 11 of the 12 cases. The clinical history of patient GBB-45 was not available. x: presence of symptom. -: absence of symptom.
Table 2.
Phenotype summary based on the diagnostic criteria for Alström syndrome according to Marshall et al. (2007) [9] for 11 of the 12 cases. The clinical history of patient GBB-45 was not available. x: presence of symptom. -: absence of symptom.
| Patient | Family | Sex | Age (Years) | Vision (History of Nystagmus in Infancy/Childhood, Legal Blindness, Cone and Rod Dystrophy by ERG) | Obesity and/or Insulin Resistance and/or T2D | History of DCM/CHF | Hearing Loss | Hepatic Dysfunction | Renal Failure | Short Stature | Males: Hypogonadism; Females: Irregular Menses and/or Hyperandrogenism | Thyroid Disorders | Predicted Protein Change |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | GBB-28 | F | 13 | x | x | x | - | - | - | - | - | - | p.(Glu929Ter)/p.(Glu929Ter) |
| 2 | RP-1232 | F | 27 | x | x | - | x | - | x | - | x | x | p.(Arg1417GlyfsTer55)/p.(Arg1417GlyfsTer55) |
| 3 | RP-1087 | F | 42 | x | x | - | x | x | x | x | x | - | p.(Tyr1714Ter)/p.(Ser615Ter) |
| 4 | GAS-37 | F | 21 | x | x | x | x | x | - | x | x | x | p.(Tyr1714Ter)/p.(Leu1424Ter) |
| 5 | GAS-37 | M | 26 | x | x | - | x | x | - | x | x | x | p.(Tyr1714Ter)/p.(Leu1424Ter) |
| 6 | GBB-46 | M | 23 | x | x | - | x | x | - | x | - | x | p.(Tyr1714Ter)/p.(Tyr1714Ter) |
| 7 | GBB-44 | M | 18 | x | x | x | x | x | - | - | - | - | p.(Tyr1714Ter)/p.(Ser3872TyrfsTer19) |
| 8 | UG-26225 | M | 3 | x | x | x | - | - | - | - | - | x | p.(His1808GlufsTer20)/p.(His1808GlufsTer20) |
| 9 | RP-2186 | M | 9 | x | x | x | - | - | - | - | - | - | p.(His2523ArgfsTer11)/p.(Glu1492Ter) |
| 10 | RP-793 | F | 11 | x | x | - | x | x | - | x | - | x | p.(Val3596GlufsTer4)/p.(Val3596GlufsTer4) |
| 12 | RP-2177 | F | 49 | x | - | x | x | - | - | - | - | - | p.(Ser3872TyrfsTer19)/p.(Arg269Ter) |
ERG: Electroretinogram; T2D: Type 2 Diabetes Mellitus; DCM: Dilated Cardiomyopathy; CHF: Congestive Heart Failure.
Table 3.
Scores obtaining from the in silico analysis of pathogenicity for the 2 novel mutations p.(Glu929Ter) and p.(His1808GlufsTer20) and their classification according to American College of Medical Genetics and Genomics (ACMG) guidelines. The 4 programs used for this analysis were: PolyPhen2, MultiPred-LOF, SIFT and PROVEAN.
Table 3.
Scores obtaining from the in silico analysis of pathogenicity for the 2 novel mutations p.(Glu929Ter) and p.(His1808GlufsTer20) and their classification according to American College of Medical Genetics and Genomics (ACMG) guidelines. The 4 programs used for this analysis were: PolyPhen2, MultiPred-LOF, SIFT and PROVEAN.
| Patient | Family | ALMS1 Pathogenic Variant c.DNA | Exon | ALMS1 Pathogenic Variant Protein | PolyPhen2 | MutPred-LOF | SIFT | PROVEAN | ACMG |
|---|---|---|---|---|---|---|---|---|---|
| 1 | GBB-28 | c.2785G>T | 8 | p.(Glu929Ter) | - | 0.432 | 0 | −3.376 | Pathogenic |
| 8 | UG-26225 | c.5420_5423del | 8 | p.(His1808GlufsTer20) | 0.852 | 0.422 | 0,05 | −2.720 | Pathogenic |
Table 4.
Haplogroup classification of Alström patients with the pathogenic variant p.(Tyr1714Ter). Analyzed SNP, predicted protein change, exon in which they are found, genotype of the study individuals and shared common allele.
Table 4.
Haplogroup classification of Alström patients with the pathogenic variant p.(Tyr1714Ter). Analyzed SNP, predicted protein change, exon in which they are found, genotype of the study individuals and shared common allele.
| Predicted Protein Change | SNPs | Exon | GBB-44 | RP-1087 | GBB-46 | Common Allele |
|---|---|---|---|---|---|---|
| p.(Phe730=) | rs7598901 | 8 | T/T | T/T | T/T | T |
| p.(Gly1415Val) | rs6546837 | 8 | G/G | C/G | G/G | G |
| p.(Ile1876Val) | rs6546838 | 8 | A/A | G/A | A/A | A |
| p.(Ser2112Arg) | rs6724782 | 8 | T/T | A/T | T/T | T |
| p.(Arg2285Leu) | rs6546839 | 8 | G/G | C/G | G/G | G |
| p.(Arg2827Ser) | rs2056486 | 10 | G/G | G/G | G/G | G |
| p.(Asn2857Ser) | rs10193972 | 10 | A/A | G/A | A/A | A |
| p.(Asn1787Asp) - | rs45608038 | 8 | A/G | A/G | G/G | G |
| p.(Tyr1714Ter) * | rs772136379 | 8 | T/G | T/G | G/G | G |
*: causal mutation. -: rare variant linked to causal mutation.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bea-Mascato, B.; Solarat, C.; Perea-Romero, I.; Jaijo, T.; Blanco-Kelly, F.; Millán, J.M.; Ayuso, C.; Valverde, D. Prevalent ALMS1 Pathogenic Variants in Spanish Alström Patients. Genes 2021, 12, 282. https://doi.org/10.3390/genes12020282
AMA Style
Bea-Mascato B, Solarat C, Perea-Romero I, Jaijo T, Blanco-Kelly F, Millán JM, Ayuso C, Valverde D. Prevalent ALMS1 Pathogenic Variants in Spanish Alström Patients. Genes. 2021; 12(2):282. https://doi.org/10.3390/genes12020282
Chicago/Turabian StyleBea-Mascato, Brais, Carlos Solarat, Irene Perea-Romero, Teresa Jaijo, Fiona Blanco-Kelly, José M. Millán, Carmen Ayuso, and Diana Valverde. 2021. "Prevalent ALMS1 Pathogenic Variants in Spanish Alström Patients" Genes 12, no. 2: 282. https://doi.org/10.3390/genes12020282
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.