
Unlocking the Complete Chloroplast Genome of a Native Tree Species from the Amazon Basin, Capirona (Calycophyllum Spruceanum, Rubiaceae), and Its Comparative Analysis with Other Ixoroideae Species

 , , ,
, , ,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Genomic DNA Extraction
2.2. DNA Sequence and Genome Assembly
2.3. Annotation and Analysis of C. spruceanum Chloroplast DNA Sequence
2.4. Comparative Analysis of Ixoroideae Chloroplast Genomes
2.5. Phylogenetic Analyses
3. Results
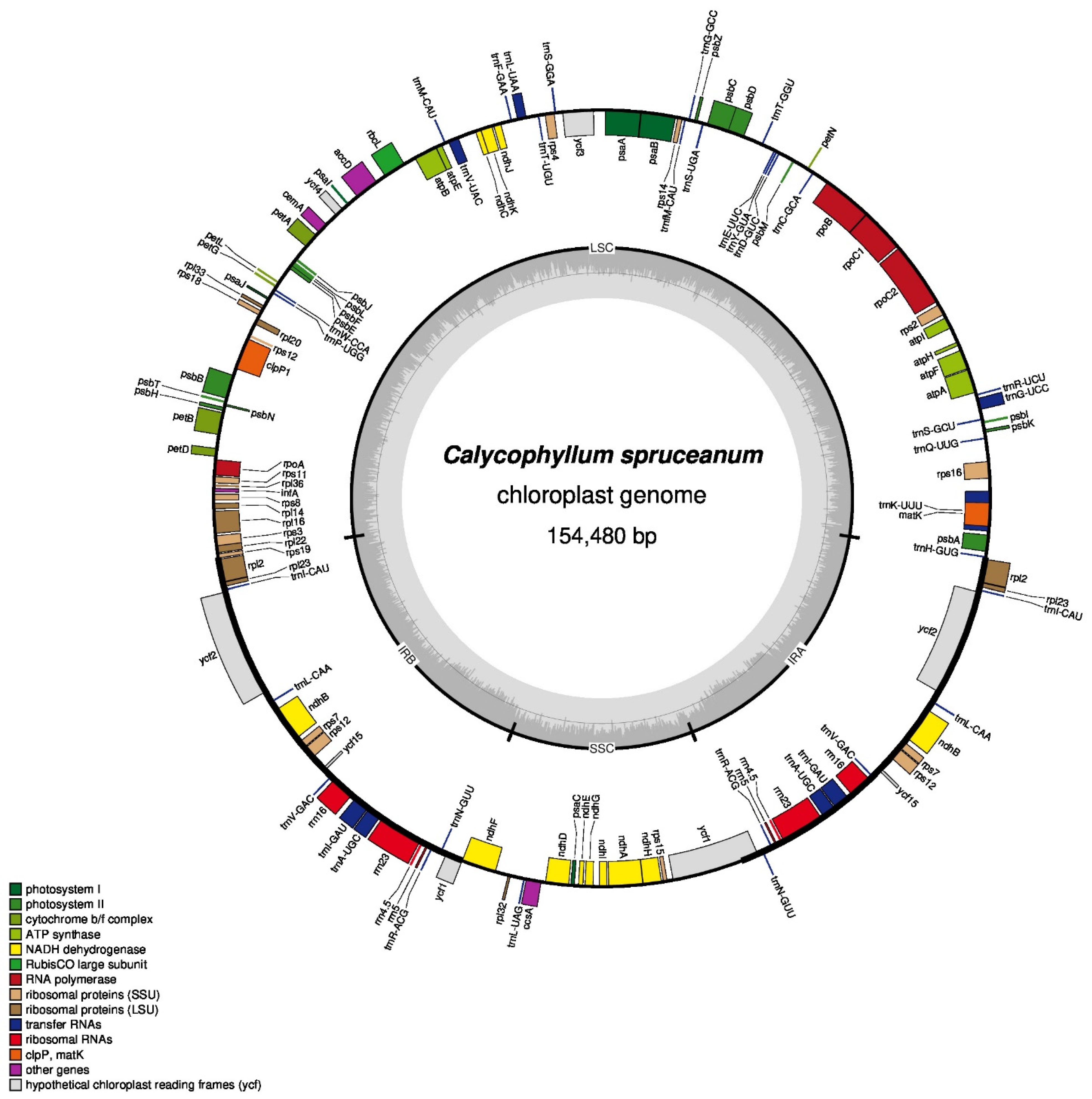
3.1. C. spruceanum Chloroplast Genome Assembly and Its Features
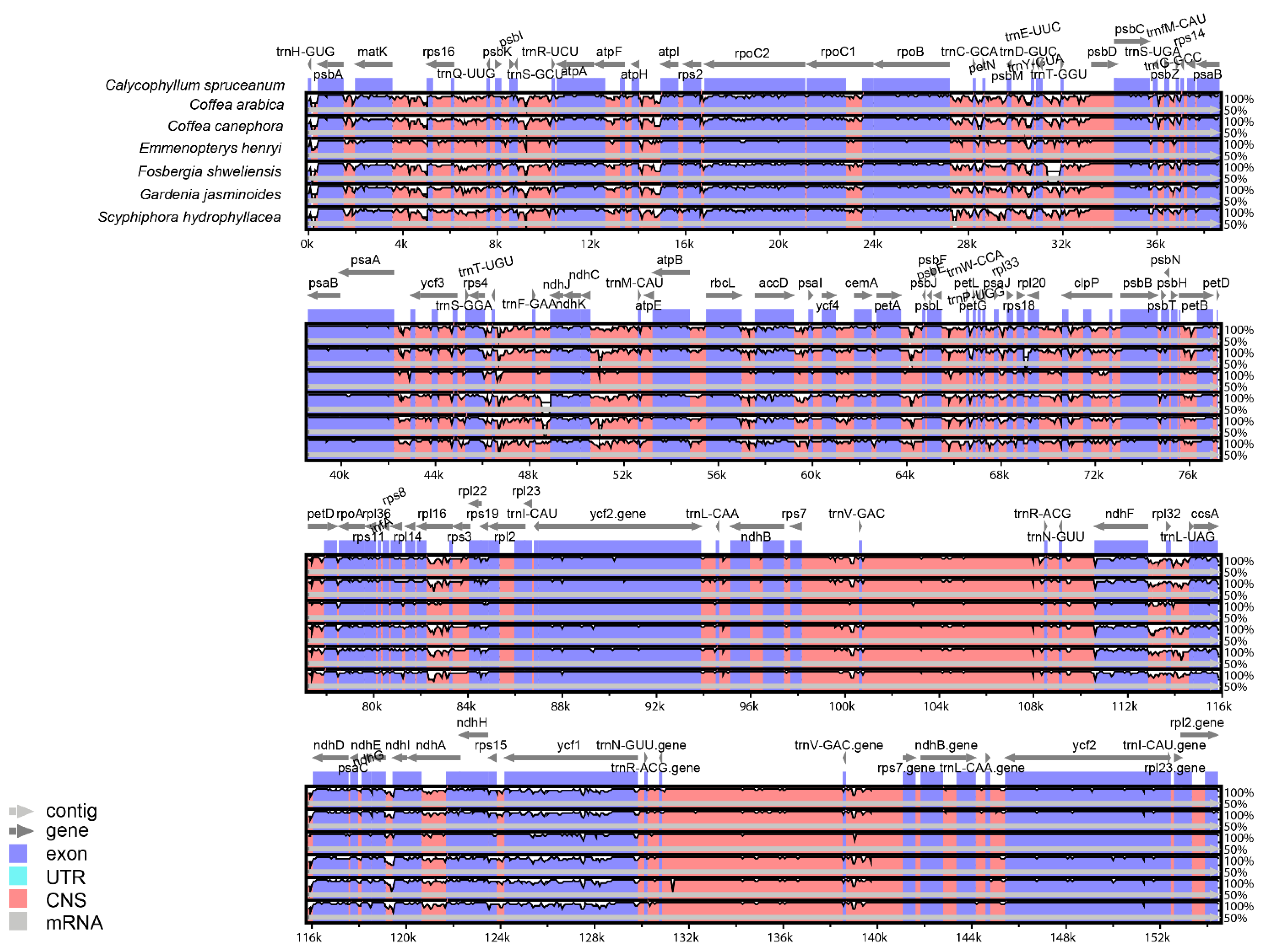
3.2. Comparative Analysis of Genome Structure
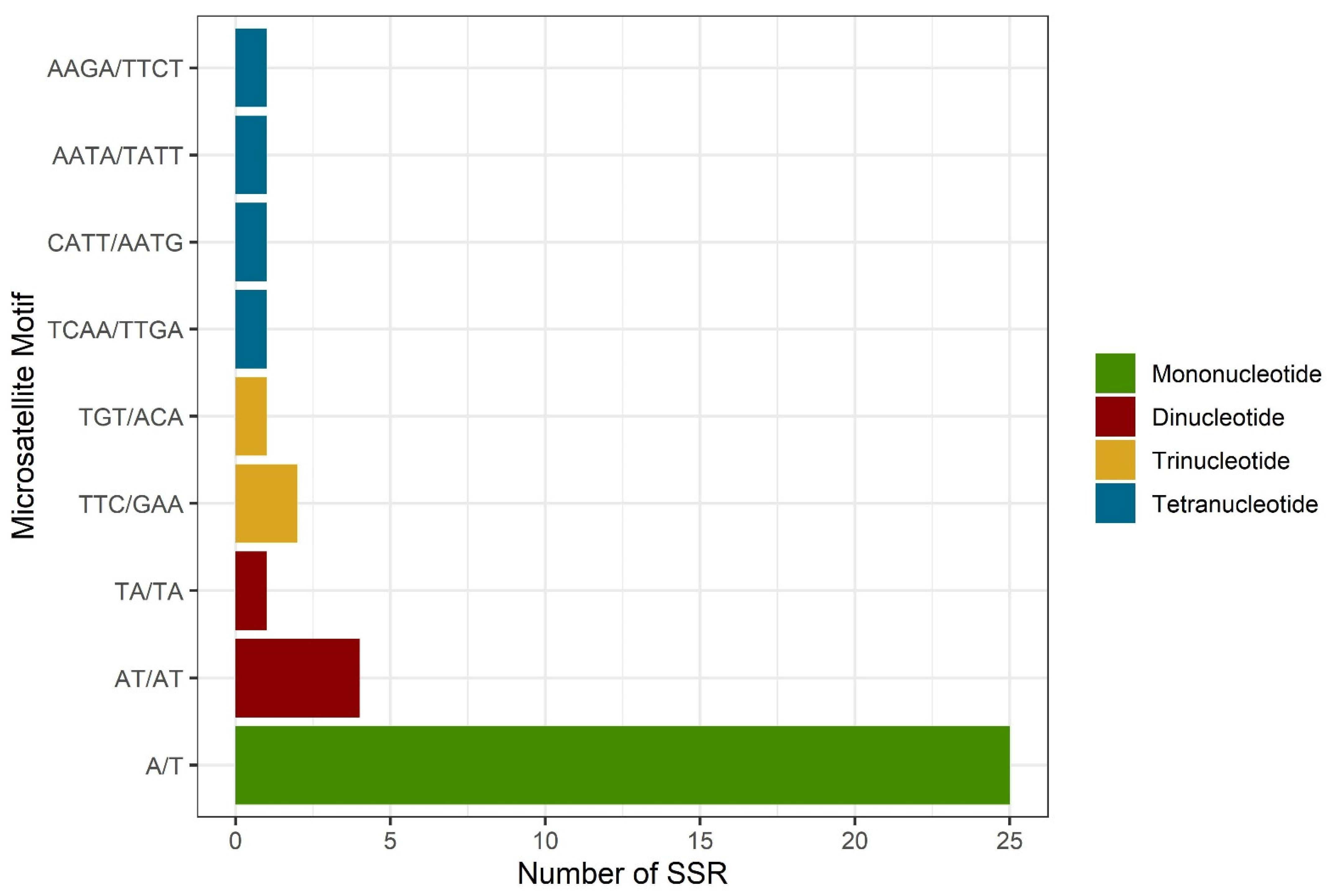
3.3. SSR Loci Identified in Ixoroideae cp Genomes
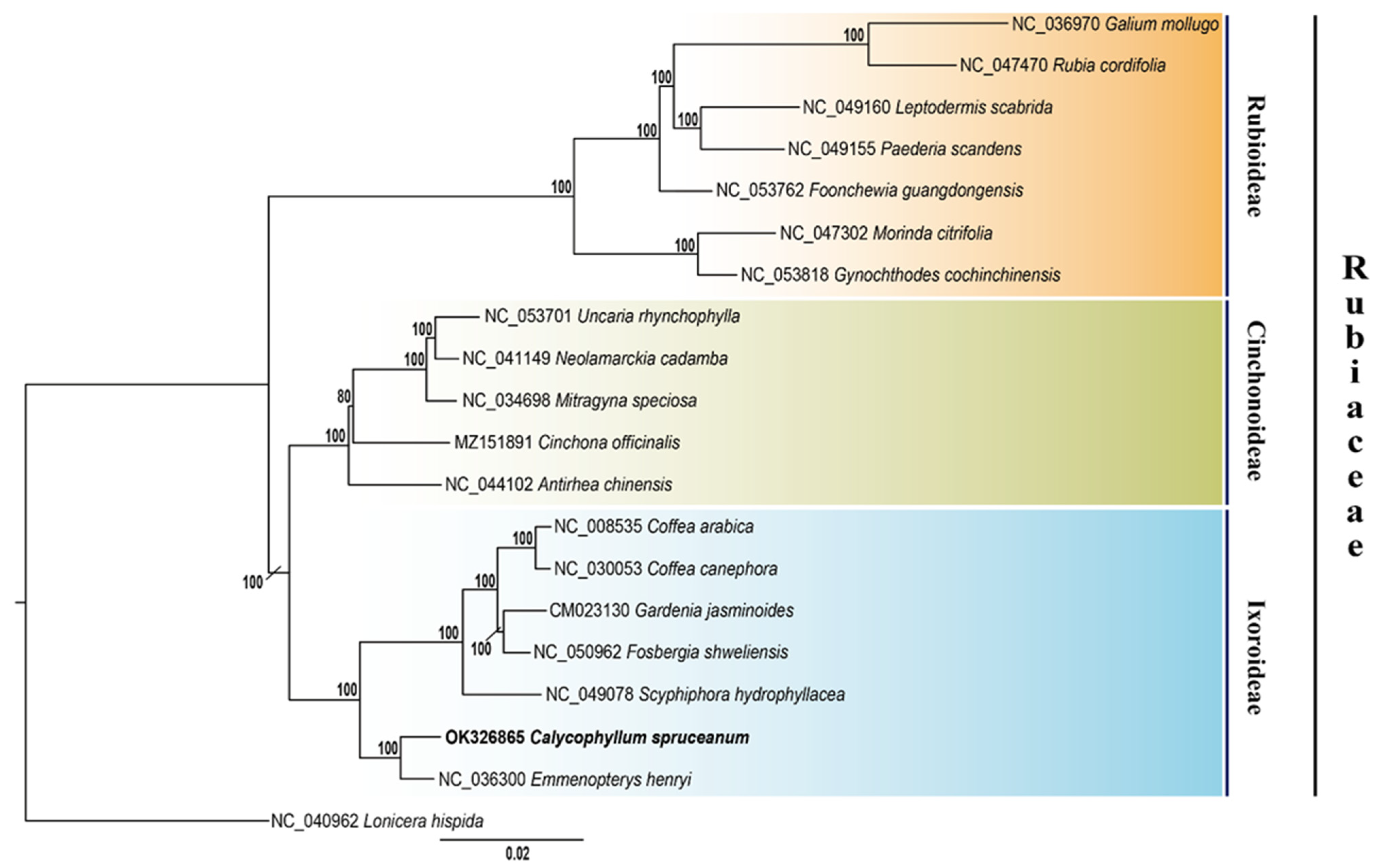
3.4. Phylogenetic Inference of C. spruceanum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kainulainen, K.; Razafimandimbison, S.G.; Bremer, B. Phylogenetic relationships and new tribal delimitations in subfamily Ixoroideae (Rubiaceae). Bot. J. Linn. Soc. 2013, 173, 387–406. [Google Scholar] [CrossRef] [Green Version]
- Ly, S.N.; Garavito, A.; De Block, P.; Asselman, P.; Guyeux, C.; Charr, J.-C.; Janssens, S.; Mouly, A.; Hamon, P.; Guyot, R. Chloroplast genomes of Rubiaceae: Comparative genomics and molecular phylogeny in subfamily Ixoroideae. PLoS ONE 2020, 15, e0232295. [Google Scholar] [CrossRef]
- Russell, J.R.; Weber, J.C.; Booth, A.; Powell, W.; Sotelo-Montes, C.; Dawson, I.K. Genetic variation of Calycophyllum spruceanum in the Peruvian Amazon Basin, revealed by amplified fragment length polymorphism (AFLP) analysis. Mol. Ecol. 1999, 8, 199–204. [Google Scholar] [CrossRef]
- Sears, R.R. New Forestry on the Floodplain: The Ecology and Management of Calycophyllum spruceanum (Rubiaceae) on the Amazon Landscape. Ph.D. Thesis, Columbia University, New York, NY, USA, 2003. [Google Scholar]
- Tauchen, J.; Lojka, B.; Hlasna-Cepkova, P.; Svobodova, E.; Dvorakova, Z.; Rollo, A. Morphological and genetic diversity of Calycophyllum spruceanum (Benth) K. Schum (Rubiaceae) in Peruvian Amazon. Agric. Trop. Subtrop. 2011, 44, 4. [Google Scholar]
- Weber, J.C.; Montes, C.S.; Vidaurre, H.; Dawson, I.K.; Simons, A.J. Participatory domestication of agroforestry trees: An example from the Peruvian Amazon. Dev. Pract. 2001, 11, 425–433. [Google Scholar] [CrossRef]
- Guariguata, M.R.; Ostertag, R. Neotropical secondary forest succession: Changes in structural and functional characteristics. For. Ecol. Manag. 2001, 148, 185–206. [Google Scholar] [CrossRef]
- Saldaña, C.L.; Cancan, J.D.; Cruz, W.; Correa, M.Y.; Ramos, M.; Cuellar, E.; Arbizu, C.I. Genetic Diversity and Population Structure of Capirona (Calycophyllum spruceanum Benth.) from the Peruvian Amazon Revealed by RAPD Markers. Forests 2021, 12, 1125. [Google Scholar] [CrossRef]
- Dávila-Lara, A.; Affenzeller, M.; Tribsch, A.; Díaz, V.; Comes, H.P. AFLP diversity and spatial structure of Calycophyllum candidissimum (Rubiaceae), a dominant tree species of Nicaragua’s critically endangered seasonally dry forest. Heredity 2017, 119, 275–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Lin, F.; Huang, P.; Guo, W.; Zheng, Y. Development of nuclear SSR and chloroplast genome markers in diverse Liriodendron chinense germplasm based on low-coverage whole genome sequencing. Biol. Res. 2020, 53, 21. [Google Scholar] [CrossRef]
- Gerwein, J.B.; Kesseli, R.V. Genetic diversity and population structure of Quercus rubra (Fagaceae) in old-growth and secondary forests in southern New England. Rhodora 2006, 108, 1–18. [Google Scholar] [CrossRef]
- Du, Q.; Wang, B.; Wei, Z.; Zhang, D.; Li, B. Genetic Diversity and Population Structure of Chinese White Poplar (Populus tomentosa) Revealed by SSR Markers. J. Hered. 2012, 103, 853–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paredes-Villanueva, K.; De Groot, G.A.; Laros, I.; Bovenschen, J.; Bongers, F.; Zuidema, P.A. Genetic differences among Cedrela odorata sites in Bolivia provide limited potential for fine-scale timber tracing. Tree Genet. Genomes 2019, 15, 33. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Singh, S.P.; Tiwari, A.K.; Sharma, B.L. Genetic diversity of sugarcane hybrid cultivars by RAPD markers. 3 Biotech 2017, 7, 222. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.W. The evolutionary origins of organelles. Trends Genet. 1989, 5, 294–299. [Google Scholar] [CrossRef]
- Howe, C.J.; Barbrook, A.C.; Koumandou, V.L.; Nisbet, R.E.R.; Symington, H.A.; Wightman, T.F.; Fray, R.; Leaver, C.J.; Walker, J.E.; Gray, J.C.; et al. Evolution of the chloroplast genome. Philos. Trans. R. Soc. B Biol. Sci. 2003, 358, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, P.C.; Zhang, Y.Z.; Geng, H.M.; Chen, S.L. The complete chloroplast genome sequence of Gentiana lawrencei var. farreri (Gentianaceae) and comparative analysis with its congeneric species. PeerJ 2016, 4, e2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; Depamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J.; et al. Methods for Obtaining and Analyzing Whole Chloroplast Genome Sequences. Methods Enzymol. 2005, 395, 348–384. [Google Scholar] [CrossRef] [PubMed]
- Raman, G.; Park, S.J. The Complete Chloroplast Genome Sequence of the Speirantha gardenii: Comparative and Adaptive Evolutionary Analysis. Agronomy 2020, 10, 1405. [Google Scholar] [CrossRef]
- Liu, Y. A code within the genetic code: Codon usage regulates co-translational protein folding. Cell Commun. Signal. 2020, 18, 145. [Google Scholar] [CrossRef]
- Behura, S.K.; Severson, D.W. Codon usage bias: Causative factors, quantification methods and genome-wide patterns: With emphasis on insect genomes. Biol. Rev. 2013, 88, 49–61. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Cruz, W.; Ramos, H.; Cuellar, J. Manual de Protocolos para el Estudio de Diversidad Genética en Especies Forestales Nativas: Tornillo (Cedrelinga cateniformis (Ducke) Ducke), Capirona (Calycophyllum spruceanum Benth.), Shihuahuaco (Dipteryx sp.), Ishpingo (Amburana sp.) y Castaña (Bertholletia excelsa); Instituto Nacional de Innovación Agraria: Lima, Perú, 2019; p. 52. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2013, 17, 10–12. [Google Scholar]
- Arbizu, C.I.; Ferro-Mauricio, R.D.; Chávez-Galarza, J.C.; Guerrero-Abad, J.C.; Vásquez, H.V.; Maicelo, J.L. The complete chloroplast genome of the national tree of Peru, quina (Cinchona officinalis L., Rubiaceae). Mitochondrial DNA Part B Resour. 2021, 6, 2781–2783. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; Depamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, 273–279. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddison, D.R.; Maddison, W.P. MacClade 4.08a: Analysis of Phylogeny and Character Evolution; Sinauer: Sunderland, MA, USA, 2005. [Google Scholar]
- Kassambara, A. ggpubr: “ggplot2” Based Publication Ready Plots. 2020. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 30 November 2021).
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, J.-W.; Yang, Y.; Li, X.-N. Structural and Comparative Analysis of the Complete Chloroplast Genome of a Mangrove Plant: Scyphiphora hydrophyllacea Gaertn. f. and Related Rubiaceae Species. Forests 2019, 10, 1000. [Google Scholar] [CrossRef] [Green Version]
- Guy, L.; Kultima, J.R.; Andersson, S.G.E.; Quackenbush, J. genoPlotR: Comparative gene and genome visualization in R. Bioinformatics 2011, 27, 2334–2335. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Wang, W.; Shao, F.; Deng, X.; Liu, Y.; Chen, S.; Li, Y.; Guo, W.; Jiang, Q.; Liang, H.; Zhang, X. Genome surveying reveals the complete chloroplast genome and nuclear genomic features of the crocin-producing plant Gardenia jasminoides Ellis. Genet. Resour. Crop. Evol. 2021, 68, 1165–1180. [Google Scholar] [CrossRef]
- Geng, Y.; Li, Y.; Yuan, X.; Luo, T.; Wang, Y. The complete chloroplast genome sequence of Fosbergia shweliensis, an endemic species to Yunnan of China. Mitochondrial DNA Part B Resour. 2020, 5, 1796–1797. [Google Scholar] [CrossRef] [Green Version]
- Ren, W.; Guo, D.; Xing, G.; Yang, C.; Zhang, Y.; Yang, J.; Niu, L.; Zhong, X.; Zhao, Q.; Cui, Y.; et al. Complete Chloroplast Genome Sequence and Comparative and Phylogenetic Analyses of the Cultivated Cyperus esculentus. Diversity 2021, 13, 405. [Google Scholar] [CrossRef]
- Kuroda, H.M.P. The plastid clpP1 protease gene is essential for plant development. Nat. Publ. 2003, 425, 30–33. [Google Scholar] [CrossRef]
- Clarke, A.K.; Schelin, J.; Porankiewicz, J. Inactivation of the clpP1 gene for the proteolytic subunit of the ATP-dependent Clp protease in the cyanobacterium Synechococcus limits growth and light acclimation. Plant Mol. Biol. 1998, 37, 791–801. [Google Scholar] [CrossRef]
- Cahoon, A.B.; Cunningham, K.A.; Stern, D.B. The Plastid clpP Gene May Not be Essential for Plant Cell Viability. Plant Cell Physiol. 2003, 44, 93–95. [Google Scholar] [CrossRef] [Green Version]
- Boudreau, E.; Takahashi, Y.; Lemieux, C.; Turmel, M.; Rochaix, J. The chloroplast ycf3 and ycf4 open reading frames of Chlamydomonas reinhardtii are required for the accumulation of the photosystem I complex. EMBO J. 1997, 16, 6095–6104. [Google Scholar] [CrossRef] [Green Version]
- Naver, H.; Boudreau, E.; Rochaix, J.-D. Functional Studies of Ycf3: Its Role in Assembly of Photosystem I and Interactions with Some of Its Subunits. Plant Cell 2001, 13, 2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G.; Muse, S.V. A Primer of Genome Science; Sinauer Associates: Sunderland, MA, USA, 2009. [Google Scholar]
- Li, W.; Graur, D. Fundamentals of Molecular Evolution; Sinauer Associates: Sunderland, MA, USA, 1991; 284p. [Google Scholar]
- Yang, J.-B.; Yang, S.-X.; Li, H.T.; Yang, J.; Li, D.-Z. Comparative Chloroplast Genomes of Camellia Species. PLoS ONE 2013, 8, e73053. [Google Scholar] [CrossRef] [Green Version]
- Raman, G.; Park, V.; Kwak, M.; Lee, B.; Park, S.J. Characterization of the complete chloroplast genome of Arabis stellari and comparisons with related species. PLoS ONE 2017, 12, e0183197. [Google Scholar] [CrossRef]
- Chen, X.; Cho, Y.G.; McCouch, S.R. Sequence divergence of rice microsatellites in Oryza and other plant species. Mol. Genet. Genom. 2002, 268, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Spooner, D.M.; Ruess, H.; Iorizzo, M.; Senalik, D.; Simon, P. Entire plastid phylogeny of the carrot genus (Daucus, Apiaceae): Concordance with nuclear data and mitochondrial and nuclear DNA insertions to the plastid. Am. J. Bot. 2017, 104, 296–312. [Google Scholar] [CrossRef] [Green Version]
- Olsson, S.; Grivet, D.; Cid-Vian, J. Species-diagnostic markers in the genus Pinus: Evaluation of the chloroplast regions matK and ycf1. For. Syst. 2018, 27, e016. [Google Scholar] [CrossRef] [Green Version]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Dong, W.; Liu, H.; Xu, C.; Zuo, Y.; Chen, Z.; Zhou, S. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: A case study on ginsengs. BMC Genet. 2014, 15, 138. [Google Scholar] [CrossRef] [Green Version]
- Nybom, H.; Weising, K.; Rotter, B. DNA fingerprinting in botany: Past, present, future. Investig. Genet. 2014, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Khayi, S.; Gaboun, F.; Pirro, S.; Tatusova, T.; El Mousadik, A.; Ghazal, H.; Mentag, R. Complete Chloroplast Genome of Argania spinosa: Structural Organization and Phylogenetic Relationships in Sapotaceae. Plants 2020, 9, 1354. [Google Scholar] [CrossRef]
- Varshney, R.K.; Sigmund, R.; Börner, A.; Korzun, V.; Stein, N.; Sorrells, M.E.; Langridge, P.; Graner, A. Interspecific transferability and comparative mapping of barley EST-SSR markers in wheat, rye and rice. Plant Sci. 2005, 168, 195–202. [Google Scholar] [CrossRef]
- Liu, H.-Y.; Yu, Y.; Deng, Y.-Q.; Li, J.; Huang, Z.-X.; Zhou, S.-D. The Chloroplast Genome of Lilium henrici: Genome Structure and Comparative Analysis. Molecules 2018, 23, 1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biju, V.C.; Shidhi, P.R.; Vijayan, S.; Rajan, V.S.; Sasi, A.; Janardhanan, A.; Nair, A.S. The Complete Chloroplast Genome of Trichopus zeylanicus, And Phylogenetic Analysis with Dioscoreales. Plant Genome 2019, 12, 190032. [Google Scholar] [CrossRef] [Green Version]
- Kuang, D.-Y.; Wu, H.; Wang, Y.-L.; Gao, L.-M.; Zhang, S.-Z.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xing, H.; Yuan, Y.; Wang, X.; Saeed, M.; Tao, J.; Feng, W.; Zhang, G.; Song, X.; Sun, X. Genome-wide analysis of codon usage bias in four sequenced cotton species. PLoS ONE 2018, 13, e0194372. [Google Scholar] [CrossRef] [Green Version]
- Duan, R.; Huang, M.; Yang, L.; Liu, Z. Characterization of the complete chloroplast genome of Emmenopterys henryi (Gentianales: Rubiaceae), an endangered relict tree species endemic to China. Conserv. Genet. Resour. 2017, 9, 459–461. [Google Scholar] [CrossRef]
- Hershberg, R.; Petrov, D.A. Selection on Codon Bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [Green Version]
- Dong, F.; Lin, Z.; Lin, J.; Ming, R.; Zhang, W. Chloroplast Genome of Rambutan and Comparative Analyses in Sapindaceae. Plants 2021, 10, 283. [Google Scholar] [CrossRef]
- Yu, X.; Zuo, L.; Lu, D.; Lu, B.; Yang, M.; Wang, J. Comparative analysis of chloroplast genomes of five Robinia species: Genome comparative and evolution analysis. Gene 2019, 689, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Spalik, K.; Downie, S.R. Intercontinental disjunctions in Cryptotaenia (Apiaceae, Oenantheae): An appraisal using molecular data. J. Biogeogr. 2007, 34, 2039–2054. [Google Scholar] [CrossRef]
- Du, Y.-P.; Bi, Y.; Yang, F.-P.; Zhang, M.-F.; Chen, X.-Q.; Xue, J.; Zhang, X.-H. Complete chloroplast genome sequences of Lilium: Insights into evolutionary dynamics and phylogenetic analyses. Sci. Rep. 2017, 7, 5751. [Google Scholar] [CrossRef] [Green Version]
- Bremer, B.; Eriksson, T. Time tree of Rubiaceae: Phylogeny and dating the family, subfamilies, and tribes. Int. J. Plant Sci. 2009, 170, 766–793. [Google Scholar] [CrossRef] [Green Version]
- Bedoya, A.M.; Ruhfel, B.R.; Philbrick, C.T.; Madriñán, S.; Bove, C.P.; Mesterházy, A.; Olmstead, R.G. Plastid Genomes of five Species of Riverweeds (Podostemaceae): Structural organization and comparative analysis in Malpighiales. Front. Plant Sci. 2019, 10, 1035. [Google Scholar] [CrossRef] [Green Version]
- Bremer, B.; Jansen, R.K.; Oxelman, B.; Backlund, M.; Lantz, H.; Kim, K. More characters or more taxa for a robust phylogeny-case study from the coffee family. Syst. Biol. 1999, 48, 413–435. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Features | Calycophyllum spruceanum | Coffea arabica | Coffea canephora | Emmenopterys henryi | Fosbergia shweliensis | Gardenia jasminoides | Scyphiphora hydrophyllacea |
|---|---|---|---|---|---|---|---|
| Genome size (bp) | 154,480 | 155,189 | 154,751 | 155,379 | 154,717 | 154,921 | 155,132 |
| SSC length (bp) | 18,101 | 18,137 | 18,133 | 18,245 | 18,230 | 18,095 | 18,165 |
| LSC length (bp) | 84,813 | 85,166 | 84,850 | 85,554 | 84,747 | 85,236 | 85,239 |
| IRA length (bp) | 25,783 | 25,908 | 23,834 | 25,790 | 25,870 | 25,795 | 25,864 |
| IRB length (bp) | 25,783 | 25,943 | 23,884 | 25,790 | 25,870 | 25,795 | 25,864 |
| No. of protein-coding genes | 87 | 85 | 86 | 87 | 85 | 87 | 88 |
| No. of different rRNA genes | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| No. of tRNA genes | 37 | 38 | 37 | 37 | 36 | 37 | 37 |
| %GC content in LSC | 35.48 | 31.28 | 31.75 | 31.90 | 35.5 | 35.3 | 31.65 |
| %GC content in SSC | 31.89 | 35.35 | 35.48 | 35.48 | 31.4 | 31.5 | 35.49 |
| %GC content in IR | 43.14 | 43.01 | 43.55 | 43.26 | 43.2 | 43.2 | 43.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saldaña, C.L.; Rodriguez-Grados, P.; Chávez-Galarza, J.C.; Feijoo, S.; Guerrero-Abad, J.C.; Vásquez, H.V.; Maicelo, J.L.; Jhoncon, J.H.; Arbizu, C.I. Unlocking the Complete Chloroplast Genome of a Native Tree Species from the Amazon Basin, Capirona (Calycophyllum Spruceanum, Rubiaceae), and Its Comparative Analysis with Other Ixoroideae Species. Genes 2022, 13, 113. https://doi.org/10.3390/genes13010113
Saldaña CL, Rodriguez-Grados P, Chávez-Galarza JC, Feijoo S, Guerrero-Abad JC, Vásquez HV, Maicelo JL, Jhoncon JH, Arbizu CI. Unlocking the Complete Chloroplast Genome of a Native Tree Species from the Amazon Basin, Capirona (Calycophyllum Spruceanum, Rubiaceae), and Its Comparative Analysis with Other Ixoroideae Species. Genes. 2022; 13(1):113. https://doi.org/10.3390/genes13010113
Chicago/Turabian StyleSaldaña, Carla L., Pedro Rodriguez-Grados, Julio C. Chávez-Galarza, Shefferson Feijoo, Juan Carlos Guerrero-Abad, Héctor V. Vásquez, Jorge L. Maicelo, Jorge H. Jhoncon, and Carlos I. Arbizu. 2022. "Unlocking the Complete Chloroplast Genome of a Native Tree Species from the Amazon Basin, Capirona (Calycophyllum Spruceanum, Rubiaceae), and Its Comparative Analysis with Other Ixoroideae Species" Genes 13, no. 1: 113. https://doi.org/10.3390/genes13010113