
Impairments of Photoreceptor Outer Segments Renewal and Phototransduction Due to a Peripherin Rare Haplotype Variant: Insights from Molecular Modeling

 , ,
, ,  and
and
Abstract
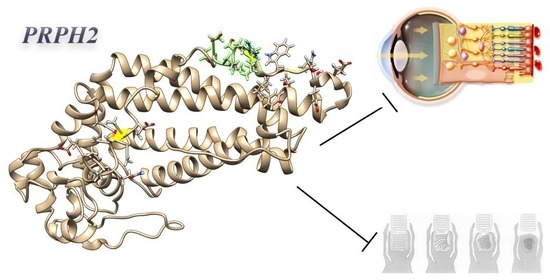
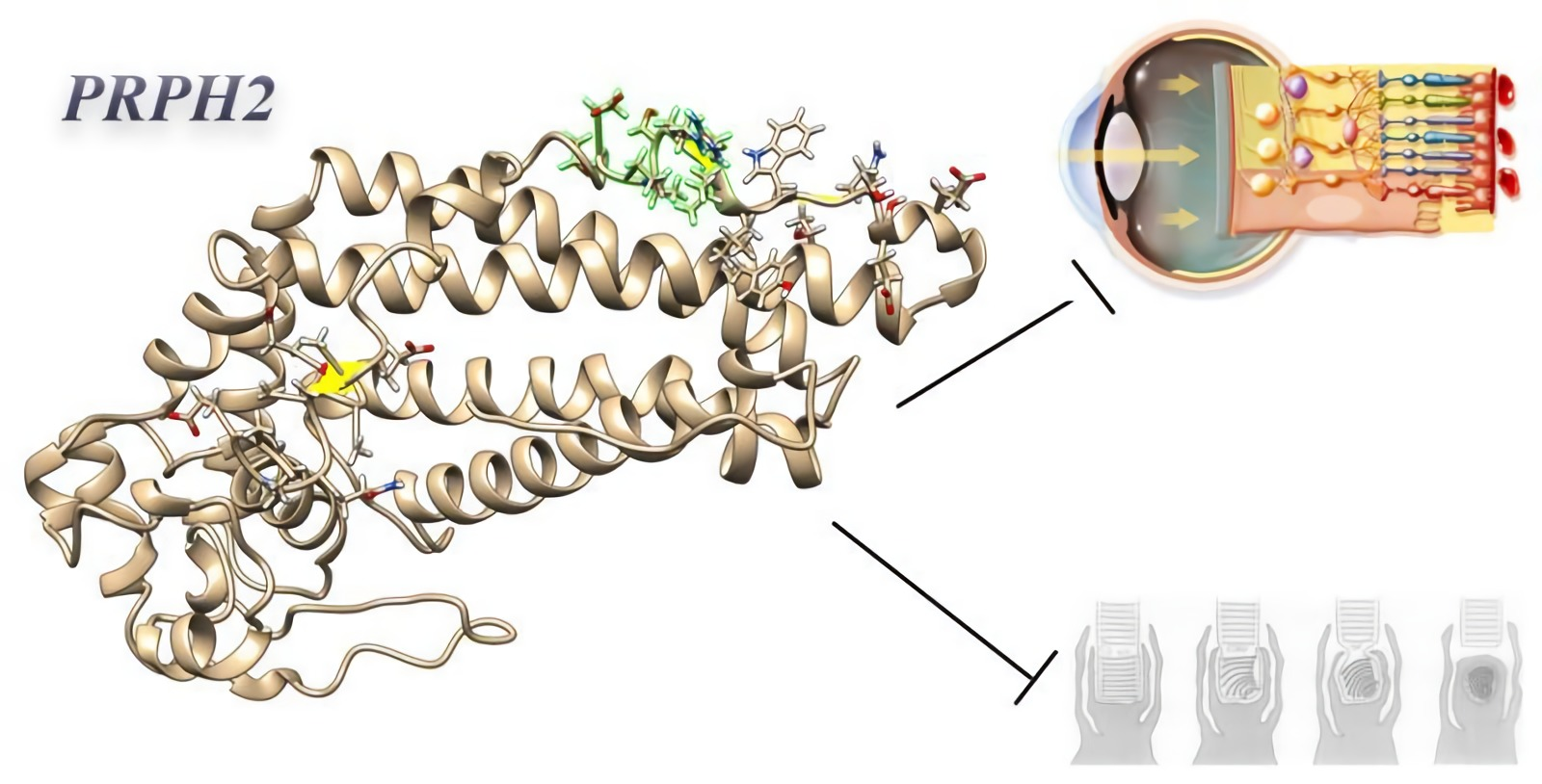
:
1. Introduction
2. Results
2.1. Genotyping of Probands Revealed Variants in PRHP2, RHO, and RLBP1
2.2. Haplotype Analyses Shed Light on a PRPH2 Candidate, A Rare Haplotype Variant Associated to RPA
2.3. Primary Structural and Biochemical Analyses Highlighted Possible Altered Chemical-Physical Features in Mutated PRPH2
2.4. Secondary Structure Analysis of Mutated PRPH2 Showed a Global Instable Protein
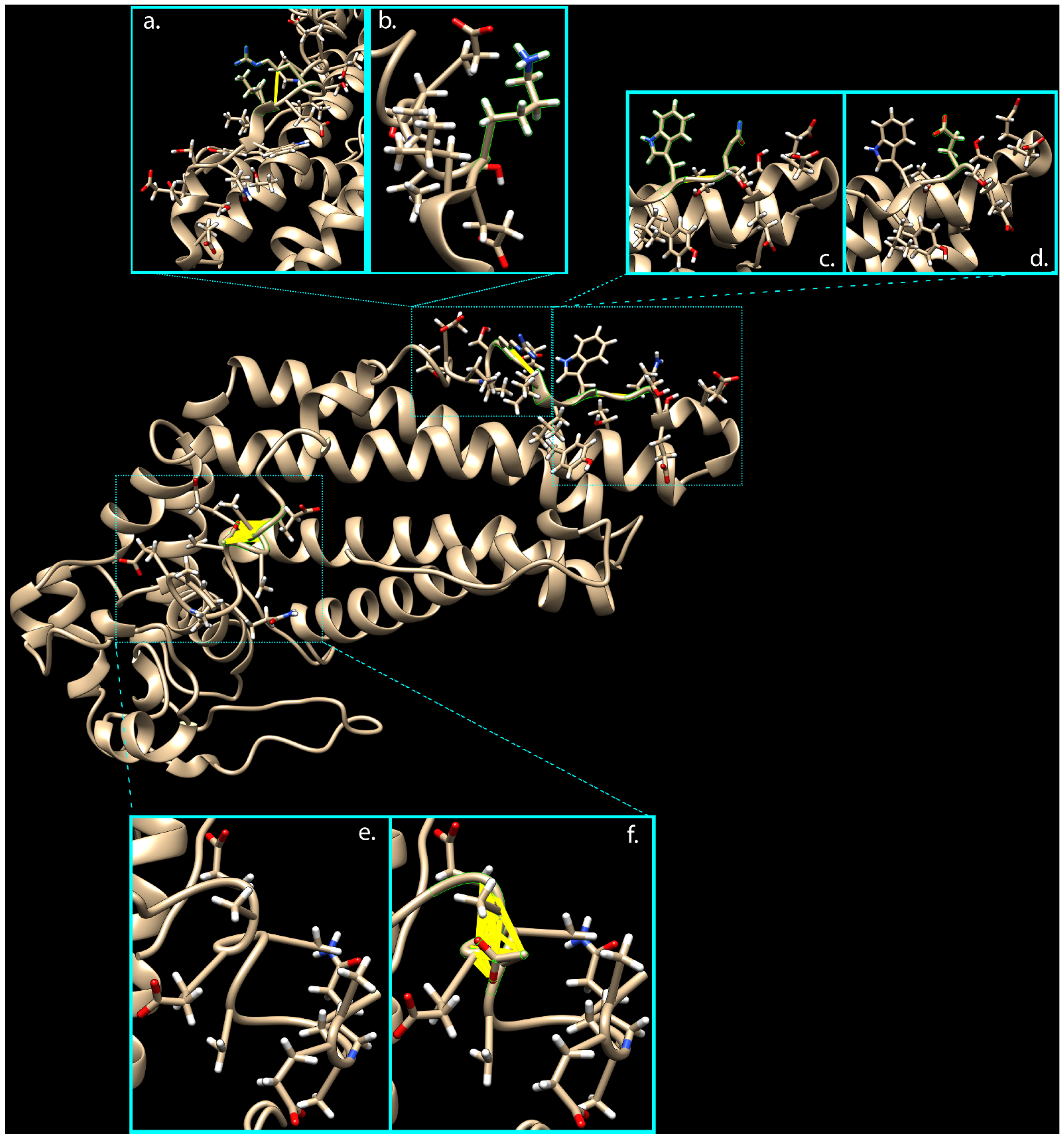
2.5. Clash Analysis of the PRPH2 Mutated 3D Predicted Structure Evidenced a Possible Misfolding in Tetraspanin and MREG Interacting Domains
2.6. Variants in RHO and RLBP1 Promoters Might Alter Their Expression, Indirectly Involving PRPH2
3. Discussion
4. Methods
4.1. Clinical Data
4.2. Mutational Analysis
4.3. Haplotype Statistical Analysis
4.4. In Silico Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- O’Neal, T.B.; Luther, E.E. Retinitis Pigmentosa; StatPearls: Treasure Island, FL, USA, 2018. [Google Scholar]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye. Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Bramanti, P.; Scimone, C.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. miRNAexpression profile of retinal pigment epithelial cells under oxidative stress conditions. FEBS Open Biol. 2018, 8, 219–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, L.; Scimone, C.; Alibrandi, S.; Rinaldi, C.; Sidoti, A.; D’Angelo, R. Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa. Antioxidants 2020, 9, 318. [Google Scholar] [CrossRef] [Green Version]
- Dias, M.F.; Joo, K.; Kemp, J.A.; Fialho, S.L.; da Silva Cunha, A., Jr.; Woo, S.J.; Kwon, Y.J. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retin. Eye. Res. 2018, 63, 107–131. [Google Scholar] [CrossRef] [PubMed]
- Scimone, C.; Alibrandi, S.; Scalinci, S.Z.; Trovato Battagliola, E.; D’Angelo, R.; Sidoti, A.; Donato, L. Expression of Pro-Angiogenic Markers Is Enhanced by Blue Light in Human RPE Cells. Antioxidants 2020, 9, 1154. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Alibrandi, S.; Abdalla, E.M.; Nabil, K.M.; D’Angelo, R.; Sidoti, A. New Omics-Derived Perspectives on Retinal Dystrophies: Could Ion Channels-Encoding or Related Genes Act as Modifier of Pathological Phenotype? Int. J. Mol. Sci. 2020, 22, 70. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Alibrandi, S.; Pitruzzella, A.; Scalia, F.; D’Angelo, R.; Sidoti, A. Possible A2E Mutagenic Effects on RPE Mitochondrial DNA from Innovative RNA-Seq Bioinformatics Pipeline. Antioxidants 2020, 9, 1158. [Google Scholar] [CrossRef]
- Ramdani, T.; Sekhsoukh, R. Retinitis punctata albescens. Pan. Afr. Med. J. 2016, 25, 39. [Google Scholar] [CrossRef]
- De Laey, J.J. Flecked retina disorders. Bull Soc. Belge Ophtalmol. 1993, 249, 11–22. [Google Scholar] [PubMed]
- Kajiwara, K.; Sandberg, M.A.; Berson, E.L.; Dryja, T.P. A null mutation in the human peripherin/RDS gene in a family with autosomal dominant retinitis punctata albescens. Nat. Genet. 1993, 3, 208–212. [Google Scholar] [CrossRef]
- Bohm, S.; Riedmayr, L.M.; Nguyen, O.N.P.; Giessl, A.; Liebscher, T.; Butz, E.S.; Schon, C.; Michalakis, S.; Wahl-Schott, C.; Biel, M.; et al. Peripherin-2 and Rom-1 have opposing effects on rod outer segment targeting of retinitis pigmentosa-linked peripherin-2 mutants. Sci. Rep. 2017, 7, 2321. [Google Scholar] [CrossRef] [Green Version]
- Molday, R.S.; Goldberg, A.F.X. Peripherin diverts ciliary ectosome release to photoreceptor disc morphogenesis. J. Cell. Biol. 2017, 216, 1227–1229. [Google Scholar] [CrossRef]
- Conley, S.M.; Stuck, M.W.; Watson, J.N.; Zulliger, R.; Burnett, J.L.; Naash, M.I. Prph2 initiates outer segment morphogenesis but maturation requires Prph2/Rom1 oligomerization. Hum. Mol. Genet. 2019, 28, 459–475. [Google Scholar] [CrossRef]
- Gill, J.S.; Georgiou, M.; Kalitzeos, A.; Moore, A.T.; Michaelides, M. Progressive cone and cone-rod dystrophies: Clinical features, molecular genetics and prospects for therapy. Br. J. Ophthalmol. 2019, 103, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Renner, A.B.; Fiebig, B.S.; Weber, B.H.; Wissinger, B.; Andreasson, S.; Gal, A.; Cropp, E.; Kohl, S.; Kellner, U. Phenotypic variability and long-term follow-up of patients with known and novel PRPH2/RDS gene mutations. Am. J. Ophthalmol. 2009, 147, 518–530.e1. [Google Scholar] [CrossRef] [PubMed]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye. Res. 2018, 62, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Noorwez, S.M.; Kuksa, V.; Imanishi, Y.; Zhu, L.; Filipek, S.; Palczewski, K.; Kaushal, S. Pharmacological chaperone-mediated in vivo folding and stabilization of the P23H-opsin mutant associated with autosomal dominant retinitis pigmentosa. J. Biol. Chem. 2003, 278, 14442–14450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Deretic, D. Molecular complexes that direct rhodopsin transport to primary cilia. Prog. Retin. Eye. Res. 2014, 38, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, L.; D’Angelo, R.; Alibrandi, S.; Rinaldi, C.; Sidoti, A.; Scimone, C. Effects of A2E-Induced Oxidative Stress on Retinal Epithelial Cells: New Insights on Differential Gene Response and Retinal Dystrophies. Antioxidants 2020, 9, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souied, E.; Soubrane, G.; Benlian, P.; Coscas, G.J.; Gerber, S.; Munnich, A.; Kaplan, J. Retinitis punctata albescens associated with the Arg135Trp mutation in the rhodopsin gene. Am. J. Ophthalmol. 1996, 121, 19–25. [Google Scholar] [CrossRef]
- Scimone, C.; Donato, L.; Esposito, T.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. A novel RLBP1 gene geographical area-related mutation present in a young patient with retinitis punctata albescens. Hum. Genom. 2017, 11, 18. [Google Scholar] [CrossRef]
- Xue, Y.; Shen, S.Q.; Jui, J.; Rupp, A.C.; Byrne, L.C.; Hattar, S.; Flannery, J.G.; Corbo, J.C.; Kefalov, V.J. CRALBP supports the mammalian retinal visual cycle and cone vision. J. Clin. Investig. 2015, 125, 727–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collery, R.; McLoughlin, S.; Vendrell, V.; Finnegan, J.; Crabb, J.W.; Saari, J.C.; Kennedy, B.N. Duplication and divergence of zebrafish CRALBP genes uncovers novel role for RPE- and Muller-CRALBP in cone vision. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3812–3820. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Scimone, C.; Nicocia, G.; Denaro, L.; Robledo, R.; Sidoti, A.; D’Angelo, R. GLO1 gene polymorphisms and their association with retinitis pigmentosa: A case-control study in a Sicilian population. Mol. Biol. Rep. 2018, 45, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Byrnes, A.E.; Li, M. To identify associations with rare variants, just WHaIT: Weighted haplotype and imputation-based tests. Am. J. Hum. Genet. 2010, 87, 728–735. [Google Scholar] [CrossRef] [Green Version]
- Spencer, K.L.; Hauser, M.A.; Olson, L.M.; Schnetz-Boutaud, N.; Scott, W.K.; Schmidt, S.; Gallins, P.; Agarwal, A.; Postel, E.A.; Pericak-Vance, M.A.; et al. Haplotypes spanning the complement factor H gene are protective against age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4277–4283. [Google Scholar] [CrossRef]
- Biswas, S.; Papachristou, C. Evaluation of logistic Bayesian LASSO for identifying association with rare haplotypes. BMC Proc. 2014, 8, S54. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.Y.; Yi, N.; Zhi, D.; Zhang, K.; Gao, G.; Tiwari, H.K.; Liu, N. Haplotype-based methods for detecting uncommon causal variants with common SNPs. Genet. Epidemiol. 2012, 36, 572–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milstein, M.L.; Kimler, V.A.; Ghatak, C.; Ladokhin, A.S.; Goldberg, A.F.X. An inducible amphipathic helix within the intrinsically disordered C terminus can participate in membrane curvature generation by peripherin-2/rds. J. Biol. Chem. 2017, 292, 7850–7865. [Google Scholar] [CrossRef] [Green Version]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Miao, Y.; Tipakornsaowapak, T.; Zheng, L.; Mu, Y.; Lewellyn, E. Phospho-regulation of intrinsically disordered proteins for actin assembly and endocytosis. FEBS J. 2018, 285, 2762–2784. [Google Scholar] [CrossRef]
- Boesze-Battaglia, K.; Song, H.; Sokolov, M.; Lillo, C.; Pankoski-Walker, L.; Gretzula, C.; Gallagher, B.; Rachel, R.A.; Jenkins, N.A.; Copeland, N.G.; et al. The tetraspanin protein peripherin-2 forms a complex with melanoregulin, a putative membrane fusion regulator. Biochemistry 2007, 46, 1256–1272. [Google Scholar] [CrossRef] [Green Version]
- Kruse, S.W.; Suino-Powell, K.; Zhou, X.E.; Kretschman, J.E.; Reynolds, R.; Vonrhein, C.; Xu, Y.; Wang, L.; Tsai, S.Y.; Tsai, M.J.; et al. Identification of COUP-TFII orphan nuclear receptor as a retinoic acid-activated receptor. PLoS Biol. 2008, 6, e227. [Google Scholar] [CrossRef]
- Ma, Z.; Jhun, B.; Jung, S.Y.; Oh, C.K. Binding of upstream stimulatory factor 1 to the E-box regulates the 4G/5G polymorphism-dependent plasminogen activator inhibitor 1 expression in mast cells. J. Allergy Clin. Immunol. 2008, 121, 1006–1012 e1002. [Google Scholar] [CrossRef]
- Viollet, B.; Lefrancois-Martinez, A.M.; Henrion, A.; Kahn, A.; Raymondjean, M.; Martinez, A. Immunochemical characterization and transacting properties of upstream stimulatory factor isoforms. J. Biol. Chem. 1996, 271, 1405–1415. [Google Scholar] [CrossRef] [Green Version]
- Felsher, D.W.; Zetterberg, A.; Zhu, J.; Tlsty, T.; Bishop, J.M. Overexpression of MYC causes p53-dependent G2 arrest of normal fibroblasts. Proc. Natl. Acad. Sci. USA 2000, 97, 10544–10548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, P.; Menssen, A.; Mayr, D.; Hermeking, H. AP4 encodes a c-MYC-inducible repressor of p21. Proc. Natl. Acad. Sci. USA 2008, 105, 15046–15051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizukami, Y.; Iwamatsu, A.; Aki, T.; Kimura, M.; Nakamura, K.; Nao, T.; Okusa, T.; Matsuzaki, M.; Yoshida, K.; Kobayashi, S. ERK1/2 regulates intracellular ATP levels through alpha-enolase expression in cardiomyocytes exposed to ischemic hypoxia and reoxygenation. J. Biol. Chem. 2004, 279, 50120–50131. [Google Scholar] [CrossRef] [Green Version]
- Popov, N.; Wanzel, M.; Madiredjo, M.; Zhang, D.; Beijersbergen, R.; Bernards, R.; Moll, R.; Elledge, S.J.; Eilers, M. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 2007, 9, 765–774. [Google Scholar] [CrossRef] [PubMed]
- de Luis, O.; Valero, M.C.; Jurado, L.A. WBSCR14, a putative transcription factor gene deleted in Williams-Beuren syndrome: Complete characterisation of the human gene and the mouse ortholog. Eur. J. Hum. Genet. 2000, 8, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Vulto-van Silfhout, A.T.; Rajamanickam, S.; Jensik, P.J.; Vergult, S.; de Rocker, N.; Newhall, K.J.; Raghavan, R.; Reardon, S.N.; Jarrett, K.; McIntyre, T.; et al. Mutations affecting the SAND domain of DEAF1 cause intellectual disability with severe speech impairment and behavioral problems. Am. J. Hum. Genet. 2014, 94, 649–661. [Google Scholar] [CrossRef] [Green Version]
- Turunen, M.M.; Dunlop, T.W.; Carlberg, C.; Vaisanen, S. Selective use of multiple vitamin D response elements underlies the 1 alpha,25-dihydroxyvitamin D3-mediated negative regulation of the human CYP27B1 gene. Nucleic Acids Res. 2007, 35, 2734–2747. [Google Scholar] [CrossRef] [Green Version]
- Hurlin, P.J.; Queva, C.; Koskinen, P.J.; Steingrimsson, E.; Ayer, D.E.; Copeland, N.G.; Jenkins, N.A.; Eisenman, R.N. Mad3 and Mad4: Novel Max-interacting transcriptional repressors that suppress c-myc dependent transformation and are expressed during neural and epidermal differentiation. EMBO J. 1995, 14, 5646–5659. [Google Scholar] [CrossRef] [PubMed]
- Oshel, K.M.; Knight, J.B.; Cao, K.T.; Thai, M.V.; Olson, A.L. Identification of a 30-base pair regulatory element and novel DNA binding protein that regulates the human GLUT4 promoter in transgenic mice. J. Biol. Chem. 2000, 275, 23666–23673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becirovic, E.; Nguyen, O.N.; Paparizos, C.; Butz, E.S.; Stern-Schneider, G.; Wolfrum, U.; Hauck, S.M.; Ueffing, M.; Wahl-Schott, C.; Michalakis, S.; et al. Peripherin-2 couples rhodopsin to the CNG channel in outer segments of rod photoreceptors. Hum. Mol. Genet. 2014, 23, 5989–5997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Giron, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [Green Version]
- Lake, S.L.; Lyon, H.; Tantisira, K.; Silverman, E.K.; Weiss, S.T.; Laird, N.M.; Schaid, D.J. Estimation and tests of haplotype-environment interaction when linkage phase is ambiguous. Hum. Hered. 2003, 55, 56–65. [Google Scholar] [CrossRef]
- Schaid, D.J.; Rowland, C.M.; Tines, D.E.; Jacobson, R.M.; Poland, G.A. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am. J. Hum. Genet. 2002, 70, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Lin, S. Logistic Bayesian LASSO for identifying association with rare haplotypes and application to age-related macular degeneration. Biometrics 2012, 68, 587–597. [Google Scholar] [CrossRef]
- CLC Main Workbench 21.0.3. Available online: https://digitalinsights.qiagen.com (accessed on 23 March 2021).
- Xie, Q.; He, X.; Yang, F.; Liu, X.; Li, Y.; Liu, Y.; Yang, Z.; Yu, J.; Zhang, B.; Zhao, W. Analysis of the Genome Sequence and Prediction of B-Cell Epitopes of the Envelope Protein of Middle East Respiratory Syndrome-Coronavirus. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 15, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protocol. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallberg, M.; Margaryan, G.; Wang, S.; Ma, J.; Xu, J. RaptorX server: A resource for template-based protein structure modeling. Methods Mol. Biol. 2014, 1137, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [Green Version]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Wingender, E.; Schoeps, T.; Haubrock, M.; Krull, M.; Donitz, J. TFClass: Expanding the classification of human transcription factors to their mammalian orthologs. Nucleic Acids Res. 2018, 46, D343–D347. [Google Scholar] [CrossRef] [Green Version]
- Donato, L.; Scimone, C.; Rinaldi, C.; Aragona, P.; Briuglia, S.; D’Ascola, A.; D’Angelo, R.; Sidoti, A. Stargardt Phenotype Associated With Two ELOVL4 Promoter Variants and ELOVL4 Downregulation: New Possible Perspective to Etiopathogenesis? Investig. Ophthalmol. Vis. Sci. 2018, 59, 843–857. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| GLOBAL SCORE STATISTICS | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| global-stat = 7.7253 | df = 2 | adj. p-val (Bonferroni) = 0.007004 | N° Controls = 152 | N° Cases = 2 | ||||||||
| Haplotype, scores, p-values, hap-frequencies (hf), and odds ratios (95%CI) --- haplo.stats | ||||||||||||
| Haplotype # | rs390659 | rs425876 | rs434102 | Hap-Score | adj. p-val (Bonferroni) | pool.hf | control.hf | case.hf | glm.eff | OR lower | OR | OR upper |
| 2 | C | G | A | −1.40512 | 0.0533 | 0.49351 | 0.50000 | NA | Base | NA | 1 | NA |
| 1 | C | A | A | −0.92901 | 0.1176 | 0.29870 | 0.30263 | NA | Eff | 1 | 1 | 1 |
| 3 | G | A | G | 2.77945 | 0.0018 | 0.20779 | 0.19737 | 1 | Eff | 31460 | 55771 | 98869 |
| Haplotype, Bayesian factors, and odds ratios (95%CI) --- LBL | ||||||||||||
| Haplotype # | rs390659 | rs425876 | rs434102 | BF | OR | CI.OR lower | CI.OR upper | CI.lambda lower | CI.lambda upper | CI.D lower | CI.D upper | |
| 2 | C | G | A | / | / | / | / | 0.5862 | 1.4365 | 0.9604 | 0.9997 | |
| 1 | C | A | A | 0.9626 | 0.5904 | 0.0213 | 5.3881 | |||||
| 3 | G | A | G | 5.0203 | 6.6699 | 1.0921 | 214.0648 | |||||
| Structural Feature | Wild-Type Protein | Mutated Protein |
|---|---|---|
| Weight (da) | 39,186 | 39,271 |
| Absorption at 280nm 0.1% (= 1 g/L) | ||
| Non-reduced cysteines | 1891 | 1895 |
| Reduced cysteines | 1873 | 1877 |
| Count of hydrophobic (A, F, G, I, L, M, P, V, W) residues | 175 | 174 |
| Count of hydrophilic (C, N, Q, S, T, Y) residues | 95 | 96 |
| Pfam domain result | Tetraspanin (aa. 16–287) | |
| Uniprot domain result | Interaction domain with Melanoregulin (MREG) (aa. 341–346) | |
| Instability index | 92, 59 | 88, 72 |
| Amphiphilicity and hydropathy | Changes in aa. 26–28, 106, 181–184, 209, 258–261, 272, 307–315, 336 | |
| Flexibility | 287–305, 309–316 | 287–306, 310–316 |
| Disorder | 286–312, 328–346 | 286–310, 327–346 |
| Surface probability | Three residues (aa. 310–312) | Two residues (aa. 311–312) |
| / | One residue (aa. 335) | |
| Three residues (aa. 341–343) | Four residues (aa. 340–343) | |
| Feature | Wild-Type | Rs390659 | Wild-Type | Rs425876 | Wild-Type | Rs434102 |
|---|---|---|---|---|---|---|
| AA change | E304Q—GLUTAMINE (GLN) | GLUTAMIC ACID (GLU) | K310R—ARGININE (ARG) | LYSINE (LYS) | G338D—ASPARTIC ACID (ASP) | GLYCINE (GLY) |
| Position | 304 | 304 | 310 | 310 | 338 | 338 |
| Type | L-PEPTIDE LINKING | L-PEPTIDE LINKING | L-PEPTIDE LINKING | L-PEPTIDE LINKING | PEPTIDE LINKING | L-PEPTIDE LINKING |
| MW [g/mol] | 147.129 | 146.144 | 147.195 | 175.209 | 75.067 | 133.103 |
| Net charge [pH = 7] | −1 | −0.01 | 0.99 | 0.99 | 0 | −1 |
| pI | 4 | 5.52 | 8.75 | 9.75 | 5.52 | 3.8 |
| Average hydropathy | −3.5 | −3.5 | −3.9 | −4.5 | −0.4 | −3.5 |
| Aliphatic index | 0 | 0 | 0 | 0 | 0 | 0 |
| A₂₈₀ (ox.) | 0 | 0 | 0 | 0 | 0 | 0 |
| A₂₈₀ (red.) | 0 | 0 | 0 | 0 | 0 | 0 |
| ε₂₈₀ [M⁻1 cm⁻1] | 0 | 0 | 0 | 0 | 0 | 0 |
| φ | −62.5° | −62.5° | −66.3° | −66.3° | −162.2° | −162.2° |
| ψ | 15.4° | 15.4° | 32.9° | 32.9° | −2.7° | −2.7° |
| ω | 111.0° | 111.0° | 123.8° | 123.8° | 161.5° | 161.5° |
| Median B-factor | 10.18 | 25 | 12.28 | 25 | 10.9 | 17.95 |
| Effect on protein structure | stabilizing | stabilizing | destabilizing | |||
| Delta_energy | −0.554 | −0.143 | 1664 | |||
| Potential | DFIRE-A | DFIRE-A | DFIRE-A |
| Gene | SNP ID | Lost/ New | Family/Matrix | TF Name | Family Info | Further Info | Opt. thresh. | Start pos. | End pos. | Strand | Core sim. | Matrix sim. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RHO | rs7984 | lost | V$NR2F/COUPTFII.01 | NR2F2 | Nuclear receptor subfamily 2 factors | Chicken ovalbumin upstream promoter transcription factor 2, NR2F2 homodimer, DR1 sites | 0.8 | 490 | 514 | + | 1 | 0.828 |
| lost | V$RXRF/RXRA.01 | RXRA | RXR heterodimer binding sites | Retinoid X receptor alpha homodimer, DR1 sites | 0.83 | 492 | 516 | + | 1 | 0.894 | ||
| lost | V$EBOX/USF.03 | USF3 | E-box binding factors | Upstream stimulating factor | 0.89 | 494 | 510 | - | 1 | 0.904 | ||
| lost | V$EBOX/MYCMAX.03 | MYC and MAX | E-box binding factors | MYC-MAX binding sites | 0.91 | 495 | 511 | + | 0.842 | 0.919 | ||
| lost | V$CHRE/CHREBP_MLX.01 | MLXIPL | Carbohydrate response elements, consist of two E box motifs separated by 5 bp | Carbohydrate response element binding protein (CHREBP) and Max-like protein X (Mlx) bind as heterodimers to glucose-responsive promoters | 0.82 | 500 | 516 | + | 1 | 0.883 | ||
| RLBP1 | rs3743384 | lost | V$DEAF/NUDR.01 | DEAF1 | Homolog to deformed epidermal autoregulatory factor-1 from D. melanogaster | NUDR (nuclear DEAF-1 related transcriptional regulator protein) | 0.75 | 186 | 204 | - | 1 | 0.801 |
| new | V$HDBP/HDBP1_2.01 | SLC2A4RG | Huntington’s disease gene regulatory region binding proteins | Huntington’s disease gene regulatory region-binding protein 1 and 2 (SLC2A4 regulator and papillomavirus binding factor) | 0.84 | 189 | 207 | - | 1 | 0.863 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donato, L.; Abdalla, E.M.; Scimone, C.; Alibrandi, S.; Rinaldi, C.; Nabil, K.M.; D'Angelo, R.; Sidoti, A. Impairments of Photoreceptor Outer Segments Renewal and Phototransduction Due to a Peripherin Rare Haplotype Variant: Insights from Molecular Modeling. Int. J. Mol. Sci. 2021, 22, 3484. https://doi.org/10.3390/ijms22073484
Donato L, Abdalla EM, Scimone C, Alibrandi S, Rinaldi C, Nabil KM, D'Angelo R, Sidoti A. Impairments of Photoreceptor Outer Segments Renewal and Phototransduction Due to a Peripherin Rare Haplotype Variant: Insights from Molecular Modeling. International Journal of Molecular Sciences. 2021; 22(7):3484. https://doi.org/10.3390/ijms22073484
Chicago/Turabian StyleDonato, Luigi, Ebtesam Mohamed Abdalla, Concetta Scimone, Simona Alibrandi, Carmela Rinaldi, Karim Mahmoud Nabil, Rosalia D'Angelo, and Antonina Sidoti. 2021. "Impairments of Photoreceptor Outer Segments Renewal and Phototransduction Due to a Peripherin Rare Haplotype Variant: Insights from Molecular Modeling" International Journal of Molecular Sciences 22, no. 7: 3484. https://doi.org/10.3390/ijms22073484