PIP2 Mediated Inhibition of TREK Potassium Currents by Bradykinin in Mouse Sympathetic Neurons

 , , and
, , and 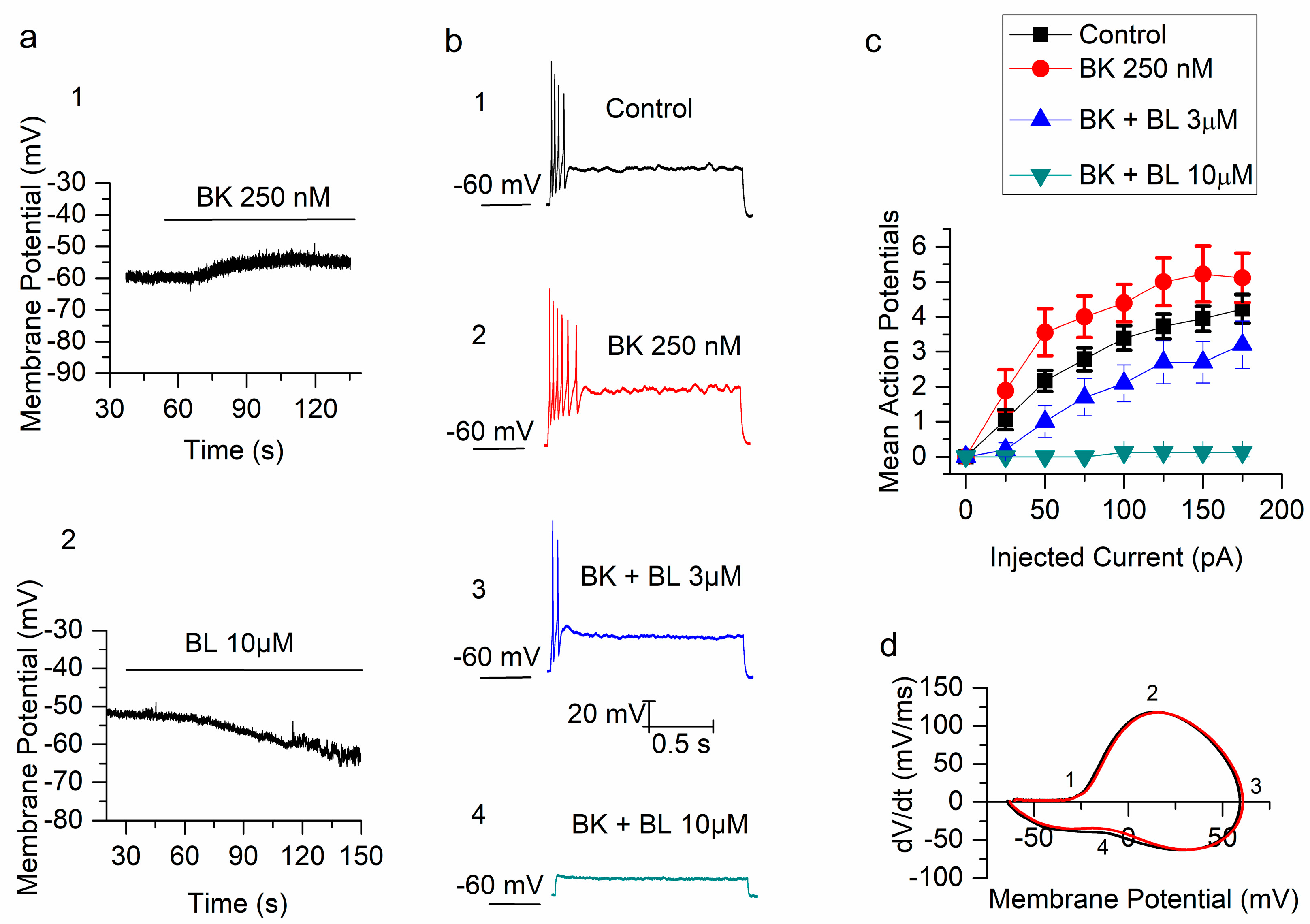{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Bradykinin Increases the Excitability of mSCG Neurons
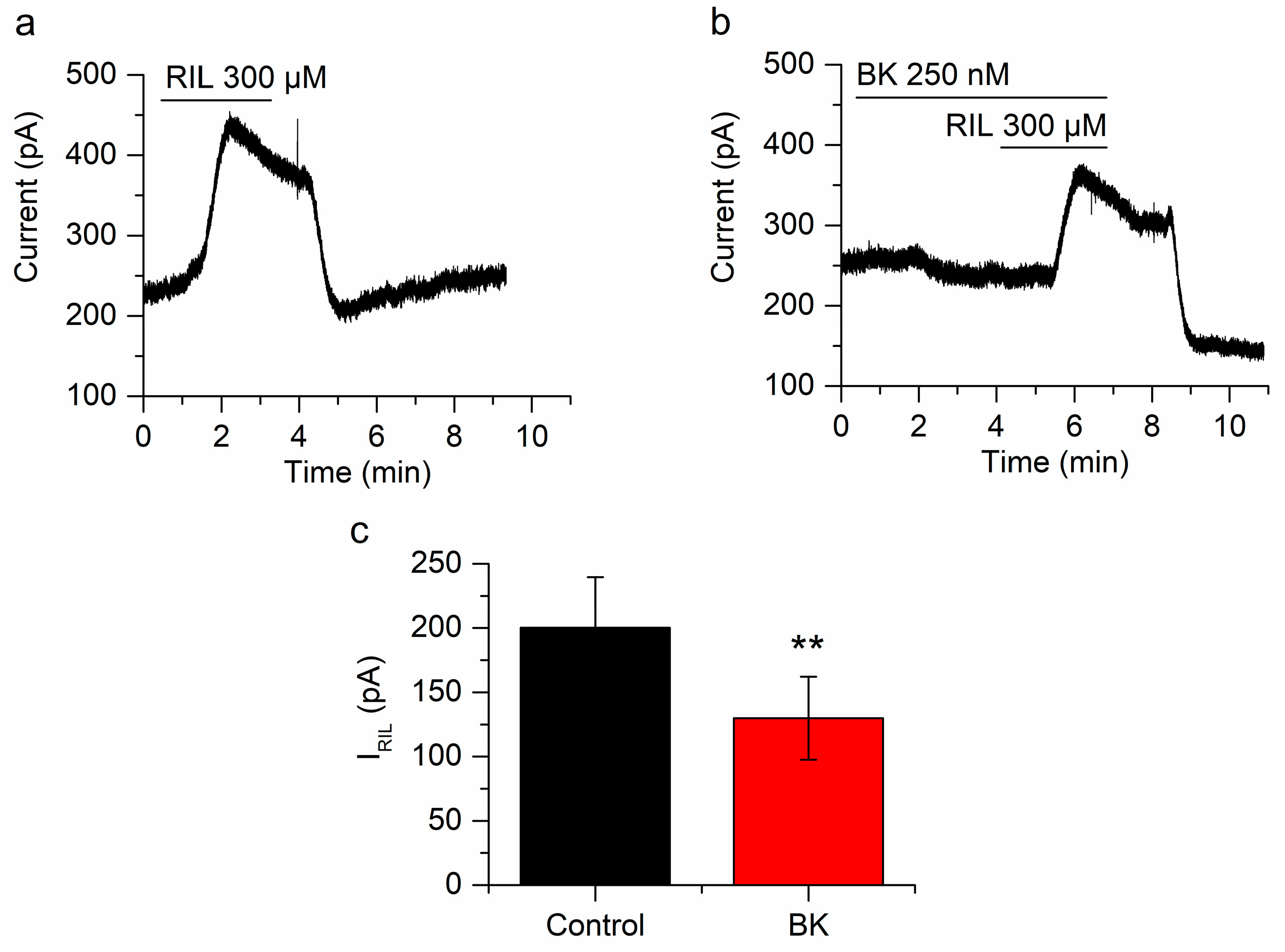
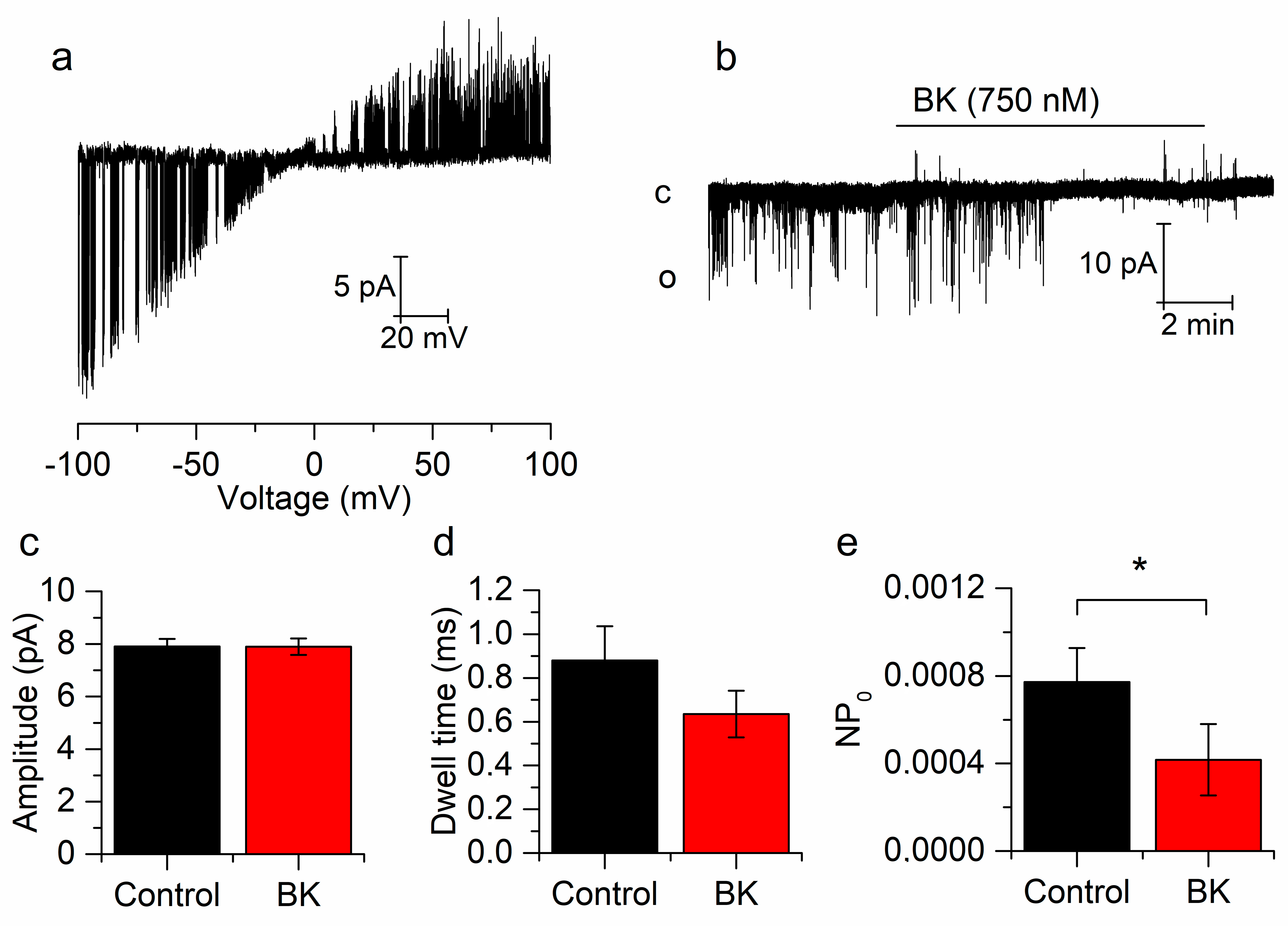
2.2. Bradykinin Inhibits Whole-Cell and Single-Channel TREK-2 Currents
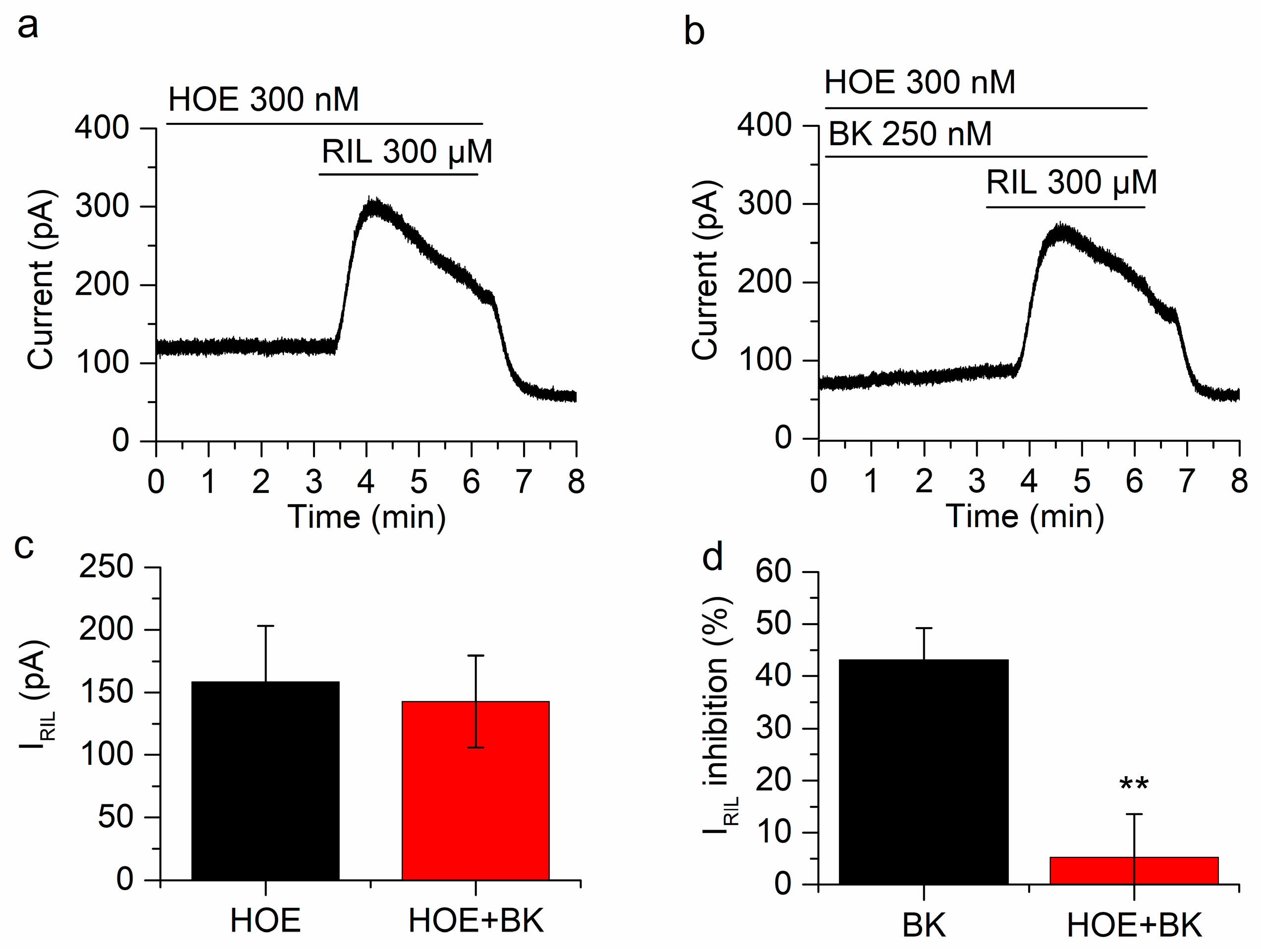
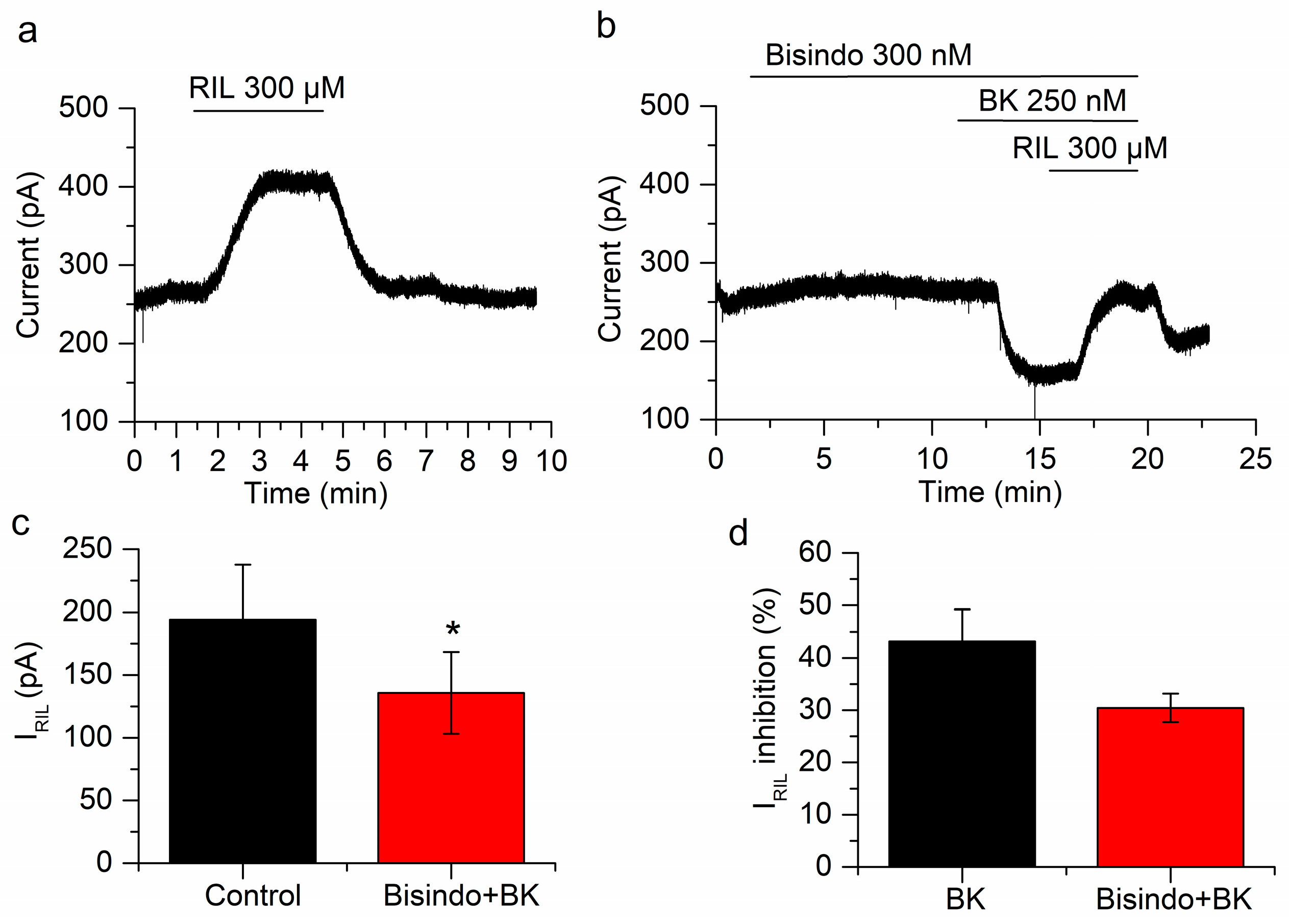
2.3. The Inhibition of IRIL by BK Is Mediated through B2 Receptors
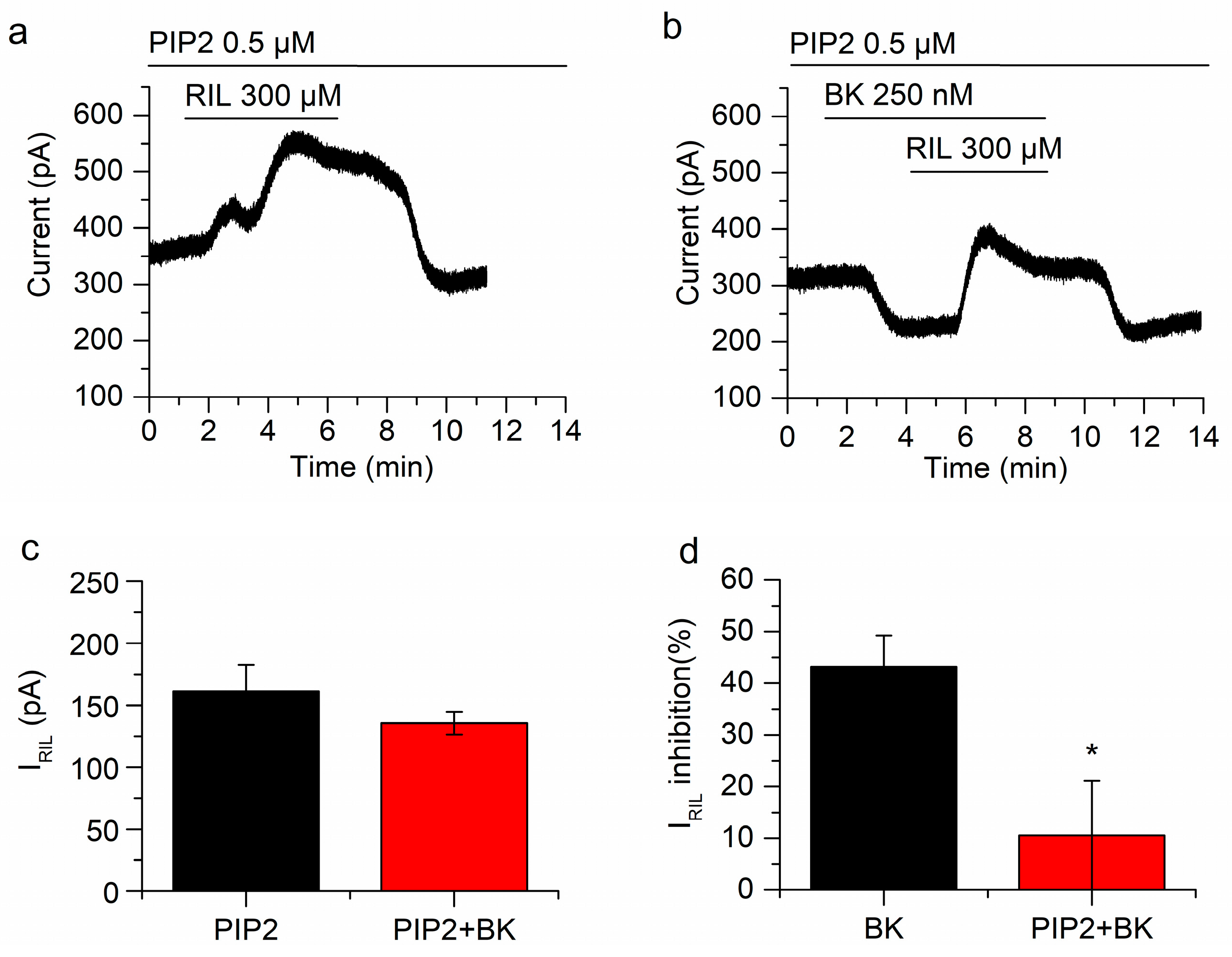
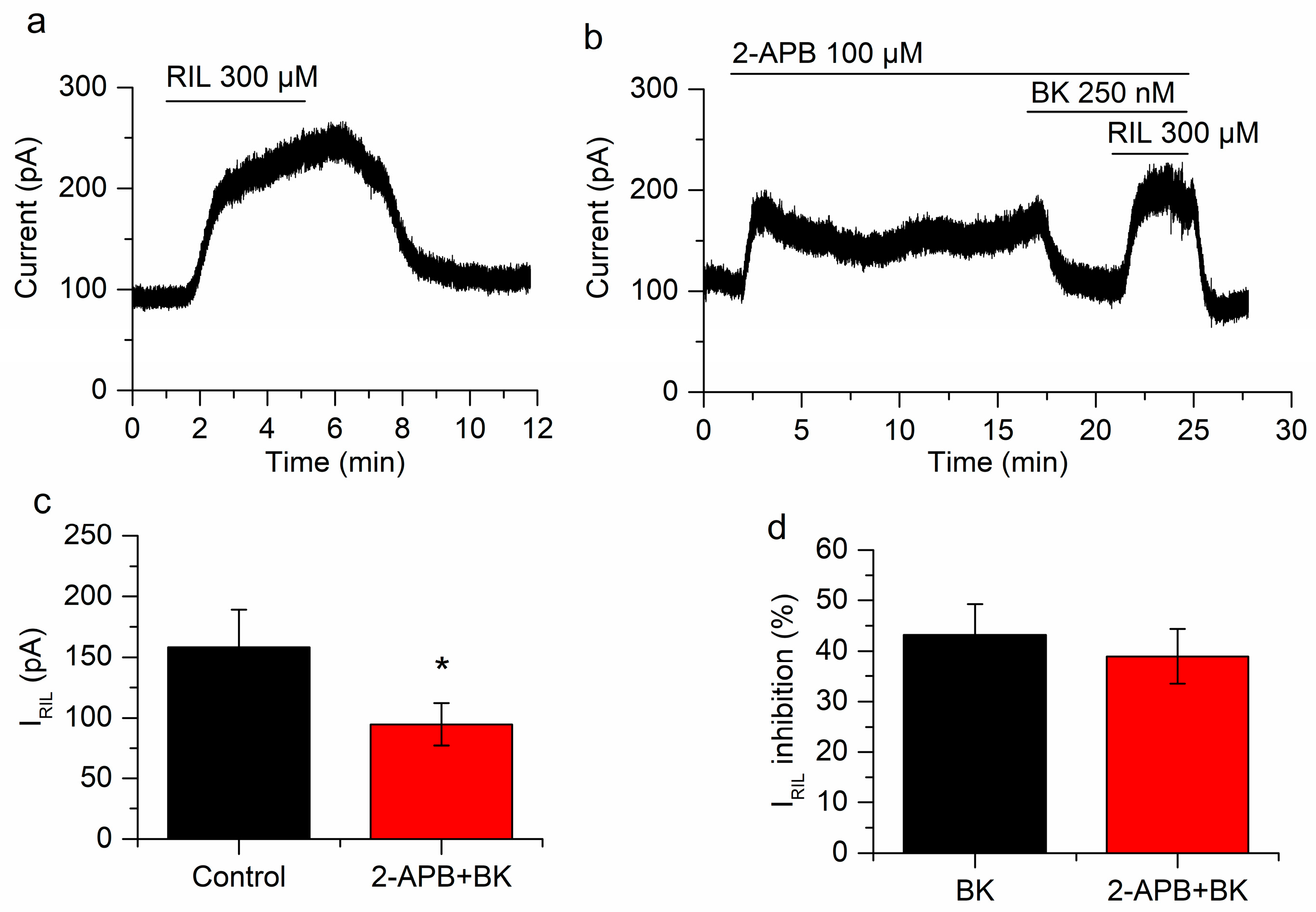
2.4. Manipulating PIP2 Concentration Affects the Inhibition of IRIL by Bradykinin
2.5. Neither Protein Kinase C nor Ca2+ Are Involved in the Inhibition of IRIL by Bradykinin
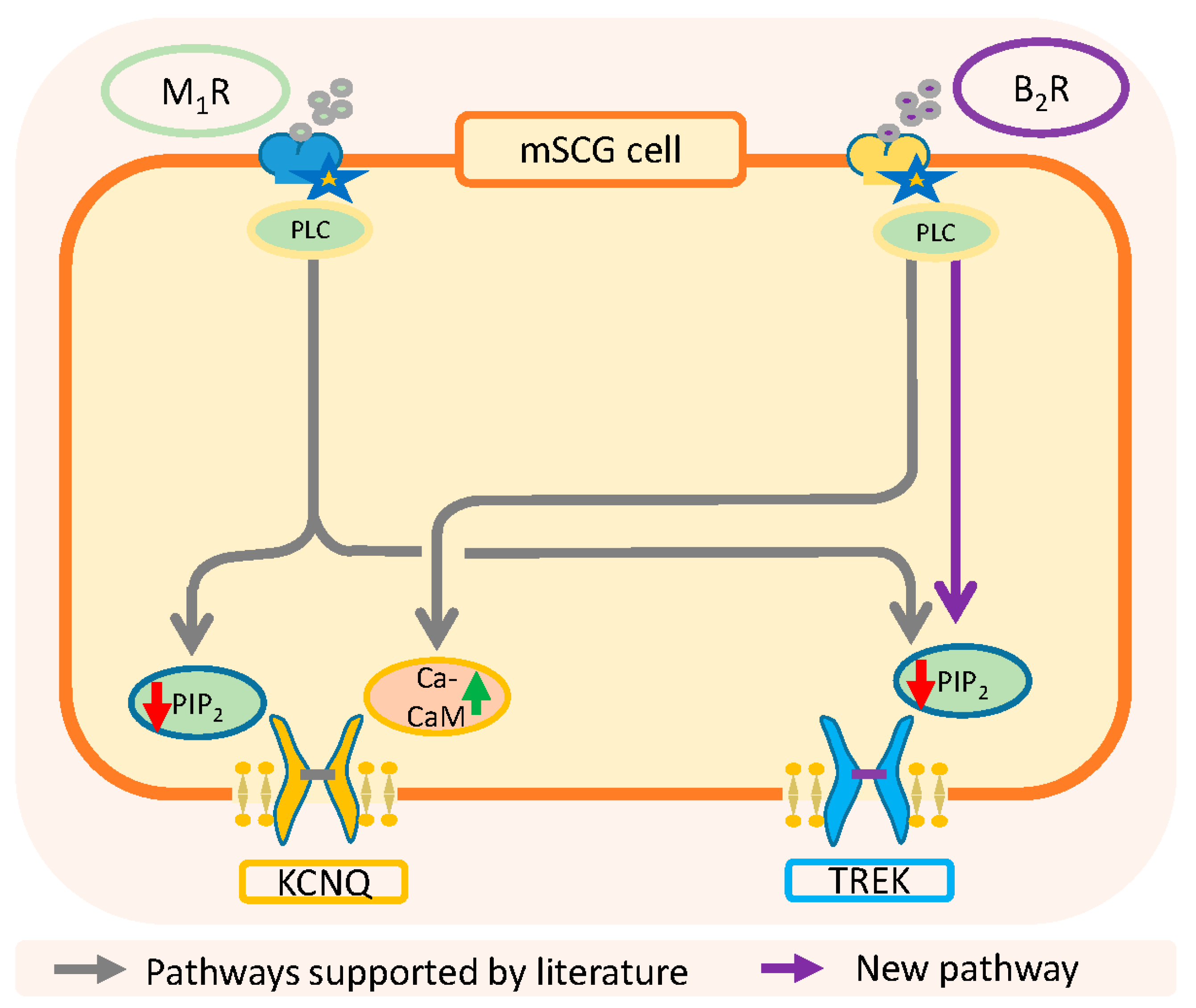
3. Discussion
3.1. I-30 Includes ITREK and IM
3.2. IRIL is Due to the Activation of ITREK
3.3. I-30 versus ITREK
3.4. Physiological Relevance and Conclusions
4. Materials and Methods
4.1. Cell Culture
4.2. Perforated-Patch Recording
4.3. Single-Channel Recording
4.4. Drugs
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2-APB | 2-aminoethoxydiphenylborane |
| BK | bradykinin |
| BL | BL1249 |
| BRs | bradykinin receptors |
| B1R | bradykinin receptor 1 |
| B2R | bradykinin receptor 2 |
| DAG | diacylglycerol |
| DRG | dorsal root ganglia |
| HOE | HOE-140. d-Arg-[Hyp3, Thi5, d-Tic7, Oic8]BK |
| Ih | hyperpolarization activated cationic current |
| IM | potassium M-current |
| IP3 | inositol triphosphate |
| IP3R | inositol triphosphate receptor |
| IRIL | riluzole-activated current |
| ITREK | potassium current through TREK channels |
| I-30 | basal outward current at -30 mV |
| K2P | two pore domain potassium channels |
| mSCG | mouse superior cervical ganglion |
| NPo | open probability |
| PIP2 | phosphatidylinositol 4,5-bisphosphate |
| PKC | protein kinase C |
| PLC | phospholipase C |
| RMP | resting membrane potential |
| rSCG | Rat superior cervical ganglion |
| SCG | superior cervical ganglion |
| SFA | spike frequency adaptation |
| TEA TRAAK | Tetraethylammonium TWIK-related arachidonic acid-stimulated potassium channel |
| TREK TRESK TWIK | TWIK-related potassium channel TWIK-related spinal cord potassium channel Tandem of pore-domains in a Weakly Inward rectifying K+ channel |
| TTX | tetrodotoxin |
References
- Greaves, M.; Shuster, S. Responses of skin blood vessels to bradykinin, histamine and 5-hydroxytryptamine. J. Physiol Lond. 1967, 193, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Dray, A.; Bettaney, J.; Forster, P.; Perkins, M.N. Activation of a bradykinin receptor in peripheral nerve and spinal cord in the neonatal rat in vitro. Br. J. Pharmacol. 1988, 95, 1008–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.L.; Naeem, S.; Phagoo, S.B.; Campbell, E.A.; Urban, L.; Burgess, G.M. B1 bradykinin receptors and sensory neurones. Br. J. Pharmacol. 1996, 118, 1469–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babbedge, R.; Dray, A.; Urban, L. Bradykinin depolarises the rat isolated superior cervical ganglion via B2 receptor activation. Neurosci. Lett. 1995, 193, 161–164. [Google Scholar] [CrossRef]
- Prado, G.N.; Taylor, L.; Zhou, X.; Ricupero, D.; Mierke, D.F.; Polgar, P. Mechanisms regulating the expression, self-maintenance, and signaling-function of the bradykinin B2 and B1 receptors. J. Cell Physiol. 2002, 193, 275–286. [Google Scholar] [CrossRef]
- Bascands, J.L.; Emond, C.; Pecher, C.; Regoli, D.; Girolami, J.P. Bradykinin stimulates production of inositol (1,4,5) trisphosphate in cultured mesangial cells of the rat via a BK2-kinin receptor. Br. J. Pharmacol. 1991, 102, 962–965. [Google Scholar] [CrossRef] [Green Version]
- Innis, R.B.; Manning, D.C.; Stewart, J.M.; Snyder, S.H. [3H]Bradykinin receptor binding in mammalian tissue membranes. Proc. Natl. Acad. Sci. USA 1981, 78, 2630–2634. [Google Scholar] [CrossRef] [Green Version]
- Regoli, D.; Barabe, J. Pharmacology of bradykinin and related kinins. Pharmacol. Rev. 1980, 32, 1–46. [Google Scholar]
- McGuire, C.; Boundouki, G.; Hockley, J.R.F.; Reed, D.; Cibert-Goton, V.; Peiris, M.; Kung, V.; Broad, J.; Aziz, Q.; Chan, C.; et al. Ex vivo study of human visceral nociceptors. Gut 2018, 67, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Brunsden, A.M.; Grundy, D. Sensitization of visceral afferents to bradykinin in rat jejunum in vitro. J. Physiol Lond. 1999, 521, 517–527. [Google Scholar] [CrossRef]
- Lewis, G.P.; Reit, E. The action of angiotensin and bradykinin on the superior cervical ganglion of the cat. J. Physiol Lond. 1965, 179, 538–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Brown, D.A.; Milligan, G.; Willer, E.; Buckley, N.J.; Caulfield, M.P. Bradykinin excites rat sympathetic neurons by inhibition of M current through a mechanism involving B2 Receptors and Gaq/11. Neuron 1995, 14, 399–405. [Google Scholar] [CrossRef] [Green Version]
- Seabrook, G.R.; Bowery, B.J.; Hill, R.G. Bradykinin receptors in mouse and rat isolated superior cervical ganglia. Br. J. Pharmacol. 1995, 115, 368–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaika, O.; Tolstykh, G.P.; Jaffe, D.B.; Shapiro, M.S. Inositol triphosphate-mediated Ca2+ signals direct purinergic P2Y receptor regulation of neuronal ion channels. J. Neurosci. 2007, 27, 8914–8926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seabrook, G.R.; Bowery, B.J.; Heavens, R.; Brown, N.; Ford, H.; Sirinathsinghi, D.J.; Borkowski, J.A.; Hess, J.F.; Strader, C.D.; Hill, R.G. Expression of B1 and B2 bradykinin receptor mRNA and their functional roles in sympathetic ganglia and sensory dorsal root ganglia neurones from wild-type and B2 receptor knockout mice. Neuropharmacology 1997, 36, 1009–1017. [Google Scholar] [CrossRef]
- Borkowski, J.A.; Ransom, R.W.; Seabrook, G.R.; Trumbauer, M.; Chen, H.; Hill, R.G.; Strader, C.D.; Hess, J.F. Targeted disruption of a B2 bradykinin receptor gene in mice eliminates bradykinin action in smooth muscle and neurons. J. Biol. Chem. 1995, 270, 13706–13710. [Google Scholar] [CrossRef] [Green Version]
- Levine, J.D.; Taiwo, Y.O.; Collins, S.D.; Tam, J.K. Noradrenaline hyperalgesia is mediated through interaction with sympathetic postganglionic neurone terminals rather than activation of primary afferent nociceptors. Nature 1986, 323, 158–160. [Google Scholar] [CrossRef]
- Haley, J.E.; Delmas, P.; Offermanns, S.; Abogadie, F.C.; Simon, M.I.; Buckley, N.J.; Brown, D.A. Muscarinic inhibition of calcium current and M current in Galpha q-deficient mice. J. Neurosci. 2000, 20, 3973–3979. [Google Scholar] [CrossRef] [Green Version]
- Cruzblanca, H.; Koh, D.S.; Hille, B. Bradykinin inhibits M current via phospholipase C and Ca2+ release from IP3-sensitive Ca2+ stores in rat sympathetic neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 7151–7156. [Google Scholar] [CrossRef] [Green Version]
- Haley, J.E.; Abogadie, F.C.; Fernandez, F.; Dayrell, M.; Vallis, Y.; Buckley, N.J.; Brown, D.A. Bradykinin, but not muscarinic, inhibition of M-current in rat sympathetic ganglion neurons involves phospholipase C-beta 4. J. Neurosci. 2000, 20, RC105. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.A.; Abogadie, F.C.; Allen, T.G.; Buckley, N.J.; Caulfield, M.P.; Delmas, P.; Haley, J.E.; Lamas, J.A.; Selyanko, A.A. Muscarinic mechanisms in nerve cells. Life Sci. 1997, 60, 1137–1144. [Google Scholar] [CrossRef]
- Romero, M.; Reboreda, A.; Sanchez, E.; Lamas, J.A. Newly developed blockers of the M-current do not reduce spike frequency adaptation in cultured mouse sympathetic neurons. Eur. J. Neurosci. 2004, 19, 2693–2702. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Constanti, A. Intracellular observations on the effects of muscarinic agonists on rat sympathetic neurones. Br. J. Pharmacol. 1980, 70, 593–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.A.; Adams, P.R. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 1980, 283, 673–676. [Google Scholar] [CrossRef]
- Rivas-Ramirez, P.; Cadaveira-Mosquera, A.; Lamas, J.A.; Reboreda, A. Muscarinic modulation of TREK currents in mouse sympathetic superior cervical ganglion neurons. Eur. J. Neurosci. 2015, 42, 1797–1807. [Google Scholar] [CrossRef]
- Cadaveira-Mosquera, A.; Ribeiro, S.J.; Reboreda, A.; Perez, M.; Lamas, J.A. Activation of TREK currents by the neuroprotective agent riluzole in mouse sympathetic neurons. J. Neurosci. 2011, 31, 1375–1385. [Google Scholar] [CrossRef]
- Cadaveira-Mosquera, A.; Perez, M.; Reboreda, A.; Rivas-Ramirez, P.; Fernandez-Fernandez, D.; Lamas, J.A. Expression of K2P channels in sensory and motor neurons of the autonomic nervous system. J. Mol. Neurosci. 2012, 48, 86–96. [Google Scholar] [CrossRef]
- Maingret, F.; Lauritzen, I.; Patel, A.J.; Heurteaux, C.; Reyes, R.; Lesage, F.; Lazdunski, M.; Honore, E. TREK-1 is a heat-activated background K+ channel. EMBO J. 2000, 19, 2483–2491. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Shepherd, N.; Creazzo, T.L. Temperature-sensitive TREK currents contribute to setting the resting membrane potential in embryonic atrial myocytes. J. Physiol Lond. 2008, 586, 3645–3656. [Google Scholar] [CrossRef]
- Lamas, J.A.; Rueda-Ruzafa, L.; Herrera-Perez, S. Ion Channels and Thermosensitivity: TRP, TREK, or Both? Int. J. Mol. Sci. 2019, 20, 2371. [Google Scholar] [CrossRef] [Green Version]
- Duprat, F.; Lesage, F.; Patel, A.J.; Fink, M.; Romey, G.; Lazdunski, M. The neuroprotective agent riluzole activates the two P domain K(+) channels TREK-1 and TRAAK. Mol. Pharmacol. 2000, 57, 906–912. [Google Scholar] [PubMed]
- Lesage, F.; Terrenoire, C.; Romey, G.; Lazdunski, M. Human TREK2, a 2P domain mechano-sensitive K+ channel with multiple regulations by polyunsaturated fatty acids, lysophospholipids, and Gs, Gi, and Gq protein-coupled receptors. J. Biol. Chem. 2000, 275, 28398–28405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, M.; Lesage, F.; Duprat, F.; Heurteaux, C.; Reyes, R.; Fosset, M.; Lazdunski, M. A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J. 1998, 17, 3297–3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamas, J.A.; Fernandez-Fernandez, D. Tandem pore TWIK-related potassium channels and neuroprotection. Neural Regen. Res. 2019, 14, 1293–1308. [Google Scholar] [CrossRef]
- Lamas, J.A. Mechanosensitive K2P channels, TREKking through the autonomic nervous system. In Mechanically Gated Channels and their Regulation; Kamkin, A., Lozinsky, I., Eds.; Springer Science & Business Media: Dordrecht, The Netherlands, 2012; Volume 6, pp. 35–68. [Google Scholar]
- Chemin, J.; Girard, C.; Duprat, F.; Lesage, F.; Romey, G.; Lazdunski, M. Mechanisms underlying excitatory effects of group I metabotropic glutamate receptors via inhibition of 2P domain K+ channels. EMBO J. 2003, 22, 5403–5411. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.; Han, J.; Kim, D. Mechanism of inhibition of TREK-2 (K2P10.1) by the Gq-coupled M3 muscarinic receptor. Am. J. Physiol. Cell Physiol. 2006, 291, C649–C656. [Google Scholar] [CrossRef]
- Cohen, A.; Sagron, R.; Somech, E.; Segal-Hayoun, Y.; Zilberberg, N. Pain-associated signals, acidosis and lysophosphatidic acid, modulate the neuronal K(2P)2.1 channel. Mol. Cell Neurosci. 2009, 40, 382–389. [Google Scholar] [CrossRef]
- Xiao, Z.; Deng, P.Y.; Rojanathammanee, L.; Yang, C.; Grisanti, L.; Permpoonputtana, K.; Weinshenker, D.; Doze, V.A.; Porter, J.E.; Lei, S. Noradrenergic depression of neuronal excitability in the entorhinal cortex via activation of TREK-2 K+ channels. J. Biol. Chem. 2009, 284, 10980–10991. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.I.; Hwang, S.W. Depolarizing Effectors of Bradykinin Signaling in Nociceptor Excitation in Pain Perception. Biomol. Ther. (Seoul) 2018, 26, 255–267. [Google Scholar] [CrossRef]
- Garry, A.; Fromy, B.; Blondeau, N.; Henrion, D.; Brau, F.; Gounon, P.; Guy, N.; Heurteaux, C.; Lazdunski, M.; Saumet, J.L. Altered acetylcholine, bradykinin and cutaneous pressure-induced vasodilation in mice lacking the TREK1 potassium channel: The endothelial link. EMBO Rep. 2007, 8, 354–359. [Google Scholar] [CrossRef]
- Lamas, J.A. A hyperpolarization-activated cation current (I-h) contributes to resting membrane potential in rat superior cervical sympathetic neurones. Pflugers Arch. Eur. J. Physiol. 1998, 436, 429–435. [Google Scholar] [CrossRef]
- Hadley, J.K.; Noda, M.; Selyanko, A.A.; Wood, I.C.; Abogadie, F.C.; Brown, D.A. Differential tetraethylammonium sensitivity of KCNQ1-4 potassium channels. Br. J. Pharmacol. 2000, 129, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Lamas, J.A.; Romero, M.; Reboreda, A.; Sanchez, E.; Ribeiro, S.J. A riluzole- and valproate-sensitive persistent sodium current contributes to the resting membrane potential and increases the excitability of sympathetic neurones. Pflugers Arch. Eur. J. Physiol. 2009, 458, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Pope, L.; Arrigoni, C.; Lou, H.; Bryant, C.; Gallardo-Godoy, A.; Renslo, A.R.; Minor, D.L., Jr. Protein and Chemical Determinants of BL-1249 Action and Selectivity for K2P Channels. ACS Chem. Neurosci. 2018, 9, 3153–3165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tertyshnikova, S.; Knox, R.J.; Plym, M.J.; Thalody, G.; Griffin, C.; Neelands, T.; Harden, D.G.; Signor, L.; Weaver, D.; Myers, R.A.; et al. BL-1249 [(5,6,7,8-tetrahydro-naphthalen-1-yl)-[2-(1H-tetrazol-5-yl)-phenyl]-amine]: A putative potassium channel opener with bladder-relaxant properties. J. Pharmacol. Exp. Ther. 2005, 313, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Veale, E.L.; Al-Moubarak, E.; Bajaria, N.; Omoto, K.; Cao, L.; Tucker, S.J.; Stevens, E.B.; Mathie, A. Influence of the N terminus on the biophysical properties and pharmacology of TREK1 potassium channels. Mol. Pharmacol. 2014, 85, 671–681. [Google Scholar] [CrossRef]
- Yano, K.; Higashida, H.; Hattori, H.; Nozawa, Y. Bradykinin-induced transient accumulation of inositol trisphosphate in neuron-like cell line NG108-15 cells. FEBS Lett. 1985, 181, 403–406. [Google Scholar] [CrossRef] [Green Version]
- Tippmer, S.; Quitterer, U.; Kolm, V.; Faussner, A.; Roscher, A.; Mosthaf, L.; Muller-Esterl, W.; Haring, H. Bradykinin induces translocation of the protein kinase C isoforms alpha, epsilon, and zeta. Eur. J. Biochem. 1994, 225, 297–304. [Google Scholar] [CrossRef]
- Delmas, P.; Brown, D.A. Pathways modulating neural KCNQ/M (Kv7) potassium. Nat. Rev. Neurosci. 2005, 6, 850–862. [Google Scholar] [CrossRef]
- Beltran, L.; Beltran, M.; Aguado, A.; Gisselmann, G.; Hatt, H. 2-Aminoethoxydiphenyl borate activates the mechanically gated human KCNK channels KCNK 2 (TREK-1), KCNK 4 (TRAAK), and KCNK 10 (TREK-2). Front. Pharmacol. 2013, 4, 63. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, C.C.; Zaika, O.; Tolstykh, G.P.; Shapiro, M.S. Regulation of neural KCNQ channels: Signalling pathways, structural motifs and functional implications. J. Physiol. 2008, 586, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Lamas, J.A. The role of calcium in M-current inhibition by muscarinic agonists in rat sympathetic neurons. Neuroreport 1999, 10, 2395–2400. [Google Scholar] [CrossRef] [PubMed]
- Kosenko, A.; Hoshi, N. A change in configuration of the calmodulin-KCNQ channel complex underlies Ca2+-dependent modulation of KCNQ channel activity. PLoS ONE 2013, 8, e82290. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, A.; Gomis-Perez, C.; Bernardo-Seisdedos, G.; Alaimo, A.; Malo, C.; Aldaregia, J.; Lopez-Robles, C.; Areso, P.; Butz, E.; Wahl-Schott, C.; et al. Uncoupling PIP2-calmodulin regulation of Kv7.2 channels by an assembly destabilizing epileptogenic mutation. J. Cell. Sci. 2015, 128, 4014–4023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmas, P.; Wanaverbecq, N.; Abogadie, F.C.; Mistry, M.; Brown, D.A. Signaling microdomains define the specificity of receptor-mediated InsP(3) pathways in neurons. Neuron 2002, 34, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Craciun, L.C.; Mirshahi, T.; Rohacs, T.; Lopes, C.M.; Jin, T.; Logothetis, D.E. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 2003, 37, 963–975. [Google Scholar] [CrossRef] [Green Version]
- Gamper, N.; Reznikov, V.; Yamada, Y.; Yang, J.; Shapiro, M.S. Phosphatidylinositol 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J. Neurosci. 2004, 24, 10980–10992. [Google Scholar] [CrossRef] [Green Version]
- Zaika, O.; Zhang, J.; Shapiro, M.S. Combined Phosphoinositide and Ca2+ Signals Mediating Receptor Specificity toward Neuronal Ca2+ Channels. J. Biol. Chem. 2011, 286, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Noel, J.; Sandoz, G.; Lesage, F. Molecular regulations governing TREK and TRAAK channel functions. Channels (Austin) 2011, 5, 402–409. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.J.; Honore, E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci. 2001, 24, 339–346. [Google Scholar] [CrossRef]
- Alloui, A.; Zimmermann, K.; Mamet, J.; Duprat, F.; Noel, J.; Chemin, J.; Guy, N.; Blondeau, N.; Voilley, N.; Rubat-Coudert, C.; et al. TREK-1, a K+ channel involved in polymodal pain perception. EMBO J. 2006, 25, 2368–2376. [Google Scholar] [CrossRef] [Green Version]
- Acosta, C.; Djouhri, L.; Watkins, R.; Berry, C.; Bromage, K.; Lawson, S.N. TREK2 expressed selectively in IB4-binding C-fiber nociceptors hyperpolarizes their membrane potentials and limits spontaneous pain. J. Neurosci. 2014, 34, 1494–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.; Hatakeyama, T.; Taniguchi, K. Immunohistochemical colocalization of TREK-1, TREK-2 and TRAAK with TRP channels in the trigeminal ganglion cells. Neurosci. Lett. 2009, 454, 129–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathie, A.; Veale, E.L. Two-pore domain potassium channels: Potential therapeutic targets for the treatment of pain. Pflugers Arch. 2015, 467, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V.; Busserolles, J.; Christin, M.; Devilliers, M.; Poupon, L.; Legha, W.; Alloui, A.; Aissouni, Y.; Bourinet, E.; Lesage, F.; et al. Role of the TREK2 potassium channel in cold and warm thermosensation and in pain perception. Pain 2014, 155, 2534–2544. [Google Scholar] [CrossRef]
- Vivier, D.; Soussia, I.B.; Rodrigues, N.; Lolignier, S.; Devilliers, M.; Chatelain, F.C.; Prival, L.; Chapuy, E.; Bourdier, G.; Bennis, K.; et al. Development of the first two-pore domain potassium channel TWIK-related K(+) channel 1-selective agonist possessing in vivo antinociceptive activity. J. Med. Chem. 2017, 60, 1076–1088. [Google Scholar] [CrossRef]
- Devilliers, M.; Busserolles, J.; Lolignier, S.; Deval, E.; Pereira, V.; Alloui, A.; Christin, M.; Mazet, B.; Delmas, P.; Noel, J.; et al. Activation of TREK-1 by morphine results in analgesia without adverse side effects. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Veale, E.L.; Mathie, A. Aristolochic acid, a plant extract used in the treatment of pain and linked to Balkan endemic nephropathy, is a regulator of K2P channels. Br. J. Pharmacol. 2016, 173, 1639–1652. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Hao, H.; Gigout, S.; Huang, D.; Yang, Y.; Li, L.; Wang, C.; Sundt, D.; Jaffe, D.B.; Zhang, H.; et al. Control of somatic membrane potential in nociceptive neurons and its implications for peripheral nociceptive transmission. Pain 2014, 155, 2306–2322. [Google Scholar] [CrossRef] [Green Version]
- Cameron, K.; Bartle, E.; Roark, R.; Fanelli, D.; Pham, M.; Pollard, B.; Borkowski, B.; Rhoads, S.; Kim, J.; Rocha, M.; et al. Neurosteroid binding to the amino terminal and glutamate binding domains of ionotropic glutamate receptors. Steroids 2012, 77, 774–779. [Google Scholar] [CrossRef]
- Kang, D.; Kim, D. TREK-2 (K2P10.1) and TRESK (K2P18.1) are major background K+ channels in dorsal root ganglion neurons. Am. J. Physiol. Cell Physiol. 2006, 291, C138–C146. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Choe, C.; Kim, D. Thermosensitivity of the two-pore domain K+ channels TREK-2 and TRAAK. J. Physiol. 2005, 564, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Chemin, J.; Patel, A.J.; Duprat, F.; Lauritzen, I.; Lazdunski, M.; Honoré, E. A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J. 2005, 24, 44–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoz, G.; Bell, S.C.; Isacoff, E.Y. Optical probing of a dynamic membrane interaction that regulates the TREK1 channel. Proc. Natl. Acad. Sci. USA 2011, 108, 2605–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Pinna, J.; Lamas, J.A.; Gallego, R. Calcium current components in intact and dissociated adult mouse sympathetic neurons. Brain Res. 2002, 951, 227–236. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivas-Ramírez, P.; Reboreda, A.; Rueda-Ruzafa, L.; Herrera-Pérez, S.; Lamas, J.A. PIP2 Mediated Inhibition of TREK Potassium Currents by Bradykinin in Mouse Sympathetic Neurons. Int. J. Mol. Sci. 2020, 21, 389. https://doi.org/10.3390/ijms21020389
Rivas-Ramírez P, Reboreda A, Rueda-Ruzafa L, Herrera-Pérez S, Lamas JA. PIP2 Mediated Inhibition of TREK Potassium Currents by Bradykinin in Mouse Sympathetic Neurons. International Journal of Molecular Sciences. 2020; 21(2):389. https://doi.org/10.3390/ijms21020389
Chicago/Turabian StyleRivas-Ramírez, Paula, Antonio Reboreda, Lola Rueda-Ruzafa, Salvador Herrera-Pérez, and J. Antonio Lamas. 2020. "PIP2 Mediated Inhibition of TREK Potassium Currents by Bradykinin in Mouse Sympathetic Neurons" International Journal of Molecular Sciences 21, no. 2: 389. https://doi.org/10.3390/ijms21020389