L-DOPA in Parkinson’s Disease: Looking at the “False” Neurotransmitters and Their Meaning


{kind=link}
{kind=link}
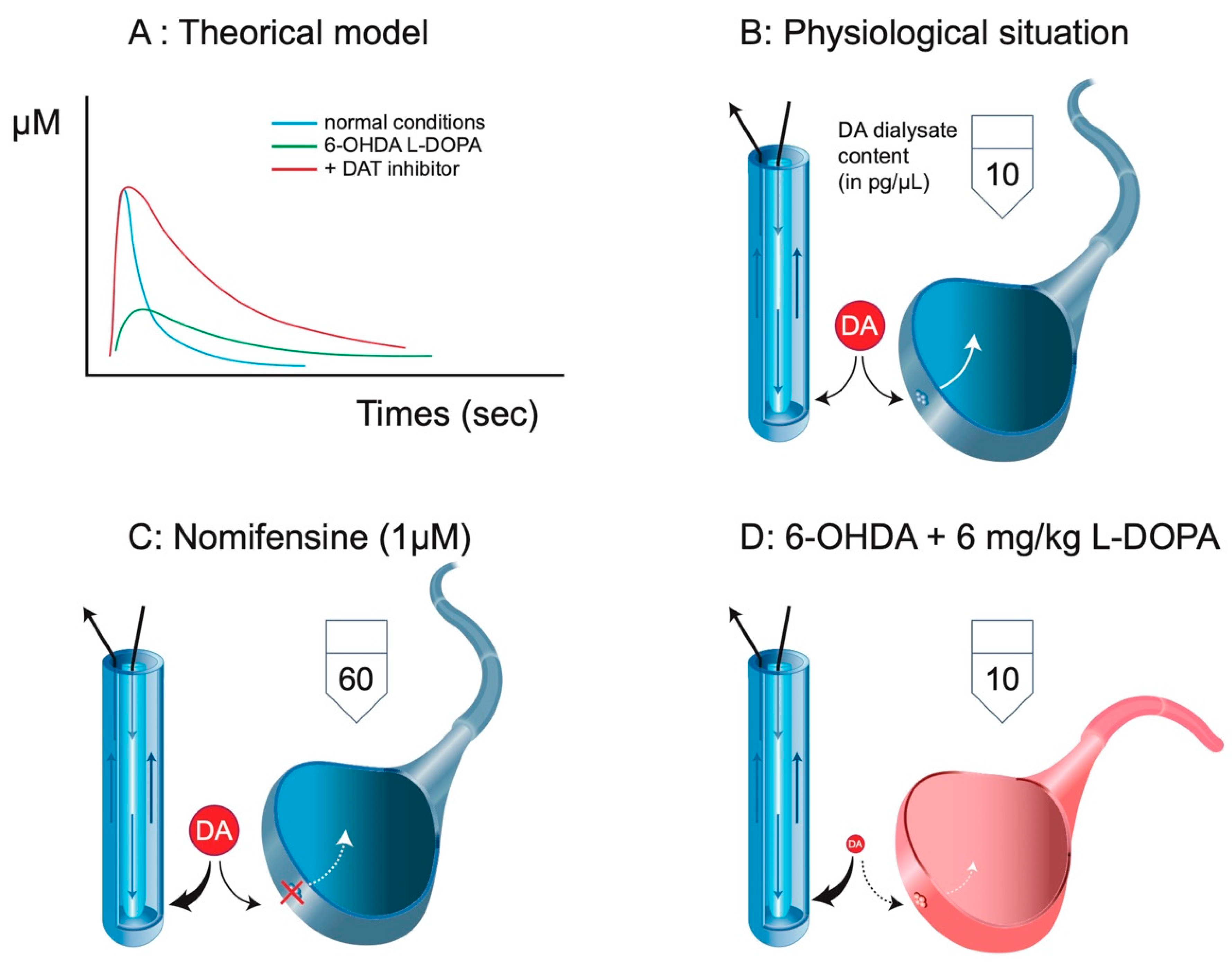{kind=link}
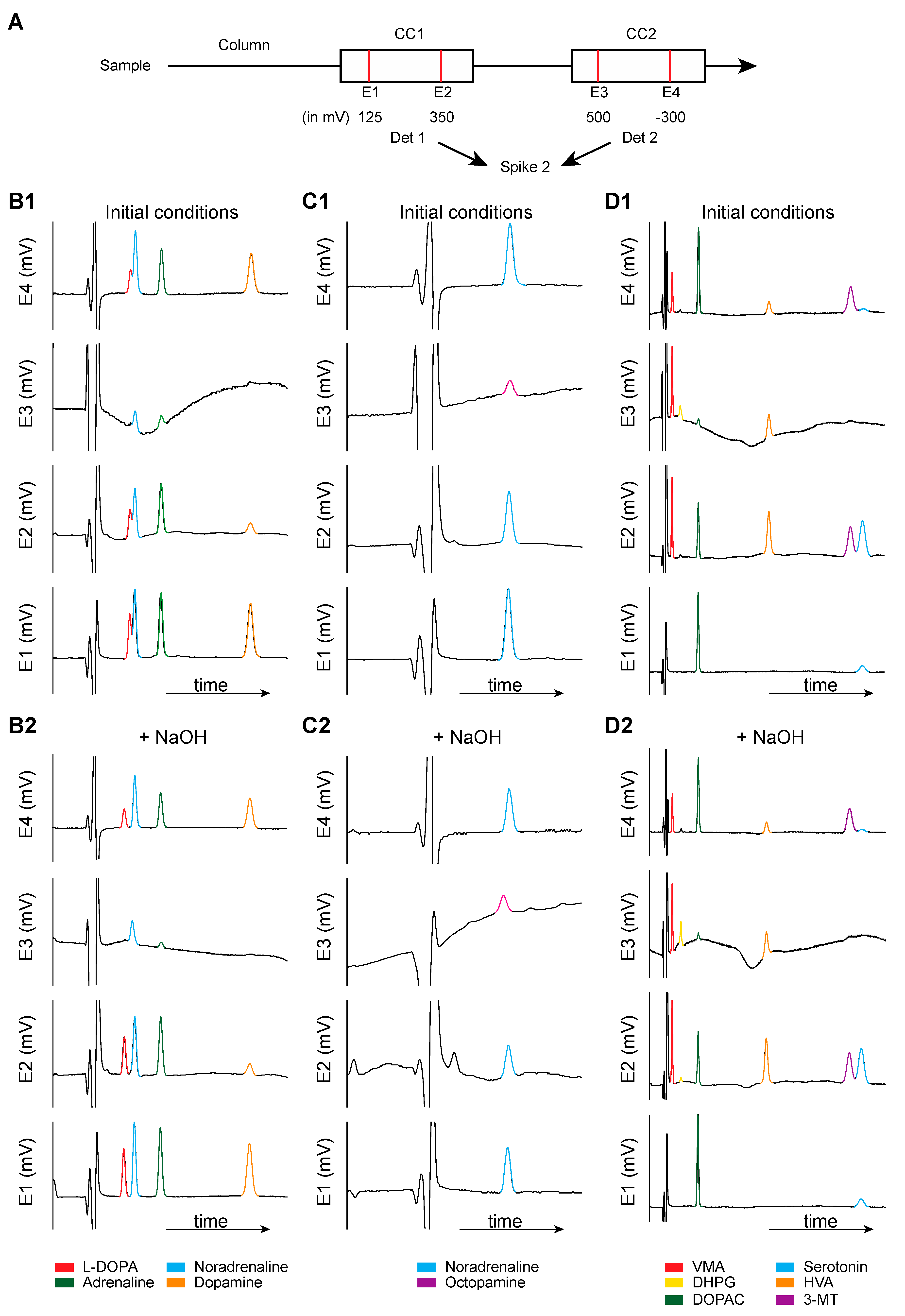{kind=link}
Abstract
:1. Introduction
2. DA Is a False Neurotransmitter in the Mechanism of Action of L-DOPA
Production of DA after L-DOPA Administration
3. DA Released as a False Neurotransmitter by Serotonergic Neurons
3.1. 5-HT Neurons Mediate L-DOPA-Induced DA Extracellular Levels
3.2. L-DOPA-Derived DA from 5-HT Neurons at the Expense of 5-HT Function
3.3. DA as a False Neurotransmitter from 5-HT Neurons: Functional Impact
3.4. Conclusion
4. DA Released as a False Neurotransmitter by Noradrenergic Neurons
4.1. Endogenous DA Can Be Released by NA at Low Levels
4.2. DA as False Transmitter on NA Function
4.3. Overall Consequences of DA as a False Neurotransmitter in 5-HT and NA Neurons
5. L-DOPA, Its Own False Neurotransmitter?
6. A Full Family of “False Transmitters”: The Trace Amines and L-DOPA Derivatives
6.1. Metabolites of L-DOPA
6.2. Trace Amines and Metabolism of DA
6.3. The Trace Amines
6.4. Impairment of Monoaminergic Transmissions by Trace Amines
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AADC | aromatic L-amino acid decarboxylase |
| AD | aldehyde dehydrogenase |
| ADH | alcohol dehydrogenase |
| COMT | catechol-O-methyltransferase |
| DA | dopamine |
| DBH | dopamine ß-hydroxylase |
| DOPAC | 3,4-ihydroxyphenylacetic acid |
| DOPAL | 3,4-dihydroxyphenylacetaldehyde |
| DOPEGAL | 3,4-methoxyphenylacetaldehyde |
| HVA | homovanillic acid |
| HMA | 4-hydroxy-3-methoxyphenylacetaldehyde |
| L-DOPA | L-3,4-dihydroxyphenylalanine |
| MAO | monoamine oxidase |
| MHPG | 3-methoxy-4-hydroxyphenylglycol |
| 3-OMD | 3-O-methyl-DOPA |
| MN | metanephrine |
| 3-MT | 3-methoxytyramine |
| MOPEGAL | 3-methoxy-4-hydroxyphenylglycolaldehyde |
| NA | norepinephrine |
| NMN | normetanephrine |
| PD | Parkinson’s disease |
| PNMT | phenolethanolamine-N-methyltransferase |
| SNc | substantia nigra pars compacta |
| TH | tyrosine hydroxylase |
| Tyr | tyrosine |
| VMA | vanillylmandelic acid |
References
- Bastide, M.F.; Meissner, W.G.; Picconi, B.; Fasano, S.; Fernagut, P.O.; Feyder, M.; Francardo, V.; Alcacer, C.; Ding, Y.; Brambilla, R.; et al. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog. Neurobiol. 2015, 132, 96–168. [Google Scholar] [CrossRef] [PubMed]
- Hauser, R.A. Levodopa: Past, present, and future. Eur. Neurol. 2009, 62, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Stern, M.B.; Sethi, K. The scientific and clinical basis for the treatment of Parkinson disease (2009). Neurology 2009, 72, S1–S136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hortnagl, H.; Pifl, C.; Hortnagl, E.; Reiner, A.; Sperk, G. Distinct gradients of various neurotransmitter markers in caudate nucleus and putamen of the human brain. J. Neurochem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kish, S.J.; Tong, J.; Hornykiewicz, O.; Rajput, A.; Chang, L.J.; Guttman, M.; Furukawa, Y. Preferential loss of serotonin markers in caudate versus putamen in Parkinson’s disease. Brain 2008, 131, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Lees, A.J.; Tolosa, E.; Olanow, C.W. Four pioneers of L-dopa treatment: Arvid Carlsson, Oleh Hornykiewicz, George Cotzias, and Melvin Yahr. Mov. Disord. 2015, 30, 19–36. [Google Scholar] [CrossRef]
- Burke, R.E.; O’Malley, K. Axon degeneration in Parkinson’s disease. Exp. Neurol. 2013, 246, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Di Giovanni, G.; Svob Strac, D.; Sole, M.; Unzeta, M.; Tipton, K.F.; Muck-Seler, D.; Bolea, I.; Della Corte, L.; Nikolac Perkovic, M.; Pivac, N.; et al. Monoaminergic and Histaminergic Strategies and Treatments in Brain Diseases. Front. Neurosci. 2016, 10, 541. [Google Scholar] [CrossRef] [Green Version]
- Delaville, C.; Deurwaerdere, P.D.; Benazzouz, A. Noradrenaline and Parkinson’s disease. Front. Syst. Neurosci. 2011, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Navailles, S.; De Deurwaerdere, P. Contribution of serotonergic transmission to the motor and cognitive effects of high-frequency stimulation of the subthalamic nucleus or levodopa in Parkinson’s disease. Mol. Neurobiol. 2012, 45, 173–185. [Google Scholar] [CrossRef]
- Politis, M.; Wu, K.; Loane, C.; Kiferle, L.; Molloy, S.; Brooks, D.J.; Piccini, P. Staging of serotonergic dysfunction in Parkinson’s disease: An in vivo 11C-DASB PET study. Neurobiol. Dis. 2010, 40, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Eskow Jaunarajs, K.L.; Angoa-Perez, M.; Kuhn, D.M.; Bishop, C. Potential mechanisms underlying anxiety and depression in Parkinson’s disease: Consequences of l-DOPA treatment. Neurosci. Biobehav. Rev. 2011, 35, 556–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, L.K.; Chase, T.N.; Colburn, R.W.; Kopin, I.J. L-dopa in Parkinsonism. A possible mechanism of action. Neurology 1972, 22, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Kannari, K.; Maeda, T.; Tomiyama, M.; Suda, T.; Matsunaga, M. Role of serotonergic neurons in L-DOPA-derived extracellular dopamine in the striatum of 6-OHDA-lesioned rats. Neuroreport 1999, 10, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Cotzias, G.C.; Papavasiliou, P.S.; Gellene, R. Modification of Parkinsonism--chronic treatment with L-dopa. N. Engl. J. Med. 1969, 280, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Kannari, K.; Yamato, H.; Arai, A.; Matsunaga, M. Effects of benserazide on L-DOPA-derived extracellular dopamine levels and aromatic L-amino acid decarboxylase activity in the striatum of 6-hydroxydopamine-lesioned rats. Tohoku J. Exp. Med. 2003, 199, 149–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navailles, S.; Lagiere, M.; Contini, A.; De Deurwaerdere, P. Multisite intracerebral microdialysis to study the mechanism of L-DOPA induced dopamine and serotonin release in the parkinsonian brain. ACS Chem. Neurosci. 2013, 4, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Zetterstrom, T.; Herrera-Marschitz, M.; Ungerstedt, U. Simultaneous measurement of dopamine release and rotational behaviour in 6-hydroxydopamine denervated rats using intracerebral dialysis. Brain Res. 1986, 376, 1–7. [Google Scholar] [CrossRef]
- Marti, M.; Trapella, C.; Viaro, R.; Morari, M. The nociceptin/orphanin FQ receptor antagonist J-113397 and L-DOPA additively attenuate experimental parkinsonism through overinhibition of the nigrothalamic pathway. J. Neurosci. 2007, 27, 1297–1307. [Google Scholar] [CrossRef]
- De Deurwaerdere, P.; Di Giovanni, G.; Millan, M.J. Expanding the repertoire of L-DOPA’s actions: A comprehensive review of its functional neurochemistry. Prog. Neurobiol. 2017, 151, 57–100. [Google Scholar] [CrossRef]
- Abercrombie, E.D.; Bonatz, A.E.; Zigmond, M.J. Effects of L-dopa on extracellular dopamine in striatum of normal and 6-hydroxydopamine-treated rats. Brain Res. 1990, 525, 36–44. [Google Scholar] [CrossRef]
- Hollister, A.S.; Breese, G.R.; Mueller, R.A. Role of monoamine neural systems in L-dihydroxyphenylalanine-stimulated activity. J. Pharm. Exp. Ther. 1979, 208, 37–43. [Google Scholar]
- De Deurwaerdere, P.; Bonhomme, N.; Le Moal, M.; Spampinato, U. d-fenfluramine increases striatal extracellular dopamine in vivo independently of serotonergic terminals or dopamine uptake sites. J. Neurochem. 1995, 65, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- De Deurwaerdere, P.; Bonhomme, N.; Lucas, G.; Le Moal, M.; Spampinato, U. Serotonin enhances striatal dopamine outflow in vivo through dopamine uptake sites. J. Neurochem. 1996, 66, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Nomikos, G.G.; Damsma, G.; Wenkstern, D.; Fibiger, H.C. In vivo characterization of locally applied dopamine uptake inhibitors by striatal microdialysis. Synapse 1990, 6, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.W.; Abercrombie, E.D. Role of high-affinity dopamine uptake and impulse activity in the appearance of extracellular dopamine in striatum after administration of exogenous L-DOPA: Studies in intact and 6-hydroxydopamine-treated rats. J. Neurochem. 1999, 72, 1516–1522. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Marschitz, M.; Arbuthnott, G.; Ungerstedt, U. The rotational model and microdialysis: Significance for dopamine signalling, clinical studies, and beyond. Prog. Neurobiol. 2010, 90, 176–189. [Google Scholar] [CrossRef]
- Navailles, S.; Carta, M.; Guthrie, M.; De Deurwaerdere, P. L-DOPA and serotonergic neurons: Functional implication and therapeutic perspectives in Parkinson’s disease. Cent. Nerv. Syst. Agents Med. Chem. 2011, 11, 305–320. [Google Scholar] [CrossRef]
- Maeda, T.; Kannari, K.; Suda, T.; Matsunaga, M. Loss of regulation by presynaptic dopamine D2 receptors of exogenous L-DOPA-derived dopamine release in the dopaminergic denervated striatum. Brain Res. 1999, 817, 185–191. [Google Scholar] [CrossRef]
- Liu, C.; Kershberg, L.; Wang, J.; Schneeberger, S.; Kaeser, P.S. Dopamine Secretion Is Mediated by Sparse Active Zone-like Release Sites. Cell 2018, 172, 706–718. [Google Scholar] [CrossRef] [Green Version]
- Omiatek, D.M.; Bressler, A.J.; Cans, A.S.; Andrews, A.M.; Heien, M.L.; Ewing, A.G. The real catecholamine content of secretory vesicles in the CNS revealed by electrochemical cytometry. Sci. Rep. 2013, 3, 1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindgren, H.S.; Andersson, D.R.; Lagerkvist, S.; Nissbrandt, H.; Cenci, M.A. L-DOPA-induced dopamine efflux in the striatum and the substantia nigra in a rat model of Parkinson’s disease: Temporal and quantitative relationship to the expression of dyskinesia. J. Neurochem. 2010, 112, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Navailles, S.; Bioulac, B.; Gross, C.; De Deurwaerdere, P. Serotonergic neurons mediate ectopic release of dopamine induced by L-DOPA in a rat model of Parkinson’s disease. Neurobiol. Dis. 2010, 38, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Bunney, B.S.; Aghajanian, G.K.; Roth, R.H. Comparison of effects of L-dopa, amphetamine and apomorphine on firing rate of rat dopaminergic neurones. Nat. New Biol. 1973, 245, 123–125. [Google Scholar] [CrossRef]
- Baldessarini, R.J. Release of catecholamines. In Biochemistry of Biogenic Amines; Plenum Press: New York, NY, USA; London, UK, 1975; Volume 3, pp. 37–137. [Google Scholar]
- Ng, K.Y.; Chase, T.N.; Colburn, R.W.; Kopin, I.J. L-Dopa-induced release of cerebral monoamines. Science 1970, 170, 76–77. [Google Scholar] [CrossRef]
- Tison, F.; Mons, N.; Geffard, M.; Henry, P. The metabolism of exogenous L-dopa in the brain: An immunohistochemical study of its conversion to dopamine in non-catecholaminergic cells of the rat brain. J. Neural Transm. Park. Dis. Dement. Sect. 1991, 3, 27–39. [Google Scholar] [CrossRef]
- Melamed, E.; Hefti, F.; Liebman, J.; Schlosberg, A.J.; Wurtman, R.J. Serotonergic neurones are not involved in action of L-dopa in Parkinson’s disease. Nature 1980, 283, 772–774. [Google Scholar] [CrossRef]
- Iderberg, H.; McCreary, A.C.; Varney, M.A.; Kleven, M.S.; Koek, W.; Bardin, L.; Depoortere, R.; Cenci, M.A.; Newman-Tancredi, A. NLX-112, a novel 5-HT1A receptor agonist for the treatment of L-DOPA-induced dyskinesia: Behavioral and neurochemical profile in rat. Exp. Neurol. 2015, 271, 335–350. [Google Scholar] [CrossRef]
- Kannari, K.; Yamato, H.; Shen, H.; Tomiyama, M.; Suda, T.; Matsunaga, M. Activation of 5-HT(1A) but not 5-HT(1B) receptors attenuates an increase in extracellular dopamine derived from exogenously administered L-DOPA in the striatum with nigrostriatal denervation. J. Neurochem. 2001, 76, 1346–1353. [Google Scholar] [CrossRef] [Green Version]
- McCreary, A.C.; Varney, M.A.; Newman-Tancredi, A. The Novel 5-HT Receptor Agonist, NLX-112 Reduces L-DOPA-induced Abnormal Involuntary Movements in Rat: A Chronic Administration Study with Microdialysis Measurements. Neuropharmacology 2016. [Google Scholar] [CrossRef]
- Fitoussi, A.; Dellu-Hagedorn, F.; De Deurwaerdere, P. Monoamines tissue content analysis reveals restricted and site-specific correlations in brain regions involved in cognition. Neuroscience 2013, 255, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Harden, D.G.; Grace, A.A. Activation of dopamine cell firing by repeated L-DOPA administration to dopamine-depleted rats: Its potential role in mediating the therapeutic response to L-DOPA treatment. J. Neurosci. 1995, 15, 6157–6166. [Google Scholar] [CrossRef] [PubMed]
- Miguelez, C.; Navailles, S.; De Deurwaerdere, P.; Ugedo, L. The acute and long-term L-DOPA effects are independent from changes in the activity of dorsal raphe serotonergic neurons in 6-OHDA lesioned rats. Br. J. Pharm. 2016. [Google Scholar] [CrossRef] [PubMed]
- Miguelez, C.; Benazzouz, A.; Ugedo, L.; De Deurwaerdere, P. Impairment of Serotonergic Transmission by the Antiparkinsonian Drug L-DOPA: Mechanisms and Clinical Implications. Front. Cell. Neurosci. 2017, 11, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellnow, R.C.; Newman, J.H.; Chambers, N.; West, A.R.; Steece-Collier, K.; Sandoval, I.M.; Benskey, M.J.; Bishop, C.; Manfredsson, F.P. Regulation of dopamine neurotransmission from serotonergic neurons by ectopic expression of the dopamine D2 autoreceptor blocks levodopa-induced dyskinesia. Acta Neuropathol. Commun. 2019, 7, 8. [Google Scholar] [CrossRef]
- Iderberg, H.; McCreary, A.C.; Varney, M.A.; Cenci, M.A.; Newman-Tancredi, A. Activity of serotonin 5-HT(1A) receptor ‘biased agonists’ in rat models of Parkinson’s disease and L-DOPA-induced dyskinesia. Neuropharmacology 2015, 93, 52–67. [Google Scholar] [CrossRef]
- Ostock, C.Y.; Bhide, N.; Goldenberg, A.A.; George, J.A.; Bishop, C. Striatal norepinephrine efflux in l-DOPA-induced dyskinesia. Neurochem. Int. 2018, 114, 85–98. [Google Scholar] [CrossRef]
- De la Fuente-Fernandez, R. Imaging of Dopamine in PD and Implications for Motor and Neuropsychiatric Manifestations of PD. Front. Neurol. 2013, 4, 90. [Google Scholar] [CrossRef] [Green Version]
- De la Fuente-Fernandez, R.; Sossi, V.; Huang, Z.; Furtado, S.; Lu, J.Q.; Calne, D.B.; Ruth, T.J.; Stoessl, A.J. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: Implications for dyskinesias. Brain 2004, 127, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Navailles, S.; Bioulac, B.; Gross, C.; De Deurwaerdere, P. Chronic L-DOPA therapy alters central serotonergic function and L-DOPA-induced dopamine release in a region-dependent manner in a rat model of Parkinson’s disease. Neurobiol. Dis. 2011, 41, 585–590. [Google Scholar] [CrossRef]
- Navailles, S.; Benazzouz, A.; Bioulac, B.; Gross, C.; De Deurwaerdere, P. High-frequency stimulation of the subthalamic nucleus and L-3,4-dihydroxyphenylalanine inhibit in vivo serotonin release in the prefrontal cortex and hippocampus in a rat model of Parkinson’s disease. J. Neurosci. 2010, 30, 2356–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miguelez, C.; Navailles, S.; Delaville, C.; Marquis, L.; Lagiere, M.; Benazzouz, A.; Ugedo, L.; De Deurwaerdere, P. L-DOPA elicits non-vesicular releases of serotonin and dopamine in hemiparkinsonian rats in vivo. Eur. Neuropsychopharmacol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.K.; Colburn, R.W.; Kopin, I.J. Effects of L-dopa on accumulation and efflux of monoamines in particles of rat brain homogenates. J. Pharm. Exp. Ther. 1972, 183, 316–325. [Google Scholar]
- Tanda, G.; Frau, R.; Di Chiara, G. Local 5HT3 receptors mediate fluoxetine but not desipramine-induced increase of extracellular dopamine in the prefrontal cortex. Psychopharmacology 1995, 119, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Navailles, S.; Milan, L.; Khalki, H.; Di Giovanni, G.; Lagiere, M.; De Deurwaerdere, P. Noradrenergic terminals regulate L-DOPA-derived dopamine extracellular levels in a region-dependent manner in Parkinsonian rats. CNS Neurosci. 2014, 20, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, K.; Lenda, T.; Konieczny, J.; Wardas, J.; Lorenc-Koci, E. Interactions of the tricyclic antidepressant drug amitriptyline with L-DOPA in the striatum and substantia nigra of unilaterally 6-OHDA-lesioned rats. Relevance to motor dysfunction in Parkinson’s disease. Neurochem. Int. 2018, 121, 125–139. [Google Scholar] [CrossRef]
- Robertson, G.S.; Robertson, H.A. Evidence that L-dopa-induced rotational behavior is dependent on both striatal and nigral mechanisms. J. Neurosci. 1989, 9, 3326–3331. [Google Scholar] [CrossRef]
- Navailles, S.; De Deurwaerdere, P. Imbalanced Dopaminergic Transmission Mediated by Serotonergic Neurons in L-DOPA-Induced Dyskinesia. Parkinsons Dis. 2012, 2012, 323686. [Google Scholar] [CrossRef]
- Carta, M.; Carlsson, T.; Kirik, D.; Bjorklund, A. Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain 2007, 130, 1819–1833. [Google Scholar] [CrossRef] [Green Version]
- Beaudoin-Gobert, M.; Epinat, J.; Metereau, E.; Duperrier, S.; Neumane, S.; Ballanger, B.; Lavenne, F.; Liger, F.; Tourvielle, C.; Bonnefoi, F.; et al. Behavioural impact of a double dopaminergic and serotonergic lesion in the non-human primate. Brain 2015, 138, 2632–2647. [Google Scholar] [CrossRef] [Green Version]
- Beaudoin-Gobert, M.; Sgambato-Faure, V. Serotonergic pharmacology in animal models: From behavioral disorders to dyskinesia. Neuropharmacology 2014, 81, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Munoz, A.; Li, Q.; Gardoni, F.; Marcello, E.; Qin, C.; Carlsson, T.; Kirik, D.; Di Luca, M.; Bjorklund, A.; Bezard, E.; et al. Combined 5-HT1A and 5-HT1B receptor agonists for the treatment of L-DOPA-induced dyskinesia. Brain 2008, 131, 3380–3394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huot, P.; Sgambato-Faure, V.; Fox, S.H.; McCreary, A.C. Serotonergic Approaches in Parkinson’s Disease: Translational Perspectives, an Update. ACS Chem. Neurosci. 2017, 8, 973–986. [Google Scholar] [CrossRef] [PubMed]
- Bezard, E.; Carta, M. Could the serotonin theory give rise to a treatment for levodopa-induced dyskinesia in Parkinson’s disease? Brain 2015, 138, 829–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanza, K.; Bishop, C. Serotonergic targets for the treatment of L-DOPA-induced dyskinesia. J. Neural Transm. 2018, 125, 1203–1216. [Google Scholar] [CrossRef] [PubMed]
- Carta, M.; Bjorklund, A. The serotonergic system in L-DOPA-induced dyskinesia: Pre-clinical evidence and clinical perspective. J. Neural Transm. 2018, 125, 1195–1202. [Google Scholar] [CrossRef]
- Sahin, G.; Thompson, L.H.; Lavisse, S.; Ozgur, M.; Rbah-Vidal, L.; Dolle, F.; Hantraye, P.; Kirik, D. Differential dopamine receptor occupancy underlies L-DOPA-induced dyskinesia in a rat model of Parkinson’s disease. PLoS ONE 2014, 9, e90759. [Google Scholar] [CrossRef]
- Acquas, E.; Carboni, E.; de Ree, R.H.; Da Prada, M.; Di Chiara, G. Extracellular concentrations of dopamine and metabolites in the rat caudate after oral administration of a novel catechol-O-methyltransferase inhibitor Ro 40-7592. J. Neurochem. 1992, 59, 326–330. [Google Scholar] [CrossRef]
- Porras, G.; De Deurwaerdere, P.; Li, Q.; Marti, M.; Morgenstern, R.; Sohr, R.; Bezard, E.; Morari, M.; Meissner, W.G. L-dopa-induced dyskinesia: Beyond an excessive dopamine tone in the striatum. Sci. Rep. 2014, 4, 3730. [Google Scholar] [CrossRef] [Green Version]
- Politis, M.; Wilson, H.; Wu, K.; Brooks, D.J.; Piccini, P. Chronic exposure to dopamine agonists affects the integrity of striatal D2 receptors in Parkinson’s patients. NeuroImage Clin. 2017, 16, 455–460. [Google Scholar] [CrossRef]
- Nevalainen, N.; Af Bjerken, S.; Lundblad, M.; Gerhardt, G.A.; Stromberg, I. Dopamine release from serotonergic nerve fibers is reduced in L-DOPA-induced dyskinesia. J. Neurochem. 2011, 118, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.; Munoz, A.; Guerra, M.J.; Labandeira-Garcia, J.L. Mechanisms of the effects of exogenous levodopa on the dopamine-denervated striatum. Neuroscience 2001, 103, 639–651. [Google Scholar] [CrossRef]
- Bastide, M.F.; Dovero, S.; Charron, G.; Porras, G.; Gross, C.E.; Fernagut, P.O.; Bezard, E. Immediate-early gene expression in structures outside the basal ganglia is associated to l-DOPA-induced dyskinesia. Neurobiol. Dis. 2013, 62C, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Meadows, S.M.; Conti, M.M.; Gross, L.; Chambers, N.E.; Avnor, Y.; Ostock, C.Y.; Lanza, K.; Bishop, C. Diverse serotonin actions of vilazodone reduce l-3,4-dihidroxyphenylalanine-induced dyskinesia in hemi-parkinsonian rats. Mov. Disord. 2018, 33, 1740–1749. [Google Scholar] [CrossRef] [PubMed]
- Charbonnier-Beaupel, F.; Malerbi, M.; Alcacer, C.; Tahiri, K.; Carpentier, W.; Wang, C.; During, M.; Xu, D.; Worley, P.F.; Girault, J.A.; et al. Gene expression analyses identify Narp contribution in the development of L-DOPA-induced dyskinesia. J. Neurosci. 2015, 35, 96–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spigolon, G.; Fisone, G. Signal transduction in L-DOPA-induced dyskinesia: From receptor sensitization to abnormal gene expression. J. Neural Transm. 2018, 125, 1171–1186. [Google Scholar] [CrossRef] [Green Version]
- Lindenbach, D.; Conti, M.M.; Ostock, C.Y.; George, J.A.; Goldenberg, A.A.; Melikhov-Sosin, M.; Nuss, E.E.; Bishop, C. The Role of Primary Motor Cortex (M1) Glutamate and GABA Signaling in l-DOPA-Induced Dyskinesia in Parkinsonian Rats. J. Neurosci. 2016, 36, 9873–9887. [Google Scholar] [CrossRef] [Green Version]
- Bastide, M.F.; de la Crompe, B.; Doudnikoff, E.; Fernagut, P.O.; Gross, C.E.; Mallet, N.; Boraud, T.; Bezard, E. Inhibiting Lateral Habenula Improves L-DOPA-Induced Dyskinesia. Biol. Psychiatry 2016, 79, 345–353. [Google Scholar] [CrossRef]
- Robertson, G.S.; Fibiger, H.C. Neuroleptics increase c-fos expression in the forebrain: Contrasting effects of haloperidol and clozapine. Neuroscience 1992, 46, 315–328. [Google Scholar] [CrossRef]
- Tremblay, P.O.; Gervais, J.; Rouillard, C. Modification of haloperidol-induced pattern of c-fos expression by serotonin agonists. Eur. J. Neurosci. 1998, 10, 3546–3555. [Google Scholar] [CrossRef]
- Wan, W.; Ennulat, D.J.; Cohen, B.M. Acute administration of typical and atypical antipsychotic drugs induces distinctive patterns of Fos expression in the rat forebrain. Brain Res. 1995, 688, 95–104. [Google Scholar] [CrossRef]
- Devoto, P.; Flore, G.; Saba, P.; Bini, V.; Gessa, G.L. The dopamine beta-hydroxylase inhibitor nepicastat increases dopamine release and potentiates psychostimulant-induced dopamine release in the prefrontal cortex. Addict. Biol. 2014, 19, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Devoto, P.; Flore, G.; Saba, P.; Fa, M.; Gessa, G.L. Co-release of noradrenaline and dopamine in the cerebral cortex elicited by single train and repeated train stimulation of the locus coeruleus. BMC Neurosci. 2005, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempadoo, K.A.; Mosharov, E.V.; Choi, S.J.; Sulzer, D.; Kandel, E.R. Dopamine release from the locus coeruleus to the dorsal hippocampus promotes spatial learning and memory. Proc. Natl. Acad. Sci. USA 2016, 113, 14835–14840. [Google Scholar] [CrossRef] [Green Version]
- Devoto, P.; Flore, G.; Saba, P.; Castelli, M.P.; Piras, A.P.; Luesu, W.; Viaggi, M.C.; Ennas, M.G.; Gessa, G.L. 6-Hydroxydopamine lesion in the ventral tegmental area fails to reduce extracellular dopamine in the cerebral cortex. J. Neurosci. Res. 2008, 86, 1647–1658. [Google Scholar] [CrossRef]
- Miguelez, C.; Berrocoso, E.; Mico, J.A.; Ugedo, L. L-DOPA modifies the antidepressant-like effects of reboxetine and fluoxetine in rats. Neuropharmacology 2013, 67, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Miguelez, C.; Aristieta, A.; Cenci, M.A.; Ugedo, L. The locus coeruleus is directly implicated in L-DOPA-induced dyskinesia in parkinsonian rats: An electrophysiological and behavioural study. PLoS ONE 2011, 6, e24679. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.S.; Wang, T.; Huang, C.; Liu, J. L-DOPA-induced dyskinesia in a rat model of Parkinson’s disease is associated with the fluctuational release of norepinephrine in the sensorimotor striatum. J. Neurosci. Res. 2014, 92, 1733–1745. [Google Scholar] [CrossRef]
- Bianco, L.E.; Wiesinger, J.; Earley, C.J.; Jones, B.C.; Beard, J.L. Iron deficiency alters dopamine uptake and response to L-DOPA injection in Sprague-Dawley rats. J. Neurochem. 2008, 106, 205–215. [Google Scholar] [CrossRef]
- Pascucci, T.; Giacovazzo, G.; Andolina, D.; Conversi, D.; Cruciani, F.; Cabib, S.; Puglisi-Allegra, S. In vivo catecholaminergic metabolism in the medial prefrontal cortex of ENU2 mice: An investigation of the cortical dopamine deficit in phenylketonuria. J. Inherit. Metab. Dis. 2012, 35, 1001–1009. [Google Scholar] [CrossRef] [Green Version]
- Dayan, L.; Finberg, J.P. L-DOPA increases noradrenaline turnover in central and peripheral nervous systems. Neuropharmacology 2003, 45, 524–533. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Kopin, I.J.; Goldstein, D.S. Catecholamine metabolism: A contemporary view with implications for physiology and medicine. Pharm. Rev. 2004, 56, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Chagraoui, A.; Whitestone, S.; Baassiri, L.; Manem, J.; Di Giovanni, G.; De Deurwaerdere, P. Neurochemical impact of the 5-HT2C receptor agonist WAY-163909 on monoamine tissue content in the rat brain. Neurochem. Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- Daws, L.C. Unfaithful neurotransmitter transporters: Focus on serotonin uptake and implications for antidepressant efficacy. Pharmacol. Ther. 2009, 121, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navailles, S.; De Deurwaerdere, P. Presynaptic control of serotonin on striatal dopamine function. Psychopharmacology 2011, 213, 213–242. [Google Scholar] [CrossRef] [PubMed]
- Sader-Mazbar, O.; Loboda, Y.; Rabey, M.J.; Finberg, J.P. Increased L-DOPA-derived dopamine following selective MAO-A or -B inhibition in rat striatum depleted of dopaminergic and serotonergic innervation. Br. J. Pharm. 2013, 170, 999–1013. [Google Scholar] [CrossRef] [Green Version]
- Gainetdinov, R.R.; Hoener, M.C.; Berry, M.D. Trace Amines and Their Receptors. Pharm. Rev. 2018, 70, 549–620. [Google Scholar] [CrossRef] [Green Version]
- Goshima, Y.; Masukawa, D.; Kasahara, Y.; Hashimoto, T.; Aladeokin, A.C. l-DOPA and Its Receptor GPR143: Implications for Pathogenesis and Therapy in Parkinson’s Disease. Front. Pharm. 2019, 10, 1119. [Google Scholar] [CrossRef] [Green Version]
- Misu, Y.; Goshima, Y. Is L-dopa an endogenous neurotransmitter? Trends Pharm. Sci. 1993, 14, 119–123. [Google Scholar]
- Misu, Y.; Goshima, Y.; Ueda, H.; Okamura, H. Neurobiology of L-DOPAergic systems. Prog. Neurobiol. 1996, 49, 415–454. [Google Scholar] [CrossRef]
- Ugrumov, M.; Taxi, J.; Pronina, T.; Kurina, A.; Sorokin, A.; Sapronova, A.; Calas, A. Neurons expressing individual enzymes of dopamine synthesis in the mediobasal hypothalamus of adult rats: Functional significance and topographic interrelations. Neuroscience 2014, 277, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Ugrumov, M.V. Non-dopaminergic neurons partly expressing dopaminergic phenotype: Distribution in the brain, development and functional significance. J. Chem. Neuroanat. 2009, 38, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Misu, Y.; Ueda, H.; Goshima, Y. Neurotransmitter-like actions of L-DOPA. Adv. Pharmacol. 1995, 32, 427–459. [Google Scholar] [PubMed]
- Goshima, Y.; Misu, Y.; Arai, N.; Misugi, K. Nanomolar L-dopa facilitates release of dopamine via presynaptic beta-adrenoceptors: Comparative studies on the actions in striatal slices from control and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated C57 black mice, an animal model for Parkinson’s disease. Jpn. J. Pharmacol. 1991, 55, 93–100. [Google Scholar] [PubMed]
- Hiroshima, Y.; Miyamoto, H.; Nakamura, F.; Masukawa, D.; Yamamoto, T.; Muraoka, H.; Kamiya, M.; Yamashita, N.; Suzuki, T.; Matsuzaki, S.; et al. The protein Ocular albinism 1 is the orphan GPCR GPR143 and mediates depressor and bradycardic responses to DOPA in the nucleus tractus solitarii. Br. J. Pharm. 2014, 171, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Lopez, V.M.; Decatur, C.L.; Stamer, W.D.; Lynch, R.M.; McKay, B.S. L-DOPA is an endogenous ligand for OA1. PLoS Biol. 2008, 6, e236. [Google Scholar] [CrossRef] [Green Version]
- Stamford, J.A.; Kruk, Z.L.; Millar, J. Striatal dopamine terminals release serotonin after 5-HTP pretreatment: In vivo voltammetric data. Brain Res. 1990, 515, 173–180. [Google Scholar] [CrossRef]
- Muller, T. Catechol-O-methyltransferase inhibitors in Parkinson’s disease. Drugs 2015, 75, 157–174. [Google Scholar] [CrossRef]
- Ries, V.; Selzer, R.; Eichhorn, T.; Oertel, W.H.; Eggert, K. Replacing a dopamine agonist by the COMT-inhibitor tolcapone as an adjunct to L-dopa in the treatment of Parkinson’s disease: A randomized, multicenter, open-label, parallel-group study. Clin. Neuropharmacol. 2010, 33, 142–150. [Google Scholar] [CrossRef]
- Fabbri, M.; Ferreira, J.J.; Lees, A.; Stocchi, F.; Poewe, W.; Tolosa, E.; Rascol, O. Opicapone for the treatment of Parkinson’s disease: A review of a new licensed medicine. Mov. Disord. 2018, 33, 1528–1539. [Google Scholar] [CrossRef]
- Lee, E.S.; Chen, H.; King, J.; Charlton, C. The role of 3-O-methyldopa in the side effects of L-dopa. Neurochem. Res. 2008, 33, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Onzawa, Y.; Kimura, Y.; Uzuhashi, K.; Shirasuna, M.; Hirosawa, T.; Taogoshi, T.; Kihira, K. Effects of 3-O-methyldopa, L-3,4-dihydroxyphenylalanine metabolite, on locomotor activity and dopamine turnover in rats. Biol. Pharm. Bull. 2012, 35, 1244–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asanuma, M.; Miyazaki, I. 3-O-Methyldopa inhibits astrocyte-mediated dopaminergic neuroprotective effects of L-DOPA. BMC Neurosci. 2016, 17, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.A.; Park, S.; Seo, O.N.; Jeong, S.W.; Lee, W.-K.; Kim, C.Y.; Kim, S.T.; Cho, M.J.; Shin, S.C. Development and validation of an LC–ESI–MS/MS method for simultaneous determination of levodopa, dopamine, L-α-methyldopa and 3-O-methyldopa in rat plasma. J. Pharm. Investig. 2012, 42, 361–368. [Google Scholar] [CrossRef]
- Sotnikova, T.D.; Beaulieu, J.M.; Espinoza, S.; Masri, B.; Zhang, X.; Salahpour, A.; Barak, L.S.; Caron, M.G.; Gainetdinov, R.R. The dopamine metabolite 3-methoxytyramine is a neuromodulator. PLoS ONE 2010, 5, e13452. [Google Scholar] [CrossRef]
- Alachkar, A.; Brotchie, J.M.; Jones, O.T. Binding of dopamine and 3-methoxytyramine as l-DOPA metabolites to human alpha(2)-adrenergic and dopaminergic receptors. Neurosci. Res. 2010, 67, 245–249. [Google Scholar] [CrossRef]
- Bunzow, J.R.; Sonders, M.S.; Arttamangkul, S.; Harrison, L.M.; Zhang, G.; Quigley, D.I.; Darland, T.; Suchland, K.L.; Pasumamula, S.; Kennedy, J.L.; et al. Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptor. Mol Pharm. 2001, 60, 1181–1188. [Google Scholar] [CrossRef]
- Charlton, C.G.; Crowell, B., Jr. Effects of dopamine metabolites on locomotor activities and on the binding of dopamine: Relevance to the side effects of L-dopa. Life Sci. 2000, 66, 2159–2171. [Google Scholar] [CrossRef]
- Nakazato, T.; Akiyama, A. Behavioral activity and stereotypy in rats induced by L-DOPA metabolites: A possible role in the adverse effects of chronic L-DOPA treatment of Parkinson’s disease. Brain Res. 2002, 930, 134–142. [Google Scholar] [CrossRef]
- Solis, O.; Garcia-Montes, J.R.; Garcia-Sanz, P.; Herranz, A.S.; Asensio, M.J.; Kang, G.; Hiroi, N.; Moratalla, R. Human COMT over-expression confers a heightened susceptibility to dyskinesia in mice. Neurobiol. Dis. 2017, 102, 133–139. [Google Scholar] [CrossRef]
- De Deurwaerdère, P.; Di Giovanni, G. Chronic L-DOPA alters the activity of monoamine oxidase in vivo. In Proceedings of the Neuropathology and Neuropharmacology of Monoaminergic System, Bordeaux, France, 8–10 October 2014. [Google Scholar]
- Grandy, D.K. Trace amine-associated receptor 1-Family archetype or iconoclast? Pharmacol. Ther. 2007, 116, 355–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, G.; Pizzolato, G.; Gucciardi, A.; Stocchero, M.; Giordano, G.; Baraldi, E.; Leon, A. Different Circulating Trace Amine Profiles in De Novo and Treated Parkinson’s Disease Patients. Sci. Rep. 2019, 9, 6151. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, G.; Nordera, G.; Pizzolato, G.; Bolner, A.; Colavito, D.; Flaibani, R.; Leon, A. Trace amine metabolism in Parkinson’s disease: Low circulating levels of octopamine in early disease stages. Neurosci. Lett. 2010, 469, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrov, A.A.; Polyakova, N.V.; Vinogradova, E.P.; Gainetdinov, R.R.; Knyazeva, V.M. The TAAR5 agonist alpha-NETA causes dyskinesia in mice. Neurosci. Lett. 2019, 704, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Alvarsson, A.; Zhang, X.; Stan, T.L.; Schintu, N.; Kadkhodaei, B.; Millan, M.J.; Perlmann, T.; Svenningsson, P. Modulation by Trace Amine-Associated Receptor 1 of Experimental Parkinsonism, L-DOPA Responsivity, and Glutamatergic Neurotransmission. J. Neurosci. 2015, 35, 14057–14069. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, S.; Manago, F.; Leo, D.; Sotnikova, T.D.; Gainetdinov, R.R. Role of catechol-O-methyltransferase (COMT)-dependent processes in Parkinson’s disease and L-DOPA treatment. CNS Neurol. Disord. Drug Targets 2012, 11, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Dyck, L.E. Release of some endogenous trace amines from rat striatal slices in the presence and absence of a monoamine oxidase inhibitor. Life Sci. 1989, 44, 1149–1156. [Google Scholar] [CrossRef]
- Beaudoin-Gobert, M.; Metereau, E.; Duperrier, S.; Thobois, S.; Tremblay, L.; Sgambato, V. Pathophysiology of levodopa-induced dyskinesia: Insights from multimodal imaging and immunohistochemistry in non-human primates. Neuroimage 2018, 183, 132–141. [Google Scholar] [CrossRef]
- Stansley, B.J.; Yamamoto, B.K. Chronic L-dopa decreases serotonin neurons in a subregion of the dorsal raphe nucleus. J. Pharm. Exp. Ther. 2014, 351, 440–447. [Google Scholar] [CrossRef] [Green Version]
- Stansley, B.J.; Yamamoto, B.K. Behavioral impairments and serotonin reductions in rats after chronic L-dopa. Psychopharmacology 2015, 232, 3203–3213. [Google Scholar] [CrossRef]
- Pregeljc, D.; Teodorescu-Perijoc, D.; Vianello, R.; Umek, N.; Mavri, J. How Important Is the Use of Cocaine and Amphetamines in the Development of Parkinson Disease? A Computational Study. Neurotox. Res. 2019. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chagraoui, A.; Boulain, M.; Juvin, L.; Anouar, Y.; Barrière, G.; Deurwaerdère, P.D. L-DOPA in Parkinson’s Disease: Looking at the “False” Neurotransmitters and Their Meaning. Int. J. Mol. Sci. 2020, 21, 294. https://doi.org/10.3390/ijms21010294
Chagraoui A, Boulain M, Juvin L, Anouar Y, Barrière G, Deurwaerdère PD. L-DOPA in Parkinson’s Disease: Looking at the “False” Neurotransmitters and Their Meaning. International Journal of Molecular Sciences. 2020; 21(1):294. https://doi.org/10.3390/ijms21010294
Chicago/Turabian StyleChagraoui, Abdeslam, Marie Boulain, Laurent Juvin, Youssef Anouar, Grégory Barrière, and Philippe De Deurwaerdère. 2020. "L-DOPA in Parkinson’s Disease: Looking at the “False” Neurotransmitters and Their Meaning" International Journal of Molecular Sciences 21, no. 1: 294. https://doi.org/10.3390/ijms21010294