Heterogeneity in Colorectal Cancer: A Challenge for Personalized Medicine?


 ,
,
Abstract
:1. Introduction
2. Inter-Patient Heterogeneity
3. Spatial Heterogeneity in Colorectal Cancer
3.1. Molecular Differences within the Primary Tumor
3.2. Molecular Differences between the Primary Tumor and Metastases
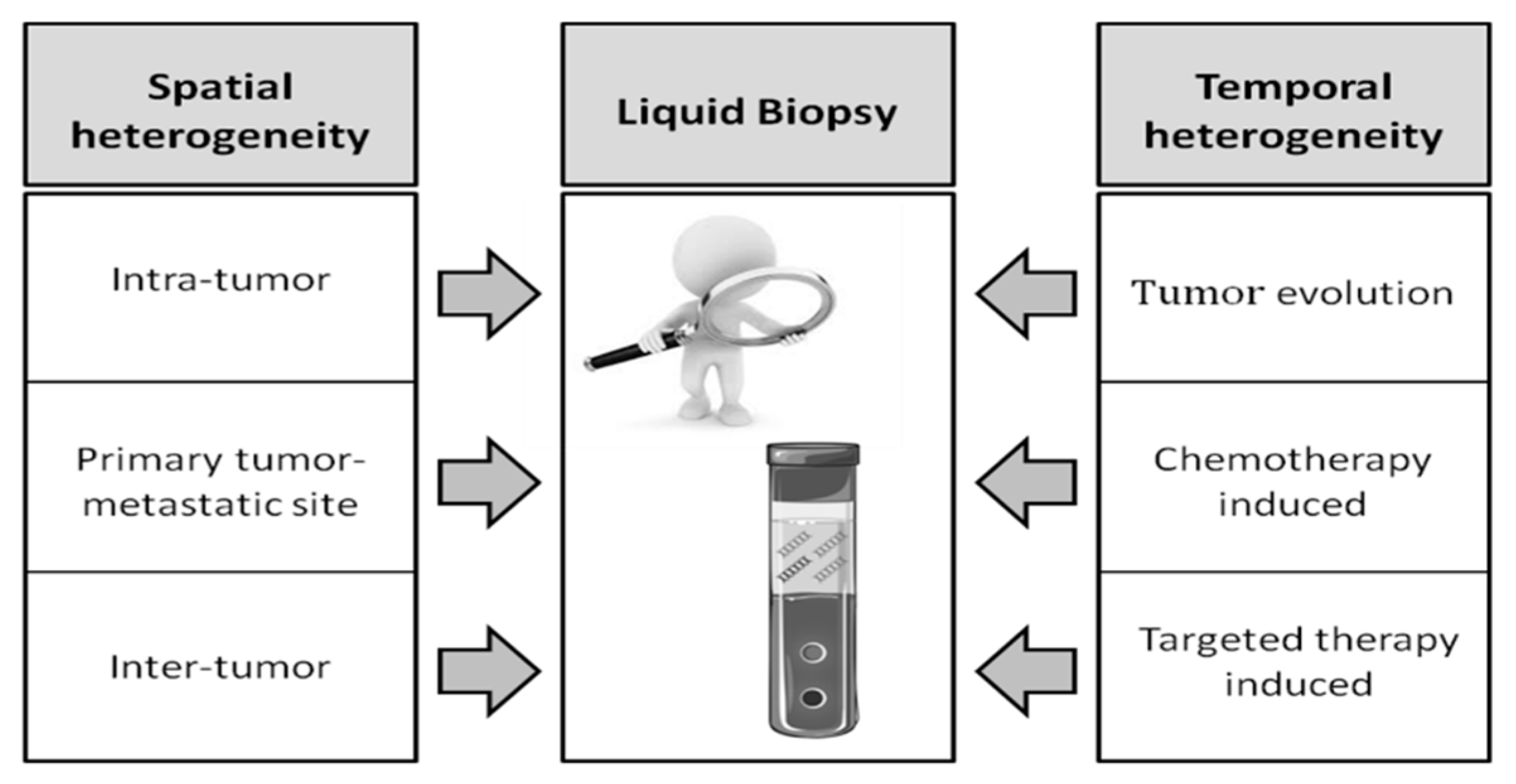
4. Liquid Biopsy to Overcome Tumor Tissue Heterogeneity
4.1. Comparison between cfDNA and Tumor Tissue
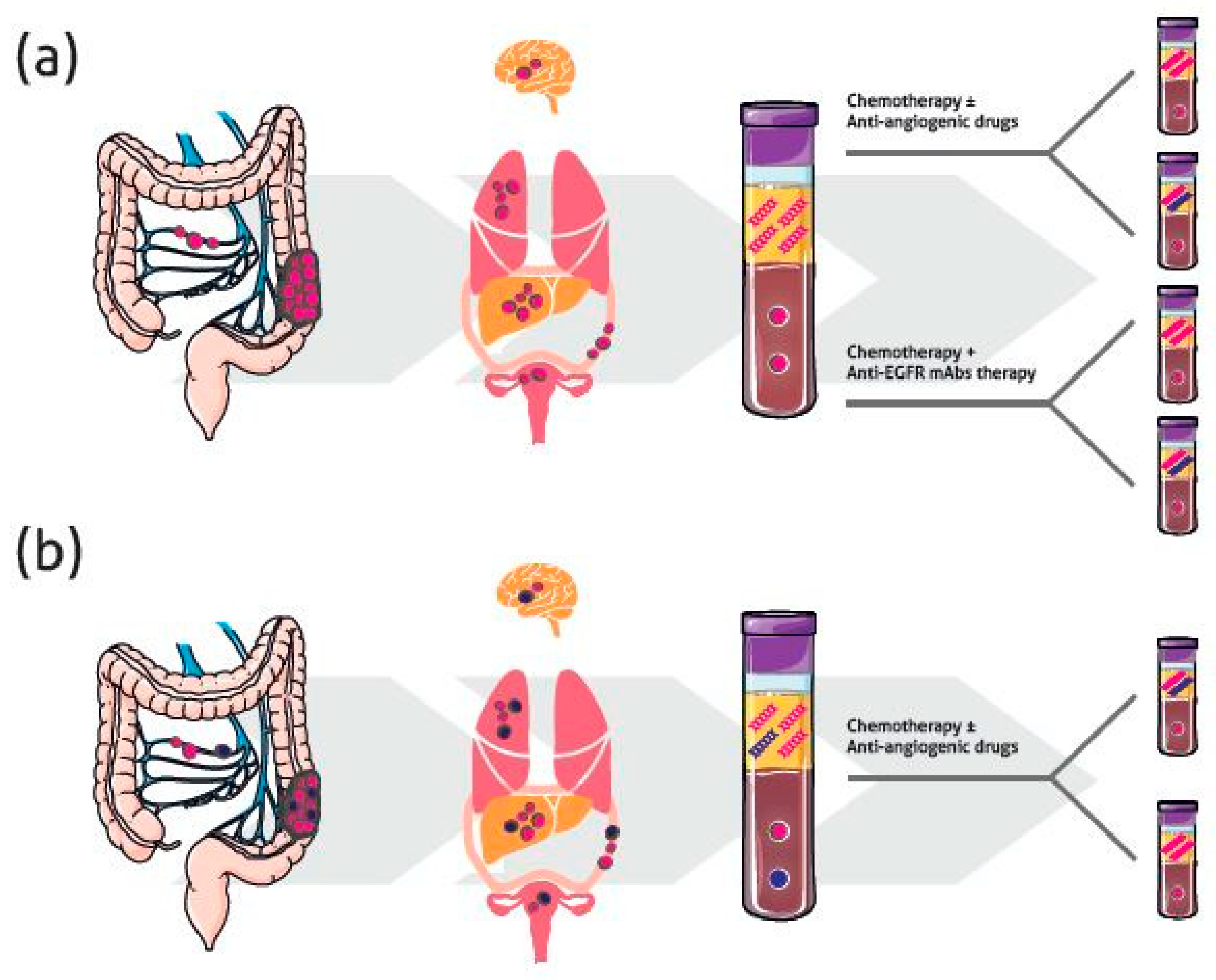
4.2. Temporal Heterogeneity and Monitoring Response to Therapy
4.2.1. Anti-EGFR Therapy
4.2.2. Chemotherapy and Other Targeted Therapies
5. Impact of Heterogeneity in Clinical Molecular Diagnostics
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moraga-Serrano, P.E. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2018, 2018, 126049. [Google Scholar]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer Statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO Consensus Guidelines for the Management of Patients with Metastatic Colorectal Cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef] [PubMed]
- Malvezzi, M.; Carioli, G.; Bertuccio, P.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E. European Cancer Mortality Predictions for the Year 2018 with Focus on Colorectal Cancer. Ann. Oncol. 2018, 29, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.; Pascal, G.; Castaing, D.; Azoulay, D.; Delvart, V.; Paule, B.; Levi, F.; Bismuth, H. Tumor Progression while on Chemotherapy: A Contraindication to Liver Resection for Multiple Colorectal Metastases? Ann. Surg. 2004, 240, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Grady, W.M.; Carethers, J.M. Genomic and Epigenetic Instability in Colorectal Cancer Pathogenesis. Gastroenterology 2008, 135, 1079–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bramsen, J.B.; Rasmussen, M.H.; Ongen, H.; Mattesen, T.B.; Orntoft, M.W.; Arnadottir, S.S.; Sandoval, J.; Laguna, T.; Vang, S.; Oster, B.; et al. Molecular-Subtype-Specific Biomarkers Improve Prediction of Prognosis in Colorectal Cancer. Cell. Rep. 2017, 19, 1268–1280. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nowak, J.A.; Hamada, T.; Phipps, A.I.; Peters, U.; Milner, D.A., Jr.; Giovannucci, E.L.; Nishihara, R.; Giannakis, M.; Garrett, W.S.; et al. Integrative Analysis of Exogenous, Endogenous, Tumour and Immune Factors for Precision Medicine. Gut 2018, 67, 1168–1180. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Chan, A.T.; Fuchs, C.S.; Giovannucci, E. Molecular Pathological Epidemiology of Colorectal Neoplasia: An Emerging Transdisciplinary and Interdisciplinary Field. Gut 2011, 60, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nowak, J.A.; Hamada, T.; Milner DA, J.; Nishihara, R. Insights into Pathogenic Interactions among Environment, Host, and Tumor at the Crossroads of Molecular Pathology and Epidemiology. Annu. Rev. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Schell, M.J.; Teer, J.K.; Greenawalt, D.M.; Yang, M.; Yeatman, T.J. Co-Evolution of Somatic Variation in Primary and Metastatic Colorectal Cancer may Expand Biopsy Indications in the Molecular Era. PLoS ONE 2015, 10, e0126670. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Wu, X.Y.; Yang, Z.Y.; Threapleton, D.E.; Yuan, J.Q.; Yu, Y.Y.; Tang, J.L. Concordant Analysis of KRAS, BRAF, PIK3CA Mutations, and PTEN Expression between Primary Colorectal Cancer and Matched Metastases. Sci. Rep. 2015, 5, 8065. [Google Scholar] [CrossRef] [PubMed]
- Linnekamp, J.F.; Hooff, S.R.V.; Prasetyanti, P.R.; Kandimalla, R.; Buikhuisen, J.Y.; Fessler, E.; Ramesh, P.; Lee, K.A.S.T.; Bochove, G.G.W.; de Jong, J.H.; et al. Consensus Molecular Subtypes of Colorectal Cancer are Recapitulated in in Vitro and in Vivo Models. Cell Death Differ. 2018, 25, 616–633. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A Genetic Model for Colorectal Tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Jass, J.R.; Smith, M. Sialic Acid and Epithelial Differentiation in Colorectal Polyps and Cancer--a Morphological, Mucin and Lectin Histochemical Study. Pathology 1992, 24, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; Sharma, P.K.; Krishnan, G.; Lockhart, A.C. Immune Checkpoints and Immunotherapy for Colorectal Cancer. Gastroenterol. Rep. 2015, 3, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T. Are KRAS/BRAF Mutations Potent Prognostic and/or Predictive Biomarkers in Colorectal Cancers? Anticancer Agents Med. Chem. 2012, 12, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Tejpar, S.; Stintzing, S.; Ciardiello, F.; Tabernero, J.; Van Cutsem, E.; Beier, F.; Esser, R.; Lenz, H.J.; Heinemann, V. Prognostic and Predictive Relevance of Primary Tumor Location in Patients with RAS Wild-Type Metastatic Colorectal Cancer: Retrospective Analyses of the CRYSTAL and FIRE-3 Trials. JAMA Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Missiaglia, E.; Jacobs, B.; D’Ario, G.; Di Narzo, A.F.; Soneson, C.; Budinska, E.; Popovici, V.; Vecchione, L.; Gerster, S.; Yan, P.; et al. Distal and Proximal Colon Cancers Differ in Terms of Molecular, Pathological, and Clinical Features. Ann. Oncol. 2014, 25, 1995–2001. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, M.B.; Rossing, M.; Ostrup, O.; Larsen, P.N.; Heiberg Engel, P.J.; Jorgensen, L.N.; Hogdall, E.V.; Eriksen, J.; Ibsen, P.; Jess, P.; et al. Genomic Alterations Accompanying Tumour Evolution in Colorectal Cancer: Tracking the Differences between Primary Tumours and Synchronous Liver Metastases by Whole-Exome Sequencing. BMC Cancer 2018, 18, 752. [Google Scholar] [CrossRef] [PubMed]
- Ulivi, P.; Scarpi, E.; Chiadini, E.; Marisi, G.; Valgiusti, M.; Capelli, L.; Casadei Gardini, A.; Monti, M.; Ruscelli, S.; Frassineti, G.L.; et al. Right- Vs. Left-Sided Metastatic Colorectal Cancer: Differences in Tumor Biology and Bevacizumab Efficacy. Int. J. Mol. Sci. 2017, 18, 1240. [Google Scholar] [CrossRef] [PubMed]
- Stintzing, S.; Tejpar, S.; Gibbs, P.; Thiebach, L.; Lenz, H.J. Understanding the Role of Primary Tumour Localisation in Colorectal Cancer Treatment and Outcomes. Eur. J. Cancer 2017, 84, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Boisen, M.K.; Johansen, J.S.; Dehlendorff, C.; Larsen, J.S.; Osterlind, K.; Hansen, J.; Nielsen, S.E.; Pfeiffer, P.; Tarpgaard, L.S.; Hollander, N.H.; et al. Primary Tumor Location and Bevacizumab Effectiveness in Patients with Metastatic Colorectal Cancer. Ann. Oncol. 2013, 24, 2554–2559. [Google Scholar] [CrossRef] [PubMed]
- Moretto, R.; Cremolini, C.; Rossini, D.; Pietrantonio, F.; Battaglin, F.; Mennitto, A.; Bergamo, F.; Loupakis, F.; Marmorino, F.; Berenato, R.; et al. Location of Primary Tumor and Benefit from Anti-Epidermal Growth Factor Receptor Monoclonal Antibodies in Patients with RAS and BRAF Wild-Type Metastatic Colorectal Cancer. Oncologist 2016, 21, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Antoniotti, C.; Lonardi, S.; Bergamo, F.; Cortesi, E.; Tomasello, G.; Moretto, R.; Ronzoni, M.; Racca, P.; Loupakis, F.; et al. Primary Tumor Sidedness and Benefit from FOLFOXIRI Plus Bevacizumab as Initial Therapy for Metastatic Colorectal Cancer. Retrospective Analysis of the TRIBE Trial by GONO. Ann. Oncol. 2018, 29, 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Komor, M.A.; Bosch, L.J.; Bounova, G.; Bolijn, A.S.; Delis-van Diemen, P.M.; Rausch, C.; Hoogstrate, Y.; Stubbs, A.P.; de Jong, M.; Jenster, G.; et al. Consensus Molecular Subtype Classification of Colorectal Adenomas. J. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-Tumour Heterogeneity: A Looking Glass for Cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Amaro, A.; Chiara, S.; Pfeffer, U. Molecular Evolution of Colorectal Cancer: From Multistep Carcinogenesis to the Big Bang. Cancer Metastasis Rev. 2016, 35, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; O’Brien, C.A.; van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable Clonal Repopulation Dynamics Influence Chemotherapy Response in Colorectal Cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang Model of Human Colorectal Tumor Growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer Heterogeneity: Implications for Targeted Therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Enriquez-Navas, P.M.; Kam, Y.; Das, T.; Hassan, S.; Silva, A.; Foroutan, P.; Ruiz, E.; Martinez, G.; Minton, S.; Gillies, R.J.; et al. Exploiting Evolutionary Principles to Prolong Tumor Control in Preclinical Models of Breast Cancer. Sci. Transl. Med. 2016, 8, 327ra24. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Swanton, C.; Taylor, B.S.; Costello, J.F. Treatment-Induced Mutagenesis and Selective Pressures Sculpt Cancer Evolution. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-Cancer Analysis of the Extent and Consequences of Intratumor Heterogeneity. Nat. Med. 2016, 22, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A.; Loes, I.M.; Alagaratnam, S.; Nilsen, G.; Holand, M.; Lingjaerde, O.C.; Sorbye, H.; Berg, K.C.; Horn, A.; Angelsen, J.H.; et al. Intra-Patient Inter-Metastatic Genetic Heterogeneity in Colorectal Cancer as a Key Determinant of Survival After Curative Liver Resection. PLoS Genet. 2016, 12, e1006225. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Kandimalla, R.; Huang, H.; Zhu, L.; Li, Y.; Gao, F.; Goel, A.; Wang, X. Molecular Subtyping of Colorectal Cancer: Recent Progress, New Challenges and Emerging Opportunities. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Suzuki, Y.; Ng, S.B.; Chua, C.; Leow, W.Q.; Chng, J.; Liu, S.Y.; Ramnarayanan, K.; Gan, A.; Ho, D.L.; Ten, R.; et al. Multiregion Ultra-Deep Sequencing Reveals Early Intermixing and Variable Levels of Intratumoral Heterogeneity in Colorectal Cancer. Mol. Oncol. 2017, 11, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Giardina, T.; Robinson, C.; Grieu-Iacopetta, F.; Millward, M.; Iacopetta, B.; Spagnolo, D.; Amanuel, B. Implementation of Next Generation Sequencing Technology for Somatic Mutation Detection in Routine Laboratory Practice. Pathology 2018, 50, 389–401. [Google Scholar] [CrossRef] [PubMed]
- D’Haene, N.; Fontanges, Q.; De Neve, N.; Blanchard, O.; Melendez, B.; Delos, M.; Dehou, M.F.; Maris, C.; Nagy, N.; Rousseau, E.; et al. Clinical Application of Targeted Next-Generation Sequencing for Colorectal Cancer Patients: A Multicentric Belgian Experience. Oncotarget 2018, 9, 20761–20768. [Google Scholar] [CrossRef] [PubMed]
- Baisse, B.; Bouzourene, H.; Saraga, E.P.; Bosman, F.T.; Benhattar, J. Intratumor Genetic Heterogeneity in Advanced Human Colorectal Adenocarcinoma. Int. J. Cancer 2001, 93, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Petaccia de Macedo, M.; Melo, F.M.; Ribeiro, H.S.C.; Marques, M.C.; Kagohara, L.T.; Begnami, M.D.; Neto, J.C.; Ribeiro, J.S.; Soares, F.A.; Carraro, D.M.; et al. KRAS Mutation Status is Highly Homogeneous between Areas of the Primary Tumor and the Corresponding Metastasis of Colorectal Adenocarcinomas: One Less Problem in Patient Care. Am. J. Cancer Res. 2017, 7, 1978–1989. [Google Scholar] [PubMed]
- Farber, L.; Efrati, E.; Elkin, H.; Peerless, Y.; Sabo, E.; Ben-Izhak, O.; Hershkovitz, D. Molecular Morphometric Analysis shows Relative Intra-Tumoural Homogeneity for KRAS Mutations in Colorectal Cancer. Virchows Arch. 2011, 459, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Jeantet, M.; Tougeron, D.; Tachon, G.; Cortes, U.; Archambaut, C.; Fromont, G.; Karayan-Tapon, L. High Intra- and Inter-Tumoral Heterogeneity of RAS Mutations in Colorectal Cancer. Int. J. Mol. Sci. 2016, 17, 2015. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.G.; Jenkins, G.; Williams, N.; Griffiths, P.; Chambers, P.; Beynon, J.; Harris, D. Genetic and Epigenetic Intra-Tumour Heterogeneity in Colorectal Cancer. World J. Surg. 2017, 41, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Buttner, J.; Johrens, K.; Klauschen, F.; Hummel, M.; Lenze, D.; Saeger, W.; Lehmann, A. Intratumoral Morphological Heterogeneity can be an Indicator of Genetic Heterogeneity in Colorectal Cancer. Exp. Mol. Pathol. 2018, 104, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Reggiani Bonetti, L.; Barresi, V.; Bettelli, S.; Caprera, C.; Manfredini, S.; Maiorana, A. Analysis of KRAS, NRAS, PIK3CA, and BRAF Mutational Profile in Poorly Differentiated Clusters of KRAS-Mutated Colon Cancer. Hum. Pathol. 2017, 62, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Puig, P.; Cayre, A.; Manceau, G.; Buc, E.; Bachet, J.B.; Lecomte, T.; Rougier, P.; Lievre, A.; Landi, B.; Boige, V.; et al. Analysis of PTEN, BRAF, and EGFR Status in Determining Benefit from Cetuximab Therapy in Wild-Type KRAS Metastatic Colon Cancer. J. Clin. Oncol. 2009, 27, 5924–5930. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Qiu, T.; Guo, L.; Ying, J. Major Challenges Related to Tumor Biological Characteristics in Accurate Mutation Detection of Colorectal Cancer by Next-Generation Sequencing. Cancer Lett. 2017, 410, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guo, X.; Li, D.; Du, X.; Yin, C.; Chen, C.; Fang, W.; Bian, Z.; Zhang, J.; Li, B.; et al. Multi-Regional Sequencing Reveals Intratumor Heterogeneity and Positive Selection of Somatic mtDNA Mutations in Hepatocellular Carcinoma and Colorectal Cancer. Int. J. Cancer 2018, 143, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Losi, L.; Baisse, B.; Bouzourene, H.; Benhattar, J. Evolution of Intratumoral Genetic Heterogeneity during Colorectal Cancer Progression. Carcinogenesis 2005, 26, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer. Cell 2018, 33, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, S.; Lepage, C.; Hatem, C.; Coatmeur, O.; Faivre, J.; Bouvier, A.M. Epidemiology and Management of Liver Metastases from Colorectal Cancer. Ann. Surg. 2006, 244, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Garden, O.J.; Rees, M.; Poston, G.J.; Mirza, D.; Saunders, M.; Ledermann, J.; Primrose, J.N.; Parks, R.W. Guidelines for Resection of Colorectal Cancer Liver Metastases. Gut 2006, 55 (Suppl. 3), iii1–iii8. [Google Scholar] [CrossRef] [PubMed]
- Mitry, E.; Guiu, B.; Cosconea, S.; Jooste, V.; Faivre, J.; Bouvier, A.M. Epidemiology, Management and Prognosis of Colorectal Cancer with Lung Metastases: A 30-Year Population-Based Study. Gut 2010, 59, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Riihimaki, M.; Hemminki, A.; Sundquist, J.; Hemminki, K. Patterns of Metastasis in Colon and Rectal Cancer. Sci. Rep. 2016, 6, 29765. [Google Scholar] [CrossRef] [PubMed]
- Lemmens, V.E.; Klaver, Y.L.; Verwaal, V.J.; Rutten, H.J.; Coebergh, J.W.; de Hingh, I.H. Predictors and Survival of Synchronous Peritoneal Carcinomatosis of Colorectal Origin: A Population-Based Study. Int. J. Cancer 2011, 128, 2717–2725. [Google Scholar] [CrossRef] [PubMed]
- Goere, D.; Allard, M.A.; Honore, C.; Dumont, F.; Elias, D. Incidence and Prognosis of Synchronous Colorectal Carcinomatosis. Future Oncol. 2013, 9, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.N.; Cohen, A.M. Ovarian Neoplasms in Patients with Colorectal Cancer: Understanding the Role of Prophylactic Oophorectomy. Clin. Colorectal Cancer 2004, 3, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Go, P.H.; Klaassen, Z.; Meadows, M.C.; Chamberlain, R.S. Gastrointestinal Cancer and Brain Metastasis: A Rare and Ominous Sign. Cancer 2011, 117, 3630–3640. [Google Scholar] [CrossRef] [PubMed]
- Engstrand, J.; Nilsson, H.; Stromberg, C.; Jonas, E.; Freedman, J. Colorectal Cancer Liver Metastases—A Population-Based Study on Incidence, Management and Survival. BMC Cancer 2018, 18, 78. [Google Scholar] [CrossRef] [PubMed]
- Artale, S.; Sartore-Bianchi, A.; Veronese, S.M.; Gambi, V.; Sarnataro, C.S.; Gambacorta, M.; Lauricella, C.; Siena, S. Mutations of KRAS and BRAF in Primary and Matched Metastatic Sites of Colorectal Cancer. J. Clin. Oncol. 2008, 26, 4217–4219. [Google Scholar] [CrossRef] [PubMed]
- Baas, J.M.; Krens, L.L.; Guchelaar, H.J.; Morreau, H.; Gelderblom, H. Concordance of Predictive Markers for EGFR Inhibitors in Primary Tumors and Metastases in Colorectal Cancer: A Review. Oncologist 2011, 16, 1239–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knijn, N.; Mekenkamp, L.J.; Klomp, M.; Vink-Borger, M.E.; Tol, J.; Teerenstra, S.; Meijer, J.W.; Tebar, M.; Riemersma, S.; van Krieken, J.H.; et al. KRAS Mutation Analysis: A Comparison between Primary Tumours and Matched Liver Metastases in 305 Colorectal Cancer Patients. Br. J. Cancer 2011, 104, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Vakiani, E.; Janakiraman, M.; Shen, R.; Sinha, R.; Zeng, Z.; Shia, J.; Cercek, A.; Kemeny, N.; D’Angelica, M.; Viale, A.; et al. Comparative Genomic Analysis of Primary Versus Metastatic Colorectal Carcinomas. J. Clin. Oncol. 2012, 30, 2956–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brannon, A.R.; Vakiani, E.; Sylvester, B.E.; Scott, S.N.; McDermott, G.; Shah, R.H.; Kania, K.; Viale, A.; Oschwald, D.M.; Vacic, V.; et al. Comparative Sequencing Analysis Reveals High Genomic Concordance between Matched Primary and Metastatic Colorectal Cancer Lesions. Genome Biol. 2014, 15, 454. [Google Scholar] [CrossRef] [PubMed]
- Tan, I.B.; Malik, S.; Ramnarayanan, K.; McPherson, J.R.; Ho, D.L.; Suzuki, Y.; Ng, S.B.; Yan, S.; Lim, K.H.; Koh, D.; et al. High-Depth Sequencing of Over 750 Genes Supports Linear Progression of Primary Tumors and Metastases in most Patients with Liver-Limited Metastatic Colorectal Cancer. Genome Biol. 2015, 16, 32. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.P.; Kim, J.E.; Hong, Y.S.; Ahn, S.M.; Chun, S.M.; Hong, S.M.; Jang, S.J.; Yu, C.S.; Kim, J.C.; Kim, T.W. Paired Primary and Metastatic Tumor Analysis of Somatic Mutations in Synchronous and Metachronous Colorectal Cancer. Cancer Res. Treat. 2017, 49, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiyoshi, K.; Yamamoto, G.; Takahashi, A.; Arai, Y.; Yamada, M.; Kakuta, M.; Yamaguchi, K.; Akagi, Y.; Nishimura, Y.; Sakamoto, H.; et al. High Concordance Rate of KRAS/BRAF Mutations and MSI-H between Primary Colorectal Cancer and Corresponding Metastases. Oncol. Rep. 2017, 37, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, V.; Geissler, A.L.; Lutz, L.; Fritsch, R.; Makowiec, F.; Wiesemann, S.; Hopt, U.T.; Passlick, B.; Werner, M.; Lassmann, S. Spatio-Temporal Mutation Profiles of Case-Matched Colorectal Carcinomas and their Metastases Reveal Unique De Novo Mutations in Metachronous Lung Metastases by Targeted Next Generation Sequencing. Mol. Cancer 2016, 15, 63. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Haq, F.; Kim, D.; Jun, C.; Jo, H.J.; Ahn, S.M.; Lee, W.S. Comparative Genomic Analysis of Primary and Synchronous Metastatic Colorectal Cancers. PLoS ONE 2014, 9, e90459. [Google Scholar] [CrossRef] [PubMed]
- Vermaat, J.S.; Nijman, I.J.; Koudijs, M.J.; Gerritse, F.L.; Scherer, S.J.; Mokry, M.; Roessingh, W.M.; Lansu, N.; de Bruijn, E.; van Hillegersberg, R.; et al. Primary Colorectal Cancers and their Subsequent Hepatic Metastases are Genetically Different: Implications for Selection of Patients for Targeted Treatment. Clin. Cancer Res. 2012, 18, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Aprile, G.; Casagrande, M.; De Maglio, G.; Fontanella, C.; Rihawi, K.; Bonotto, M.; Pisa, F.E.; Tuniz, F.; Pizzolitto, S.; Fasola, G. Comparison of the Molecular Profile of Brain Metastases from Colorectal Cancer and Corresponding Primary Tumors. Future Oncol. 2017, 13, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Crobach, S.; Ruano, D.; van Eijk, R.; Schrumpf, M.; Fleuren, G.; van Wezel, T.; Morreau, H.; PALGA Group. Somatic Mutation Profiles in Primary Colorectal Cancers and Matching Ovarian Metastases: Identification of Driver and Passenger Mutations. J. Pathol. Clin. Res. 2016, 2, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Tortola, S.; Steinert, R.; Hantschick, M.; Peinado, M.A.; Gastinger, I.; Stosiek, P.; Lippert, H.; Schlegel, W.; Reymond, M.A. Discordance between K-Ras Mutations in Bone Marrow Micrometastases and the Primary Tumor in Colorectal Cancer. J. Clin. Oncol. 2001, 19, 2837–2843. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kobunai, T.; Yamamoto, Y.; Matsuda, K.; Ishihara, S.; Nozawa, K.; Iinuma, H.; Shibuya, H.; Eshima, K. Heterogeneity of KRAS Status may Explain the Subset of Discordant KRAS Status between Primary and Metastatic Colorectal Cancer. Dis. Colon Rectum 2011, 54, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Beije, N.; Helmijr, J.C.; Weerts, M.J.A.; Beaufort, C.M.; Wiggin, M.; Marziali, A.; Verhoef, C.; Sleijfer, S.; Jansen, M.P.H.M.; Martens, J.W.M. Somatic Mutation Detection using various Targeted Detection Assays in Paired Samples of Circulating Tumor DNA, Primary Tumor and Metastases from Patients Undergoing Resection of Colorectal Liver Metastases. Mol. Oncol. 2016, 10, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Kleist, B.; Meurer, T.; Poetsch, M. Mitochondrial DNA Alteration in Primary and Metastatic Colorectal Cancer: Different Frequency and Association with Selected Clinicopathological and Molecular Markers. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Naxerova, K.; Reiter, J.G.; Brachtel, E.; Lennerz, J.K.; van de Wetering, M.; Rowan, A.; Cai, T.; Clevers, H.; Swanton, C.; Nowak, M.A.; et al. Origins of Lymphatic and Distant Metastases in Human Colorectal Cancer. Science 2017, 357, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Lee, H.S.; Kim, J.H.; Kim, Y.J.; Kwon, J.H.; Lee, J.O.; Bang, S.M.; Park, K.U.; Kim, D.W.; Kang, S.B.; et al. Different Metastatic Pattern According to the KRAS Mutational Status and Site-Specific Discordance of KRAS Status in Patients with Colorectal Cancer. BMC Cancer 2012, 12, 347. [Google Scholar] [CrossRef] [PubMed]
- Christensen, T.D.; Palshof, J.A.; Larsen, F.O.; Poulsen, T.S.; Hogdall, E.; Pfeiffer, P.; Jensen, B.V.; Yilmaz, M.K.; Nielsen, D. Associations between Primary Tumor RAS, BRAF and PIK3CA Mutation Status and Metastatic Site in Patients with Chemo-Resistant Metastatic Colorectal Cancer. Acta Oncol. 2018, 57, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Hench, I.B.; Hench, J.; Tolnay, M. Liquid Biopsy in Clinical Management of Breast, Lung, and Colorectal Cancer. Front. Med. 2018, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, F.; Carloni, S.; Zoli, W.; Ulivi, P.; Gallerani, G.; Fici, P.; Chiadini, E.; Passardi, A.; Frassineti, G.L.; Ragazzini, A.; et al. Detection and Recovery of Circulating Colon Cancer Cells using a Dielectrophoresis-Based Device: KRAS Mutation Status in Pure CTCs. Cancer Lett. 2013, 335, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Hayashi, K.; Kawakami, K.; Miwa, Y.; Hayashi, H.; Yamamoto, M. KRAS Mutation Analysis of Single Circulating Tumor Cells from Patients with Metastatic Colorectal Cancer. BMC Cancer 2017, 17, 311. [Google Scholar] [CrossRef] [PubMed]
- Buim, M.E.; Fanelli, M.F.; Souza, V.S.; Romero, J.; Abdallah, E.A.; Mello, C.A.; Alves, V.; Ocea, L.M.; Mingues, N.B.; Barbosa, P.N.; et al. Detection of KRAS Mutations in Circulating Tumor Cells from Patients with Metastatic Colorectal Cancer. Cancer Biol. Ther. 2015, 16, 1289–1295. [Google Scholar] [CrossRef] [PubMed]
- Denis, J.A.; Patroni, A.; Guillerm, E.; Pepin, D.; Benali-Furet, N.; Wechsler, J.; Manceau, G.; Bernard, M.; Coulet, F.; Larsen, A.K.; et al. Droplet Digital PCR of Circulating Tumor Cells from Colorectal Cancer Patients can Predict KRAS Mutations before Surgery. Mol. Oncol. 2016, 10, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Gasch, C.; Bauernhofer, T.; Pichler, M.; Langer-Freitag, S.; Reeh, M.; Seifert, A.M.; Mauermann, O.; Izbicki, J.R.; Pantel, K.; Riethdorf, S. Heterogeneity of Epidermal Growth Factor Receptor Status and Mutations of KRAS/PIK3CA in Circulating Tumor Cells of Patients with Colorectal Cancer. Clin. Chem. 2013, 59, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Zhang, R.; Li, Z.; Li, J. Clinical and Biological Significance of Circulating Tumor Cells, Circulating Tumor DNA, and Exosomes as Biomarkers in Colorectal Cancer. Oncotarget 2017, 8, 55632–55645. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.X.; Li, Y.M.; Ye, M.; Guo, Y.Y.; Li, Q.W.; Peng, X.M.; Wang, Q.; Zhang, S.F.; Zhao, H.X.; Zhang, H.; et al. KRAS and BRAF Mutations in Serum Exosomes from Patients with Colorectal Cancer in a Chinese Population. Oncol. Lett. 2017, 13, 3608–3616. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Lopez, L.; Blancas, I.; Garrido, J.M.; Mut-Salud, N.; Moya-Jodar, M.; Osuna, A.; Rodriguez-Serrano, F. The Role of Exosomes on Colorectal Cancer: A Review. J. Gastroenterol. Hepatol. 2018, 33, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Spindler, K.L.; Pallisgaard, N.; Vogelius, I.; Jakobsen, A. Quantitative Cell-Free DNA, KRAS, and BRAF Mutations in Plasma from Patients with Metastatic Colorectal Cancer during Treatment with Cetuximab and Irinotecan. Clin. Cancer Res. 2012, 18, 1177–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spindler, K.L.; Pallisgaard, N.; Appelt, A.L.; Andersen, R.F.; Schou, J.V.; Nielsen, D.; Pfeiffer, P.; Yilmaz, M.; Johansen, J.S.; Hoegdall, E.V.; et al. Clinical Utility of KRAS Status in Circulating Plasma DNA Compared to Archival Tumour Tissue from Patients with Metastatic Colorectal Cancer Treated with Anti-Epidermal Growth Factor Receptor Therapy. Eur. J. Cancer 2015, 51, 2678–2685. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; El Messaoudi, S.; Mollevi, C.; Raoul, J.L.; Guimbaud, R.; Pezet, D.; Artru, P.; Assenat, E.; Borg, C.; Mathonnet, M.; et al. Clinical Utility of Circulating DNA Analysis for Rapid Detection of Actionable Mutations to Select Metastatic Colorectal Patients for Anti-EGFR Treatment. Ann. Oncol. 2017, 28, 2149–2159. [Google Scholar] [CrossRef] [PubMed]
- Taly, V.; Pekin, D.; Benhaim, L.; Kotsopoulos, S.K.; Le Corre, D.; Li, X.; Atochin, I.; Link, D.R.; Griffiths, A.D.; Pallier, K.; et al. Multiplex Picodroplet Digital PCR to Detect KRAS Mutations in Circulating DNA from the Plasma of Colorectal Cancer Patients. Clin. Chem. 2013, 59, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodriguez, C.; Brozos, E.; et al. Plasma ctDNA RAS Mutation Analysis for the Diagnosis and Treatment Monitoring of Metastatic Colorectal Cancer Patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, J.; Elez, E.; Caratu, G.; Matito, J.; Santos, C.; Macarulla, T.; Vidal, J.; Garcia, M.; Vieitez, J.M.; Paez, D.; et al. Concordance of Blood- and Tumor-Based Detection of RAS Mutations to Guide Anti-EGFR Therapy in Metastatic Colorectal Cancer. Ann. Oncol. 2017, 28, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; Mouliere, F.; El Messaoudi, S.; Mollevi, C.; Lopez-Crapez, E.; Rolet, F.; Gillet, B.; Gongora, C.; Dechelotte, P.; Robert, B.; et al. Clinical Validation of the Detection of KRAS and BRAF Mutations from Circulating Tumor DNA. Nat. Med. 2014, 20, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Schmiegel, W.; Scott, R.J.; Dooley, S.; Lewis, W.; Meldrum, C.J.; Pockney, P.; Draganic, B.; Smith, S.; Hewitt, C.; Philimore, H.; et al. Blood-Based Detection of RAS Mutations to Guide Anti-EGFR Therapy in Colorectal Cancer Patients: Concordance of Results from Circulating Tumor DNA and Tissue-Based RAS Testing. Mol. Oncol. 2017, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Esposito Abate, R.; Lambiase, M.; Forgione, L.; Cardone, C.; Iannaccone, A.; Sacco, A.; Rachiglio, A.M.; Martinelli, E.; Rizzi, D.; et al. RAS Testing of Liquid Biopsy Correlates with the Outcome of Metastatic Colorectal Cancer Patients Treated with First-Line FOLFIRI Plus Cetuximab in the CAPRI-GOIM Trial. Ann. Oncol. 2018, 29, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Bachet, J.B.; Bouche, O.; Taieb, J.; Dubreuil, O.; Garcia, M.L.; Meurisse, A.; Normand, C.; Gornet, J.M.; Artru, P.; Louafi, S.; et al. RAS Mutation Analysis in Circulating Tumor DNA from Patients with Metastatic Colorectal Cancer: The AGEO RASANC Prospective Multicenter Study. Ann. Oncol. 2018, 29, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS Mutations and Acquired Resistance to Anti-EGFR Therapy in Colorectal Cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Arena, S.; Lamba, S.; Siravegna, G.; Lallo, A.; Hobor, S.; Russo, M.; Buscarino, M.; Lazzari, L.; Sartore-Bianchi, A.; et al. Blockade of EGFR and MEK Intercepts Heterogeneous Mechanisms of Acquired Resistance to Anti-EGFR Therapies in Colorectal Cancer. Sci. Transl. Med. 2014, 6, 224ra26. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.P.; Overman, M.J.; Dasari, A.; Kazmi, S.M.; Mazard, T.; Vilar, E.; Morris, V.K.; Lee, M.S.; Herron, D.; Eng, C.; et al. Characterizing the Patterns of Clonal Selection in Circulating Tumor DNA from Patients with Colorectal Cancer Refractory to Anti-EGFR Treatment. Ann. Oncol. 2015, 26, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Vernieri, C.; Siravegna, G.; Mennitto, A.; Berenato, R.; Perrone, F.; Gloghini, A.; Tamborini, E.; Lonardi, S.; Morano, F.; et al. Heterogeneity of Acquired Resistance to Anti-EGFR Monoclonal Antibodies in Patients with Metastatic Colorectal Cancer. Clin. Cancer Res. 2017, 23, 2414–2422. [Google Scholar] [CrossRef] [PubMed]
- Strickler, J.H.; Loree, J.M.; Ahronian, L.G.; Parikh, A.R.; Niedzwiecki, D.; Pereira, A.A.L.; McKinney, M.; Korn, W.M.; Atreya, C.E.; Banks, K.C.; et al. Genomic Landscape of Cell-Free DNA in Patients with Colorectal Cancer. Cancer. Discov. 2018, 8, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Siena, S.; Sartore-Bianchi, A.; Garcia-Carbonero, R.; Karthaus, M.; Smith, D.; Tabernero, J.; Van Cutsem, E.; Guan, X.; Boedigheimer, M.; Ang, A.; et al. Dynamic Molecular Analysis and Clinical Correlates of Tumor Evolution within a Phase II Trial of Panitumumab-Based Therapy in Metastatic Colorectal Cancer. Ann. Oncol. 2018, 29, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Takahashi, G.; Iwai, T.; Takeda, K.; Furuki, H.; Koizumi, M.; Shinji, S.; Matsuda, A.; Yokoyama, Y.; Hotta, M.; et al. PD-009Emergence of KRAS Mutation may Play a Major Role in the Secondary Resistance to EGFR Blockade. Ann. Oncol. 2018, 29, mdy150.008. [Google Scholar] [CrossRef]
- Gazzaniga, P.; Raimondi, C.; Nicolazzo, C.; Gradilone, A.; Cortesi, E. CtDNA might Expand Therapeutic Options for Second Line Treatment of KRAS Mutant mCRC. Ann. Oncol. 2017, 28 (Suppl. 5), v573–v594. [Google Scholar] [CrossRef]
- Nelson, A.C.; Boone, J.; Cartwright, D.; Thyagarajan, B.; Kincaid, R.; Lambert, A.P.; Karnuth, K.; Henzler, C.; Yohe, S. Optimal Detection of Clinically Relevant Mutations in Colorectal Carcinoma: Sample Pooling Overcomes Intra-Tumoral Heterogeneity. Mod. Pathol. 2018, 31, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Furuki, H.; Yamada, T.; Takahashi, G.; Iwai, T.; Koizumi, M.; Shinji, S.; Yokoyama, Y.; Takeda, K.; Taniai, N.; Uchida, E. Evaluation of Liquid Biopsies for Detection of Emerging Mutated Genes in Metastatic Colorectal Cancer. Eur. J. Surg. Oncol. 2018, 44, 975–982. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Reference | No. Patients | Methodology | Gene | Mutations in cfDNA/Tumor Tissue | Sensitivity/Specificity | Concordance |
|---|---|---|---|---|---|---|
| Spindler et al. [95] | 98 | qPCR | KRAS | 34%/43% | 78%/100% | NA |
| Taly et al. [98] | 50 | ddPCR | KRAS | 28%/38% | 74%/89% | NA |
| Thierry et al. [101] | 95 | Intplex qPCR | KRAS BRAF | 39%/38% 6%/6% | 92%/98% 100%/100% | 96% 100% |
| Spindler et al. [96] | 140 | qPCR | KRAS | 23%/34% | NA | NA |
| Vidal et al. [99] | 115 | Beaming | RAS | 47.8%/51.3% | 96.4%/90% | 93% |
| Bachet et al. [104] | 406 | NGS | RAS | 42%/55% | 92%/94% | 93% |
| Thierry et al. [97] | 34 97 | Intplex qPCR | RAS BRAF | 12%/9% 14%/7% | 67%/94% 57%/89% | 92% 87% |
| Grasselli et al. [100] | 146 | Beaming | RAS | 39%/46% | 85%/91% | 90% |
| Schmiegel et al. [102] | 98 | Beaming | RAS | 51%/53% | 90%/94% | 91.8% |
| Normanno et al. [103] | 92 | NGS | RAS | 36%/36% | 70%/83% | 78% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molinari, C.; Marisi, G.; Passardi, A.; Matteucci, L.; De Maio, G.; Ulivi, P. Heterogeneity in Colorectal Cancer: A Challenge for Personalized Medicine? Int. J. Mol. Sci. 2018, 19, 3733. https://doi.org/10.3390/ijms19123733
Molinari C, Marisi G, Passardi A, Matteucci L, De Maio G, Ulivi P. Heterogeneity in Colorectal Cancer: A Challenge for Personalized Medicine? International Journal of Molecular Sciences. 2018; 19(12):3733. https://doi.org/10.3390/ijms19123733
Chicago/Turabian StyleMolinari, Chiara, Giorgia Marisi, Alessandro Passardi, Laura Matteucci, Giulia De Maio, and Paola Ulivi. 2018. "Heterogeneity in Colorectal Cancer: A Challenge for Personalized Medicine?" International Journal of Molecular Sciences 19, no. 12: 3733. https://doi.org/10.3390/ijms19123733