Comparison of Bisulfite Pyrosequencing and Methylation-Specific qPCR for Methylation Assessment

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. p16/INK4A Methylation Analysis Based on Bisulfite Pyrosequencing
2.2. p16/INK4A Methylation Analysis Based on MS-qPCR
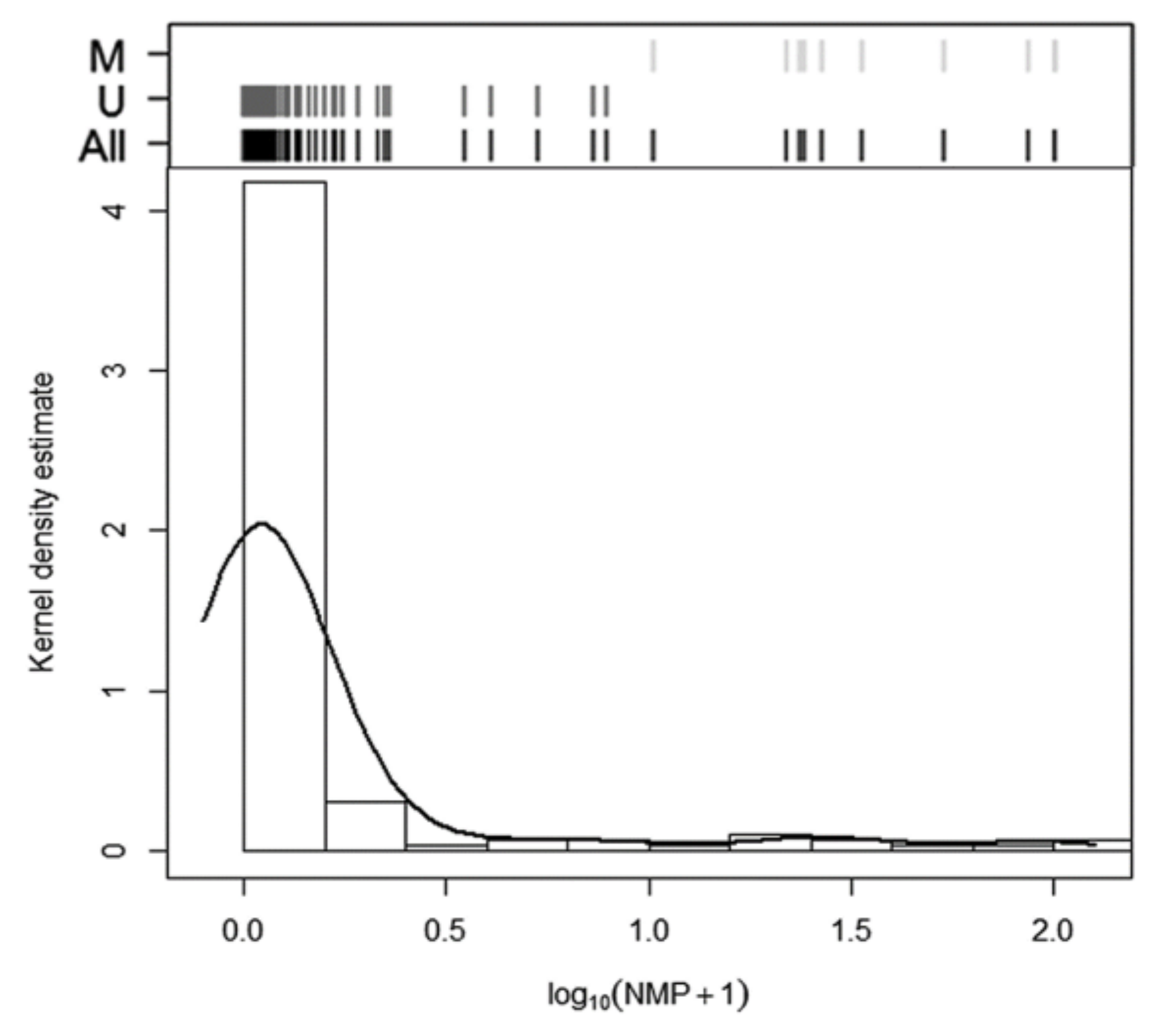
2.3. Comparison of the Methylation Approaches for the Study of p16/INK4A
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. DNA Extraction and Sodium Bisulfite Modification
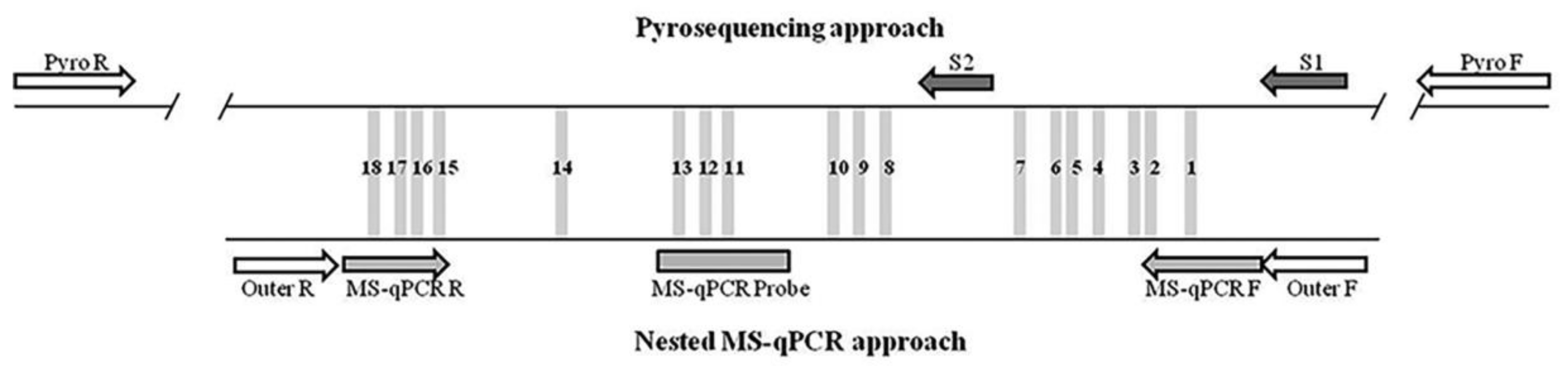
4.3. Bisulfite Pyrosequencing
4.4. Methylation-Specific qPCR (MS-qPCR)
4.5. Analysis of the MS-qPCR Data
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EBUS-TBNA | Endobronchial ultrasound transbronchial needle aspiration |
| MS-qPCR | Methylation-specific qPCR |
| NMP | Normalized methylation percentage |
| NSCLC | Non-small cell lung cancer |
| PYRmean | Mean methylation percentage |
References
- Pan, Y.; Liu, G.; Zhou, F.; Su, B.; Li, Y. DNA methylation profiles in cancer diagnosis and therapeutics. Clin. Exp. Med. 2018, 18, 1–14. [Google Scholar] [CrossRef]
- Duruisseaux, M.; Esteller, M. Lung cancer epigenetics: From knowledge to applications. Semin. Cancer Biol. 2018, 51, 116–128. [Google Scholar] [CrossRef]
- Mehta, A.; Dobersch, S.; Romero-Olmedo, A.J.; Barreto, G. Epigenetics in lung cancer diagnosis and therapy. Cancer Metastasis Rev. 2015, 34, 229–241. [Google Scholar] [CrossRef]
- Liloglou, T.; Bediaga, N.G.; Brown, B.R.B.; Field, J.K.; Davies, M.P. Epigenetic biomarkers in lung cancer. Cancer Lett. 2014, 342, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Hulbert, A.; Jusue-Torres, I.; Stark, A.; Hulbert, A.; Jusue-Torres, I.; Stark, A.; Chen, C.; Rodgers, K.; Lee, B.; Griffin, C.; et al. Early detection of lung cancer using DNA promoter hypermethylation in plasma and sputum. Clin. Cancer Res. 2017, 23, 1998–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiro-Fernandez, V.; De Chiara, L.; Rodríguez-Girondo, M.; Botana-Rial, M.; Valverde, D.; Núñez-Delgado, M.; Fernández-Villar, A. Methylation Assessment for the Prediction of Malignancy in Mediastinal Adenopathies Obtained by Endobronchial Ultrasound-Guided Transbronchial Needle Aspiration in Patients with Lung Cancer. Cancers 2019, 11, 1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestri, G.A.; Gonzalez, A.V.; Jantz, M.A.; Margolis, M.L.; Gould, M.K.; Tanoue, L.T.; Harris, L.J.; Detterbeck, F.C. Methods for staging non-small cell lung cancer: Diagnosis and management of lung cancer: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013, 143, 211–250. [Google Scholar] [CrossRef] [Green Version]
- Micames, C.G.; McCrory, D.C.; Pavey, D.A.; Jowell, P.S.; Gress, F.G. Endoscopic ultrasound-guided fine-needle aspiration for non-small cell lung cancer staging: A systematic review and metaanalysis. Chest 2007, 131, 539–548. [Google Scholar] [CrossRef]
- Tuo, L.; Sha, S.; Huayu, Z.; Du, K. P16(INK4a) gene promoter methylation as a biomarker for the diagnosis of non-small cell lung cancer: An updated meta-analysis. Thorac. Cancer 2018, 9, 1032–1040. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, T.; Zhang, H.; Wang, X.; Liu, X.; Huang, Q.; Li, L. Clinical Significance of P16 Gene Methylation in Lung Cancer. Adv. Exp. Med. Biol. 2020, 1255, 133–142. [Google Scholar]
- Merlo, A.; Herman, J.G.; Mao, L.; Lee, D.J.; Gabrielson, E.; Burger, P.C.; Baylin, S.B.; Sidransky, D. 5′CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat. Med. 1995, 1, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Liggett, W.H.; Sidransky, D. Role of the p16 tumor suppressor gene in cancer. J. Clin. Oncol. 1998, 16, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Kurdyukov, S.; Bullock, M. DNA methylation analysis: Choosing the right method. Biology 2016, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Redshaw, N.; Huggett, J.F.; Taylor, M.S.; Foy, C.A.; Devonshire, A.S. Quantification of epigenetic biomarkers: An evaluation of established and emerging methods for DNA methylation analysis. BMC Genom. 2014, 15, 1174. [Google Scholar] [CrossRef] [Green Version]
- Singer, B.D. A Practical Guide to the Measurement and Analysis of DNA Methylation. Am. J. Respir. Cell Mol. Biol. 2019, 61, 417–428. [Google Scholar] [CrossRef]
- Tost, J.; Gut, I.G. Analysis of gene-specific DNA methylation patterns by pyrosequencing technology. Methods Mol. Biol. 2007, 373, 89–102. [Google Scholar]
- Ogino, S.; Kawasaki, T.; Brahmandam, M.; Cantor, M.; Kirkner, G.J.; Spiegelman, D.; Makrigiorgos, G.M.; Weisenberger, D.J.; Laird, P.W.; Fuchs, C.S.; et al. Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. J. Mol. Diagn. 2006, 8, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Migheli, F.; Stoccoro, A.; Coppedè, F.; Omar, W.A.W.; Failli, A.; Consolini, R.; Seccia, M.; Spisni, R.; Miccoli, P.; Mathers, J.C.; et al. Comparison study of MS-HRM and pyrosequencing techniques for quantification of APC and CDKN2A gene methylation. PLoS ONE 2013, 8, e52501. [Google Scholar] [CrossRef]
- Bock, C.; Halbritter, F.; Carmona, F.J.; Tierling, S.; Datlinger, P.; Assenov, Y.; Berdasco, M.; Bergmann, A.K.; Booher, K.; Busato, F.; et al. Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat. Biotechnol. 2016, 34, 726–737. [Google Scholar]
- Pharo, H.D.; Honne, H.; Vedeld, H.M.; Dahl, C.; Andresen, K.; Liestol, K.; Jeanmougin, M.; Guldberg, P.; Lind, G.E. Experimental factors affecting the robustness of DNA methylation analysis. Sci. Rep. 2016, 6, 33936. [Google Scholar] [CrossRef] [Green Version]
- Claus, R.; Wilop, S.; Hielscher, T.; Sonnet, M.; Dahl, E.; Galm, O.; Jost, E.; Plass, C. A systematic comparison of quantitative high-resolution DNA methylation analysis and methylation-specific PCR. Epigenetics 2012, 7, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alnaes, G.I.; Ronneberg, J.A.; Kristensen, V.N.; Tost, J. Heterogeneous DNA methylation patterns in the GSTP1 promoter lead to discordant results between assay technologies and impede its implementation as epigenetic biomarkers in breast cancer. Genes 2015, 6, 878–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, H.E.; Durston, V.J.; Harvey, J.F.; Cross, N.C. Quantitative analysis of SNRPN (correction of SRNPN) gene methylation by pyrosequencing as a diagnostic test for Prader-Willi syndrome and Angelman syndrome. Clin. Chem. 2006, 52, 1005–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- House, M.G.; Guo, M.; Iacobuzio-Donahue, C.; Herman, J.G. Molecular progression of promoter methylation in intraductal papillary mucinous neoplasms (IPMN) of the pancreas. Carcinogenesis 2003, 24, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.G.; Graff, J.R.; Myöhänen, S.B.D.N.; Nelkin, B.D.; Baylin, S.B. Methylation specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harden, S.V.; Tokumaru, Y.; Westra, W.H.; Goodman, S.; Ahrendt, S.A.; Yang, S.C.; Sidransky, D. Gene promoter hypermethylation in tumors and lymph nodes of stage I lung cancer patients. Clin. Cancer Res. 2003, 9, 1370–1375. [Google Scholar]
- Šestáková, S.; Šálek, C.; Remešová, H. DNA Methylation Validation Methods: A Coherent Review with Practical Comparison. Biol. Proced. Online 2019, 21, 19. [Google Scholar] [CrossRef]
- Van Wesenbeeck, L.; Janssens, L.; Meeuws, H.; Lagatie, O.; Stuyver, L. Droplet digital PCR is an accurate method to assess methylation status on FFPE samples. Epigenetics 2018, 13, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Quillien, V.; Lavenu, A.; Karayan-Tapon, L.; Carpentier, C.; Labussière, M.; Lesimple, T.; Chinot, O.; Wager, M.; Honnorat, J.; Saikali, S.; et al. Comparative assessment of 5 methods (methylation-specific polymerase chain reaction, MethyLight, pyrosequencing, methylation-sensitive high-resolution melting, and immunohistochemistry) to analyze O6-methylguanine-DNA-methyltranferase in a series of 100 glioblastoma patients. Cancer 2012, 118, 4201–4211. [Google Scholar]
- Assenov, Y.; Brocks, D.; Gerhäuser, C. Intratumor heterogeneity in epigenetic patterns. Semin. Cancer Biol. 2018, 51, 12–21. [Google Scholar] [CrossRef]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Peter, V.; Laird, P.W. MethyLight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28, e32-00. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azalini, A.; Torelli, N. Clustering via nonparametric density estimation. Stat. Comput. 2007, 17, 71–80. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| METHYLATED n (Number of Samples) (Median and Range of Methylation Percentage) | UNMETHYLATED n (Number of Samples) (Median and Range of Methylation Percentage) | |||||||
|---|---|---|---|---|---|---|---|---|
| PYRmean * | NMP † | NMP ‡ | NMP § | PYRmean * | NMP † | NMP ‡ | NMP § | |
| All samples | n = 10 22.53 (15.33–36.46) | n = 10 29.27 (9.22–100) | n = 14 22.95 (3.10–100) | n = 15 22.63 (2.52–100) | n = 147 4.16 (1.99–12.54) | n = 135 0.08 (0.00–6.85) | n = 131 0.07 (0.00–2.52) | n = 130 0.07 (0.00–1.28) |
| Metastatic lymph nodes | n = 10 22.53 (15.33–36.46) | n = 9 25.97 (9.22–100) | n = 12 22.95 (3.10–100) | n = 13 22.63 (2.52–100) | n = 67 4.17 (2.21–12.54) | n = 61 0.09 (0.00–6.85) | n = 58 0.09 (0.00–2.52) | n = 57 0.08 (0.00–1.28) |
| Non-metastatic lymph nodes | n = 0 - - | n = 1 52.83 - | n = 2 28.58 (4.33–52.83) | n = 2 28.58 (4.33–52.83) | n = 80 4.10 (1.99–10.35) | n = 74 0.06 (0.00–4.33) | n = 73 0.06 (0.00–1.24) | n = 73 0.06 (0.00–1.24) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Chiara, L.; Leiro-Fernandez, V.; Rodríguez-Girondo, M.; Valverde, D.; Botana-Rial, M.I.; Fernández-Villar, A. Comparison of Bisulfite Pyrosequencing and Methylation-Specific qPCR for Methylation Assessment. Int. J. Mol. Sci. 2020, 21, 9242. https://doi.org/10.3390/ijms21239242
De Chiara L, Leiro-Fernandez V, Rodríguez-Girondo M, Valverde D, Botana-Rial MI, Fernández-Villar A. Comparison of Bisulfite Pyrosequencing and Methylation-Specific qPCR for Methylation Assessment. International Journal of Molecular Sciences. 2020; 21(23):9242. https://doi.org/10.3390/ijms21239242
Chicago/Turabian StyleDe Chiara, Loretta, Virginia Leiro-Fernandez, Mar Rodríguez-Girondo, Diana Valverde, María Isabel Botana-Rial, and Alberto Fernández-Villar. 2020. "Comparison of Bisulfite Pyrosequencing and Methylation-Specific qPCR for Methylation Assessment" International Journal of Molecular Sciences 21, no. 23: 9242. https://doi.org/10.3390/ijms21239242