
Senescent CD4+CD28− T Lymphocytes as a Potential Driver of Th17/Treg Imbalance and Alveolar Bone Resorption during Periodontitis

 , , and
, , and
Abstract
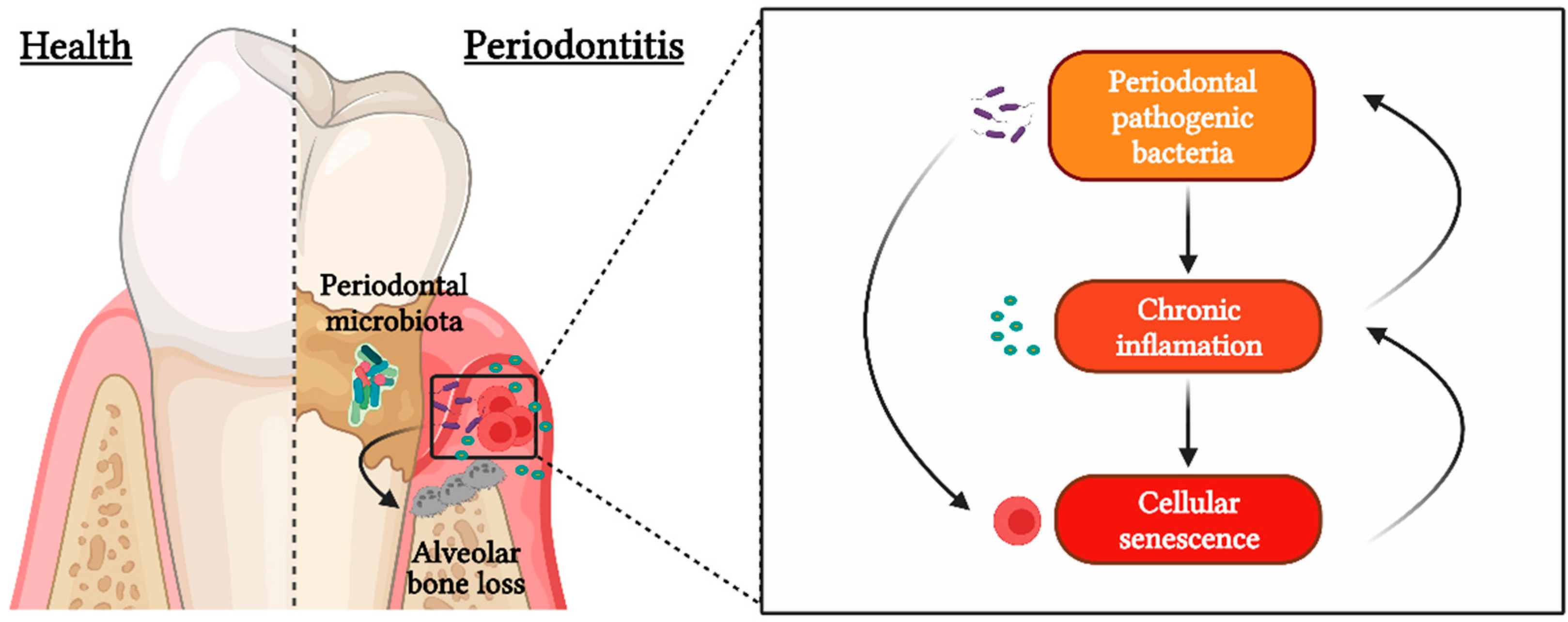
:1. Introduction
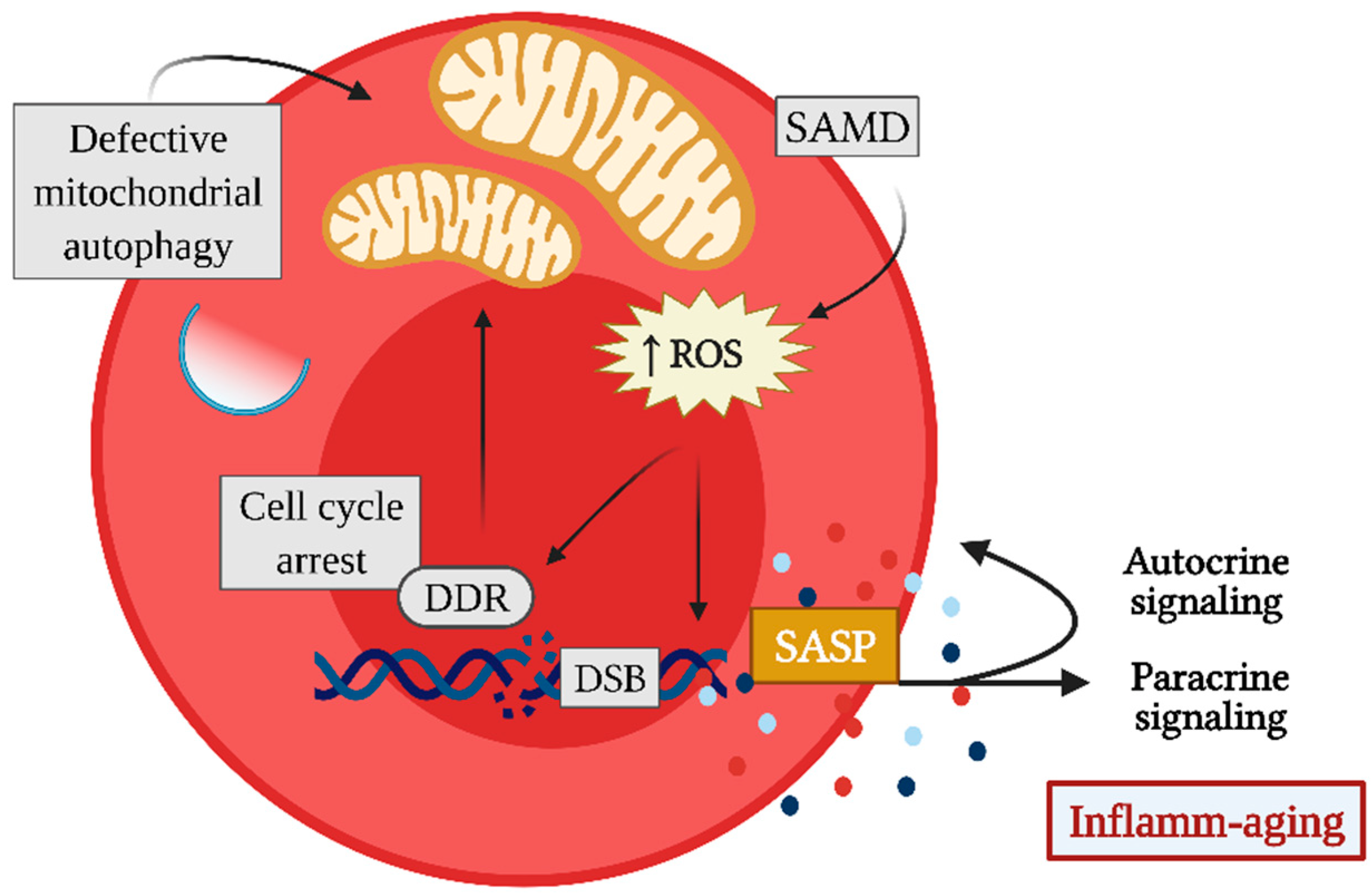
2. Cellular Senescence
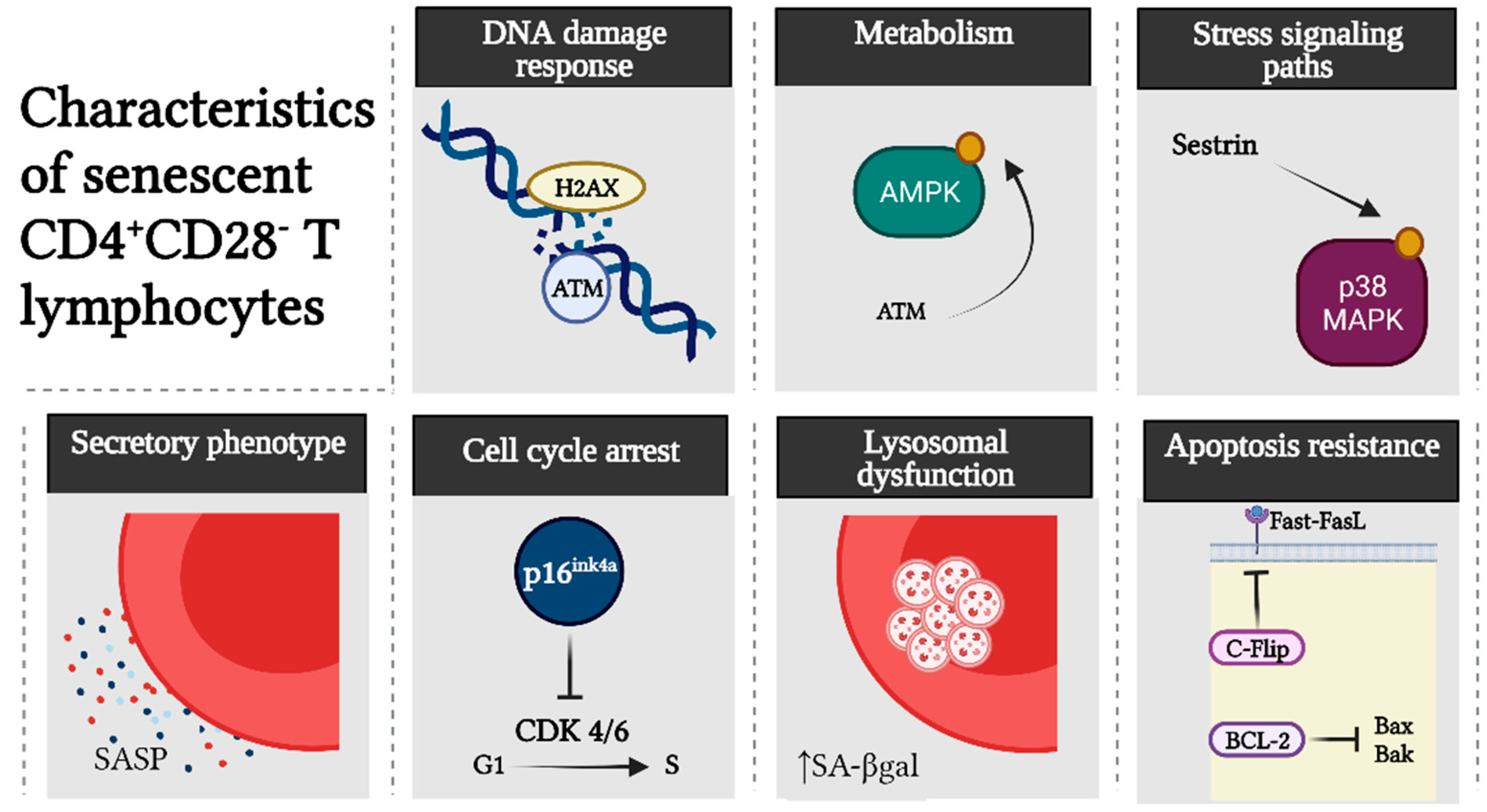
3. Senescent CD4+CD28− T Lymphocytes
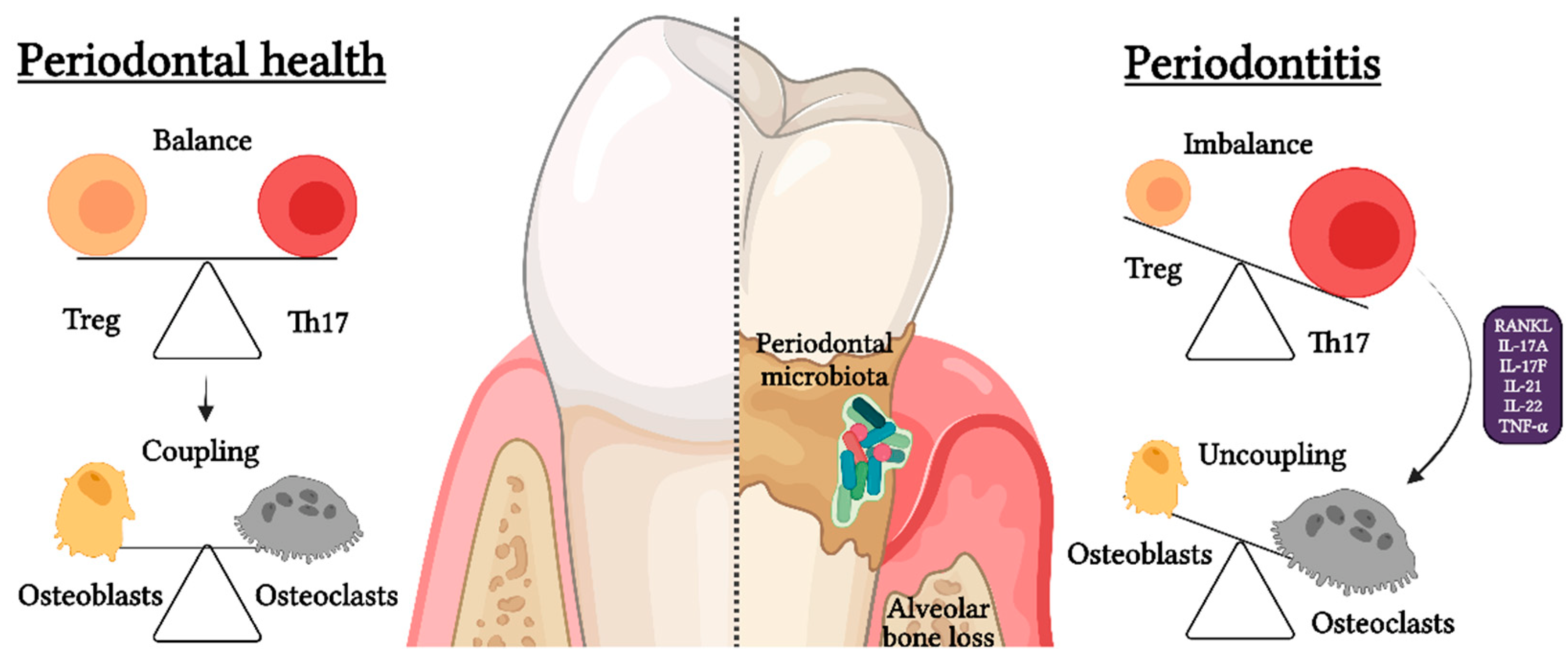
4. Osteoimmunology and Th17/Treg Imbalance
5. Senescent T Lymphocytes and Th17/Treg Imbalance during Periodontitis
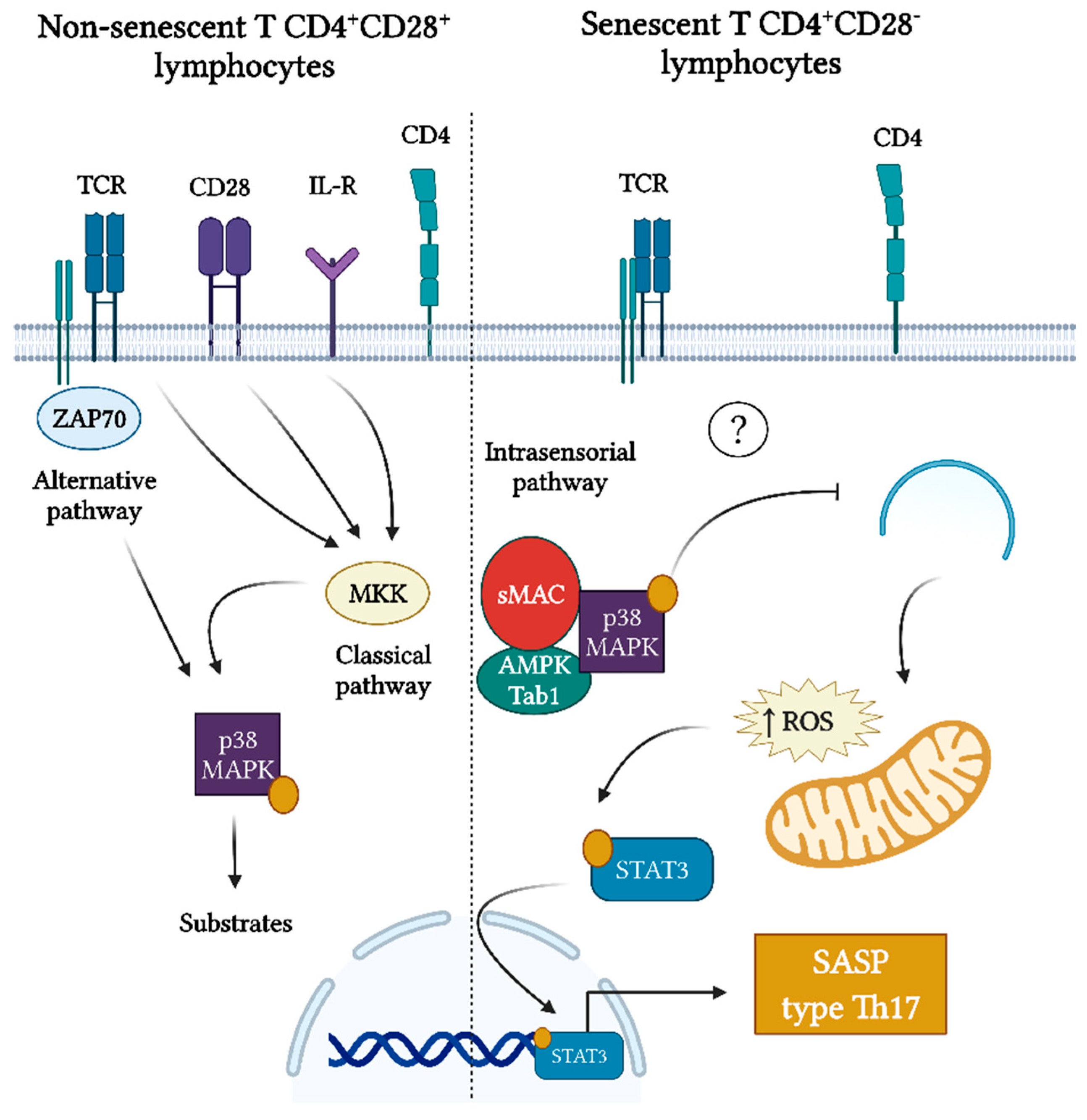
6. p38 MAPK as a Driver of SASP Development in Senescent CD4+CD28− T Cells
7. Autophagic Dysfunction during Senescence and Th17/Treg Imbalance
8. Towards the Encounter of Periodontitis-Related Senescent T Lymphocytes
9. T Lymphocyte Senescence as a Potential Therapeutic Approach during Periodontitis
10. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, Y.; Dong, G.; Xiao, W.; Xiao, E.; Miao, F.; Syverson, A.; Missaghian, N.; Vafa, R.; Ortega, A.A.C.; Rossa, J.C.; et al. Effect of aging on periodontal inflammation, microbial colonization, and disease susceptibility. J. Dent. Res. 2016, 95, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graves, D.T.; Oates, T.; Garlet, G.P. Review of osteoimmunology and the host response in endodontic and periodontal lesions. J. Oral Microbiol. 2011, 3, 5304. [Google Scholar] [CrossRef] [PubMed]
- Bartold, P.M.; Van Dyke, T.E. Host modulation: Controlling the inflammation to control the infection. Periodontol. 2000 2017, 75, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. The inflammophilic character of the periodontitis-associated microbiota. Mol. Oral Microbiol. 2014, 29, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loos, B.G.; Van Dyke, T.E. The role of inflammation and genetics in periodontal disease. Periodontol. 2000 2020, 83, 26–39. [Google Scholar] [CrossRef]
- Aquino-Martinez, R.; Khosla, S.; Farr, J.N.; Monroe, D.G. Periodontal disease and senescent cells: New players for an old oral health problem? Int. J. Mol. Sci. 2020, 21, 7441. [Google Scholar] [CrossRef]
- González-Osuna, L.; Sierra-Cristancho, A.; Rojas, C.; Cafferata, E.; Melgar-Rodríguez, S.; Cárdenas, M.; Vernal, R. Premature senescence of T-cells favors bone loss during osteolytic diseases. A new concern in the osteoimmunology arena. Aging Dis. 2021, 12, 1150–1161. [Google Scholar] [CrossRef]
- Humphreys, D.; ElGhazaly, M.; Frisan, T. Senescence and host-pathogen interactions. Cells 2020, 9, 1747. [Google Scholar] [CrossRef]
- Vallejo, A.N.; Weyand, C.M.; Goronzy, J.J. T-cell senescence: A culprit of immune abnormalities in chronic inflammation and persistent infection. Trends Mol. Med. 2004, 10, 119–124. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Quax, W.J.; Demaria, M. Regulation of survival networks in senescent cells: From mechanisms to interventions. J. Mol. Biol. 2019, 431, 2629–2643. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Ben Ameur, R.; Bauwens, E.; Dumortier, E.; Toutfaire, M.; Toussaint, O. Stress-induced (premature) senescence. In Cellular Ageing and Replicative Senescence; Rattan, S.I., Hayflick, L., Eds.; Springer: Cham, Switzerland, 2016; pp. 243–262. [Google Scholar]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; Di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Schank, M.; Zhao, J.; Wang, L.; Li, Z.; Cao, D.; Nguyen, L.N.; Dang, X.; Khanal, S.; Thakuri, B.K.C.; Ogbu, S.C.; et al. Telomeric injury by KML001 in human T cells induces mitochondrial dysfunction through the p53-PGC-1α pathway. Cell Death Dis. 2020, 11, 1030. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; Von Zglinicki, T. Mitochondria in cell senescence: Is mitophagy the weakest link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuire, P.J. Mitochondrial dysfunction and the aging immune system. Biology 2019, 8, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Park, J.T.; Lee, Y.S.; Cho, K.A.; Park, S.C. Adjustment of the lysosomal-mitochondrial axis for control of cellular senescence. Ageing Res. Rev. 2018, 47, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial dysfunction and cell senescence: Deciphering a complex relationship. FEBS Lett. 2019, 593, 1566–1579. [Google Scholar] [CrossRef] [Green Version]
- Cuollo, L.; Antonangeli, F.; Santoni, A.; Soriani, A. The senescence-associated secretory phenotype (SASP) in the challenging future of cancer therapy and age-related diseases. Biology 2020, 9, 485. [Google Scholar] [CrossRef]
- Aquino-Martinez, R.; Rowsey, J.L.; Fraser, D.G.; Eckhardt, B.A.; Khosla, S.; Farr, J.N.; Monroe, D.G. LPS-induced premature osteocyte senescence: Implications in inflammatory alveolar bone loss and periodontal disease pathogenesis. Bone 2020, 132, 115220. [Google Scholar] [CrossRef]
- Elsayed, R.; Elashiry, M.; Liu, Y.; El-Awady, A.; Hamrick, M.; Cutler, C.W. Exosome secretion and paracrine immune senescence in bystander dendritic cells. Front. Cell. Infect. Microbiol. 2021, 11, 669989. [Google Scholar] [CrossRef]
- Elashiry, M.; Elsayed, R.; Cutler, C.W. Exogenous and endogenous dendritic cell-derived exosomes: Lessons learned for immunotherapy and disease pathogenesis. Cells 2022, 11, 115. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Patil, C.K.; Campisi, J. P38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callender, L.; Carroll, E.C.; Beal, R.W.J.; Chambers, E.; Nourshargh, S.; Akbar, A.; Henson, S.M. Human CD8+ EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell 2018, 17, e12675. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, J.L.; Tchkonia, T. Cellular senescence: A translational perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Acuto, O.; Michel, F. CD28-mediated co-stimulation: A quantitative support for TCR signalling. Nat. Rev. Immunol. 2003, 3, 939–951. [Google Scholar] [CrossRef]
- Boesteanu, A.C.; Katsikis, P.D. Memory T cells need CD28 costimulation to remember. Semin. Immunol. 2009, 21, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Vallejo, A.N. CD28 extinction in human T cells: Altered functions and the program of T-cell senescence. Immunol. Rev. 2005, 205, 158–169. [Google Scholar] [CrossRef]
- Vallejo, A.N.; Bryl, E.; Klarskov, K.; Naylor, S.; Weyand, C.M.; Goronzy, J.J. Molecular basis for the loss of CD28 expression in senescent T cells. J. Biol. Chem. 2002, 277, 46940–46949. [Google Scholar] [CrossRef] [Green Version]
- Wagner, U.G.; Koetz, K.; Weyand, C.M.; Goronzy, J.J. Perturbation of the T cell repertoire in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 1998, 95, 14447–14452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, D.; Martens, P.B.; Weyand, C.M.; Goronzy, J.J. The repertoire of CD4+CD28− T cells in rheumatoid arthritis. Mol. Med. 1996, 2, 608–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzuela, H.F.; Effros, R.B. Divergent telomerase and CD28 expression patterns in human CD4 and CD8 T cells following repeated encounters with the same antigenic stimulus. Clin. Immunol. 2002, 105, 117–125. [Google Scholar] [CrossRef]
- Ma, S.; Ochi, H.; Cui, L.; Zhang, J.; He, W. Hydrogen peroxide induced down-regulation of CD28 expression of Jurkat cells is associated with a change of site α-specific nuclear factor binding activity and the activation of caspase-3. Exp. Gerontol. 2003, 38, 1109–1118. [Google Scholar] [CrossRef]
- Lanna, A.; Coutavas, E.; Levati, L.; Seidel, J.; Rustin, M.H.A.; Henson, S.; Akbar, A.N.; Franzese, O. IFN-α inhibits telomerase in human CD8+ T cells by both hTERT downregulation and induction of p38 MAPK Signaling. J. Immunol. 2013, 191, 3744–3752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryl, E.; Vallejo, A.N.; Weyand, C.M.; Goronzy, J.J. Down-regulation of CD28 expression by TNF-α. J. Immunol. 2001, 167, 3231–3238. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.P.; Ramirez, C.M.; Ryba, D.M.; Koduri, M.P.; Effros, R.B. Prostaglandin E2 promotes features of replicative senescence in chronically activated human CD8+ T cells. PLoS ONE 2014, 9, e99432. [Google Scholar] [CrossRef]
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Lanna, A.; O Gomes, D.C.; Muller-Durovic, B.; McDonnell, T.; Escors, D.; Gilroy, D.W.; Lee, J.H.; Karin, M.; Akbar, A.N. A sestrin-dependent Erk-Jnk-p38 MAPK activation complex inhibits immunity during aging. Nat. Immunol. 2017, 18, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Maly, K.; Schirmer, M. The story of CD4+CD28− T cells revisited: Solved or still ongoing? J. Immunol. Res. 2015, 2015, 348746. [Google Scholar] [CrossRef] [Green Version]
- Seyda, M.; Elkhal, A.; Quante, M.; Falk, C.S.; Tullius, S.G. T cells going innate. Trends Immunol. 2016, 37, 546–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, M.; Ruan, L.; Huang, Y.; Wang, J.; Yan, J.; Sang, Y.; Li, S.; Wang, G.; Wu, X. Premature CD4+ T cells senescence induced by chronic infection in patients with acute coronary syndrome. Aging Dis. 2020, 11, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Fessler, J.; Raicht, A.; Husic, R.; Ficjan, A.; Schwarz, C.; Duftner, C.; Schwinger, W.; Graninger, W.; Stradner, M.H.; Dejaco, C. Novel senescent regulatory T-cell subset with impaired suppressive function in rheumatoid arthritis. Front. Immunol. 2017, 8, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirmer, M.; Vallejo, A.N.; Weyand, C.M.; Goronzy, J.J. Resistance to apoptosis and elevated expression of Bcl-2 in clonally expanded CD4+CD28− T cells from rheumatoid arthritis patients. J. Immunol. 1998, 161, 1018–1025. [Google Scholar]
- Kovalcsik, E.; Antunes, R.F.; Baruah, P.; Kaski, J.C.; Dumitriu, I.E. Proteasome-mediated reduction in proapoptotic molecule bim renders CD4+CD28null T cells resistant to apoptosis in acute coronary syndrome. Circulation 2015, 131, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, C.; Monasterio, G.; Cavalla, F.; Córdova, L.A.; Hernández, M.; Heymann, D.; Garlet, G.P.; Sorsa, T.; Pärnänen, P.; Lee, H.-M.; et al. Osteoimmunology of oral and maxillofacial diseases: Translational applications based on biological mechanisms. Front. Immunol. 2019, 10, 1664. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Li, S.; Pacios, S.; Wang, Y.; Graves, D.T. Bone remodeling under pathological conditions. Front. Oral Biol. 2016, 18, 17–27. [Google Scholar] [CrossRef]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [Green Version]
- Dutzan, N.; Kajikawa, T.; Abusleme, L.; Greenwell-Wild, T.; Zuazo, C.E.; Ikeuchi, T.; Brenchley, L.; Abe, T.; Hurabielle, C.; Martin, D.; et al. A dysbiotic microbiome triggers Th17 cells to mediate oral mucosal immunopathology in mice and humans. Sci. Transl. Med. 2018, 10, eaat0797. [Google Scholar] [CrossRef] [Green Version]
- Monasterio, G.; Castillo, F.; Ibarra, J.P.; Guevara, J.; Rojas, L.; Alvarez, C.; Fernández, B.; Agüero, A.; Betancur, D.; Vernal, R. Alveolar bone resorption and Th1/Th17-associated immune response triggered during Aggregatibacter actinomycetemcomitans-induced experimental periodontitis are serotype-dependent. J. Periodontol. 2018, 89, 1249–1261. [Google Scholar] [CrossRef]
- Okamoto, K.; Takayanagi, H. Osteoclasts in arthritis and Th17 cell development. Int. Immunopharmacol. 2011, 11, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Zúñiga, J.; Melgar-Rodríguez, S.; Alvarez, C.; Monasterio, G.; Benítez, A.; Ciuchi, P.; Díaz, C.; Mardones, J.; Escobar, A.; Sanz, M.; et al. T-lymphocyte phenotype and function triggered by Aggregatibacter actinomycetemcomitans is serotype-dependent. J. Periodontal. Res. 2015, 50, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Vernal, R.; Diaz-Guerra, E.; Silva, A.; Sanz, M.; Garcia-Sanz, J.A. Distinct human T-lymphocyte responses triggered by Porphyromonas gingivalis capsular serotypes. J. Clin. Periodontol. 2014, 41, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernal, R.; Díaz-Zúñiga, J.; Melgar-Rodríguez, S.; Pujol, M.; Guerra, E.D.; Silva, A.; Sanz, M.; Garcia-Sanz, J.A. Activation of RANKL-induced osteoclasts and memory T lymphocytes by Porphyromonas gingivalis is serotype dependant. J. Clin. Periodontol. 2014, 41, 451–459. [Google Scholar] [CrossRef]
- Melgar-Rodríguez, S.; Díaz-Zúñiga, J.; Alvarez, C.; Rojas, L.; Monasterio, G.; Carvajal, P.; Escobar, A.; Sanz, M.; Vernal, R. Serotype b of Aggregatibacter actinomycetemcomitans increases osteoclast and memory T-lymphocyte activation. Mol. Oral Microbiol. 2016, 31, 162–174. [Google Scholar] [CrossRef]
- Ponzetti, M.; Rucci, N. Updates on osteoimmunology: What’s new on the cross-talk between bone and immune system. Front. Endocrinol. 2019, 10, 236. [Google Scholar] [CrossRef]
- E Adamopoulos, I.; Chao, C.-C.; Geissler, R.; Laface, D.; Blumenschein, W.; Iwakura, Y.; McClanahan, T.; Bowman, E.P. Interleukin-17A upregulates receptor activator of NF-κB on osteoclast precursors. Arthritis Res. Ther. 2010, 12, R29. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Li, F.; Li, X.; Wang, Z.G.; Zhang, B. TNF-α and RANKL promote osteoclastogenesis by upregulating RANK via the NF-κB pathway. Mol. Med. Rep. 2018, 17, 6605–6611. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.W.; Kim, H.R.; Kim, B.M.; Cho, M.; Lee, S.H. Th17 cytokines regulate osteoclastogenesis in rheumatoid arthritis. Am. J. Pathol. 2015, 185, 3011–3024. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor necrosis factor α stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 2000, 191, 275–286. [Google Scholar] [CrossRef]
- Bertoldo, E.; Adami, G.; Rossini, M.; Giollo, A.; Orsolini, G.; Viapiana, O.; Gatti, D.; Fassio, A. The emerging roles of endocrine hormones in different arthritic disorders. Front. Endocrinol. 2021, 12, 510. [Google Scholar] [CrossRef]
- Zhang, J.R.; Pang, D.D.; Tong, Q.; Liu, X.; Su, D.F.; Dai, S.M. Different modulatory effects of IL-17, IL-22, and IL-23 on osteoblast differentiation. Mediat. Inflamm. 2017, 2017, 5950395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, L.; Sun, J.; Han, J.; Jiang, X.; Wang, Z.; Chen, L. Interleukin-17 induces pyroptosis in osteoblasts through the NLRP3 inflammasome pathway in vitro. Int. Immunopharmacol. 2021, 96, 107781. [Google Scholar] [CrossRef] [PubMed]
- Knochelmann, H.M.; Dwyer, C.; Bailey, S.; Amaya, S.M.; Elston, D.M.; Mazza-McCrann, J.M.; Paulos, C.M. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell. Mol. Immunol. 2018, 15, 458–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, C.; Rojas, C.; Rojas, L.; Cafferata, E.A.; Monasterio, G.; Vernal, R. Regulatory T lymphocytes in periodontitis: A translational view. Mediat. Inflamm. 2018, 2018, 7806912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaiss, M.; Axmann, R.; Zwerina, J.; Polzer, K.; Gückel, E.; Skapenko, A.; Schulze-Koops, H.; Horwood, N.; Cope, A.; Schett, G. Treg cells suppress osteoclast formation: A new link between the immune system and bone. Arthritis Rheum. 2007, 56, 4104–4112. [Google Scholar] [CrossRef]
- Bozec, A.; Zaiss, M.M.; Kagwiria, R.; Voll, R.; Rauh, M.; Chen, Z.; Mueller-Schmucker, S.; Kroczek, R.A.; Heinzerling, L.; Moser, M.; et al. T cell costimulation molecules CD80/86 inhibit osteoclast differentiation by inducing the IDO/tryptophan pathway. Sci. Transl. Med. 2014, 6, 235ra60. [Google Scholar] [CrossRef]
- Tyagi, A.M.; Yu, M.; Darby, T.M.; Vaccaro, C.; Li, J.-Y.; Owens, J.A.; Hsu, E.; Adams, J.; Weitzmann, M.N.; Jones, R.M.; et al. The microbial metabolite butyrate stimulates bone formation via T regulatory cell-mediated regulation of wnt10b expression. Immunity 2018, 49, 1116–1131.e7. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; D’Amelio, P.; Tyagi, A.M.; Vaccaro, C.; Malik, A.; Hsu, E.; Buondonno, I.; Sassi, F.; Adams, J.; Weitzmann, M.N.; et al. Regulatory T cells are expanded by teriparatide treatment in humans and mediate intermittent PTH-induced bone anabolism in mice. EMBO Rep. 2018, 19, 156–171. [Google Scholar] [CrossRef]
- Tsukasaki, M.; Komatsu, N.; Nagashima, K.; Nitta, T.; Pluemsakunthai, W.; Shukunami, C.; Iwakura, Y.; Nakashima, T.; Okamoto, K.; Takayanagi, H. Host defense against oral microbiota by bone-damaging T cells. Nat. Commun. 2018, 9, 701. [Google Scholar] [CrossRef]
- Alvarez, C.; Suliman, S.; Almarhoumi, R.; Vega, M.E.; Rojas, C.; Monasterio, G.; Galindo, M.; Vernal, R.; Kantarci, A. Regulatory T cell phenotype and anti-osteoclastogenic function in experimental periodontitis. Sci. Rep. 2020, 10, 19018. [Google Scholar] [CrossRef] [PubMed]
- Patlán, M.; Páez, A.; Massó, F.; Amezcua-Guerra, L.M. Relative increase of Th17 phenotype in senescent CD4+CD28null T cells from peripheral blood of patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2021, 39, 925–926. [Google Scholar] [PubMed]
- Fessler, J.; Husic, R.; Schwetz, V.; Lerchbaum, E.; Aberer, F.; Fasching, P.; Ficjan, A.; Obermayer-Pietsch, B.; Duftner, C.; Graninger, W.; et al. Senescent T-cells promote bone loss in rheumatoid arthritis. Front. Immunol. 2018, 9, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phoksawat, W.; Jumnainsong, A.; Sornkayasit, K.; Srisak, K.; Komanasin, N.; Leelayuwat, C. IL-17 and IFN-γ productions by CD4+ T cells and T cell subsets expressing NKG2D associated with the number of risk factors for cardiovascular diseases. Mol. Immunol. 2020, 122, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.M.; Srivastava, K.; Sharan, K.; Yadav, D.; Maurya, R.; Singh, D. Daidzein prevents the increase in CD4+CD28null T cells and B lymphopoesis in ovariectomized mice: A key mechanism for anti-osteoclastogenic effect. PLoS ONE 2011, 6, e21216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, A.M.; Srivastava, K.; Kureel, J.; Kumar, A.; Raghuvanshi, A.; Yadav, D.; Maurya, R.; Goel, A.; Singh, D. Premature T cell senescence in Ovx mice is inhibited by repletion of estrogen and medicarpin: A possible mechanism for alleviating bone loss. Osteoporos. Int. 2012, 23, 1151–1161. [Google Scholar] [CrossRef]
- Kumar, G.; Roger, P.; Ticchioni, M.; Trojani, C.; de Dompsur, R.B.; Bronsard, N.; Carles, M.; Bernard, E. T cells from chronic bone infection show reduced proliferation and a high proportion of CD28−CD4 T cells. Clin. Exp. Immunol. 2014, 176, 49–57. [Google Scholar] [CrossRef]
- Dapunt, U.; Giese, T.; Prior, B.; Gaida, M.M.; Hänsch, G.M. Infectious versus non-infectious loosening of implants: Activation of T lymphocytes differentiates between the two entities. Int. Orthop. 2014, 38, 1291–1296. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Yang, J.; Niu, Z. Association of periodontitis with leukocyte telomere length in US adults: A cross-sectional analysis of NHANES 1999 to 2002. J. Periodontol. 2021, 92, 833–843. [Google Scholar] [CrossRef]
- Masi, S.; Salpea, K.D.; Li, K.; Parkar, M.; Nibali, L.; Donos, N.; Patel, K.; Taddei, S.; Deanfield, J.E.; D’Aiuto, F.; et al. Oxidative stress, chronic inflammation, and telomere length in patients with periodontitis. Free Radic. Biol. Med. 2011, 50, 730–735. [Google Scholar] [CrossRef]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-Hora, M.; Kodama, T.; Tanaka, S.; A Bluestone, J.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into Th17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Mathiasen, S.L.; Gall-Mas, L.; Pateras, I.S.; Theodorou, S.D.; Namini, M.R.; Hansen, M.B.; Martin, O.C.; Vadivel, C.K.; Ntostoglou, K.; Butter, D.; et al. Bacterial genotoxins induce T cell senescence. Cell Rep. 2021, 35, 109220. [Google Scholar] [CrossRef] [PubMed]
- Travan, S.; Li, F.; D’Silva, N.J.; Slate, E.H.; Kirkwood, K.L. Differential expression of mitogen activating protein kinases in periodontitis. J. Clin. Periodontol. 2013, 40, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Kirkwood, K.; Li, F.; Rogers, J.E.; Otremba, J.; Coatney, D.D.; Kreider, J.M.; D’Silva, N.J.; Chakravarty, S.; Dugar, S.; Higgins, L.S.; et al. A p38α selective mitogen-activated protein kinase inhibitor prevents periodontal bone loss. J. Pharmacol. Exp. Ther. 2007, 320, 56–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, J.E.; Li, F.; Coatney, D.D.; Otremba, J.; Kriegl, J.M.; A Protter, T.A.; Higgins, L.S.; Medicherla, S.; Kirkwood, K. A p38 mitogen-activated protein kinase inhibitor arrests active alveolar bone loss in a rat periodontitis model. J. Periodontol. 2007, 78, 1992–1998. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 2020, 32, 44–55.e6. [Google Scholar] [CrossRef]
- Akbar, A.N.; Henson, S.M.; Lanna, A. Senescence of T lymphocytes: Implications for enhancing human immunity. Trends Immunol. 2016, 37, 866–876. [Google Scholar] [CrossRef]
- Henson, S.M.; Lanna, A.; Riddell, N.E.; Franzese, O.; Macaulay, R.; Griffiths, S.J.; Puleston, D.J.; Watson, A.S.; Simon, A.K.; Tooze, S.A.; et al. P38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J. Clin. Investig. 2014, 124, 4004–4016. [Google Scholar] [CrossRef]
- Kovacs, J.R.; Li, C.; Yang, Q.; Li, G.; Garcia, I.G.; Ju, S.; Roodman, D.; Windle, J.; Zhang, X.; Lu, B. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ. 2012, 19, 144–152. [Google Scholar] [CrossRef]
- Cluxton, D.; Petrasca, A.; Moran, B.; Fletcher, J.M. Differential regulation of human treg and Th17 cells by fatty acid synthesis and glycolysis. Front. Immunol. 2019, 10, 115. [Google Scholar] [CrossRef] [Green Version]
- Park, M.-J.; Lee, S.-Y.; Moon, S.-J.; Son, H.-J.; Lee, S.-H.; Kim, E.-K.; Byun, J.-K.; Shin, D.Y.; Park, S.-H.; Yang, C.-W.; et al. Metformin attenuates graft-versus-host disease via restricting mammalian target of rapamycin/signal transducer and activator of transcription 3 and promoting adenosine monophosphate-activated protein kinase-autophagy for the balance between T helper 17 and Tregs. Transl Res. 2016, 173, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Carriche, G.M.; Almeida, L.; Stüve, P.; Velasquez, L.; Dhillon-LaBrooy, A.; Roy, U.; Lindenberg, M.; Strowig, T.; Plaza-Sirvent, C.; Schmitz, I.; et al. Regulating T cell differentiation through the polyamine spermidine. J. Allergy Clin. Immunol. 2020, 147, 335–348.e11. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Long, L.; Yang, K.; Guy, C.; Shrestha, S.; Chen, Z.; Wu, C.; Vogel, P.; Neale, G.; Green, D.R.; et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 2016, 17, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabat, A.M.; Harrison, O.; Riffelmacher, T.; Moghaddam, A.; Pearson, C.F.; Laing, A.; Abeler-Dörner, L.; Forman, S.P.; Grencis, R.K.; Sattentau, Q.; et al. The autophagy gene Atg16l1 differentially regulates Treg and Th2 cells to control intestinal inflammation. eLife 2016, 5, e12444. [Google Scholar] [CrossRef]
- Ren, W.; Yin, J.; Duan, J.; Liu, G.; Tan, B.; Yang, G.; Wu, G.; Bazer, F.W.; Peng, Y.; Yin, Y. mTORC1 signaling and IL-17 expression: Defining pathways and possible therapeutic targets. Eur. J. Immunol. 2016, 46, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of Th17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, S.; Singer, B.; Steinert, E.M.; Martinez, C.A.; Mehta, M.M.; Martinez-Reyes, I.; Gao, P.; Helmin, K.A.; Abdala-Valencia, H.; Sena, L.A.; et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 2019, 565, 495–499. [Google Scholar] [CrossRef]
- Beier, U.H.; Angelin, A.; Akimova, T.; Wang, L.; Liu, Y.; Xiao, H.; Koike, M.A.; Hancock, S.A.; Bhatti, T.R.; Han, R.; et al. Essential role of mitochondrial energy metabolism in Foxp3+ T-regulatory cell function and allograft survival. FASEB J. 2015, 29, 2315–2326. [Google Scholar] [CrossRef] [Green Version]
- Gerriets, V.A.; Kishton, R.J.; Johnson, M.O.; Cohen, S.; Siska, P.J.; Nichols, A.G.; Warmoes, M.O.; De Cubas, A.A.; Maciver, N.J.; Locasale, J.W.; et al. Foxp3 and Toll-like receptor signaling balance T reg cell anabolic metabolism for suppression. Nat. Immunol. 2016, 17, 1459–1466. [Google Scholar] [CrossRef]
- Araujo, L.; Khim, P.; Mkhikian, H.; Mortales, C.L.; Demetriou, M. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. eLife 2017, 6, e21330. [Google Scholar] [CrossRef]
- Cabral, J.; Hanley, S.A.; Gerlach, J.Q.; O’Leary, N.; Cunningham, S.; Ritter, T.; Ceredig, R.; Joshi, L.; Griffin, M.D. Distinctive surface glycosylation patterns associated with mouse and human CD4+ regulatory T cells and their suppressive function. Front. Immunol. 2017, 8, 987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Paul, W.E. Heterogeneity and plasticity of T helper cells. Cell Res. 2010, 20, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Mittelbrunn, M.; Kroemer, G. Hallmarks of T cell aging. Nat. Immunol. 2021, 22, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef]
- Di Mitri, D.; Azevedo, R.I.; Henson, S.M.; Libri, V.; Riddell, N.E.; Macaulay, R.; Kipling, D.; Soares, M.V.D.; Battistini, L.; Akbar, A.N. Reversible senescence in human CD4+CD45RA+CD27− memory T cells. J. Immunol. 2011, 187, 2093–2100. [Google Scholar] [CrossRef] [Green Version]
- Najeeb, S.; Zafar, M.S.; Khurshid, Z.; Zohaib, S.; Madathil, S.A.; Mali, M.; Almas, K. Efficacy of metformin in the management of periodontitis: A systematic review and meta-analysis. Saudi Pharm. J. 2018, 26, 634–642. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Evidence | Findings | References | Evidence Suggests That |
|---|---|---|---|
| Association of periodontitis with leukocyte telomere length. | Patients with periodontitis show leukocytes with significantly shorter telomeres than age-matched healthy subjects, which is associated with disease severity. | [80,81] | Periodontitis causes early replicative senescence in leukocytes. |
| Pro-inflammatory mediators induce T lymphocyte senescence. | Inflammatory mediators, such as interferon (IFN)-α, tumor necrosis factor (TNF)-α, prostaglandin E2 (PGE2), and ROS, are able to induce CD28 loss and senescence of T lymphocytes in vitro. | [35,36,37,38] | These mediators, being present in periodontitis, may trigger senescence of CD4+ T lymphocytes. |
| Bacterial genotoxins induce CD4+ T lymphocyte senescence. | The cytolethal distending toxin (CDT), a virulence factor present in the Gram-negative bacterium Aggregatibacter actinomycetemcomitans closely related to periodontitis etiology, induces premature senescence in CD4+ T lymphocytes in vitro and in vivo models. In this context, it is assumed that the produced SASP is induced by the activation of p38 MAPK signaling. | [83] | Periodontal pathogenic bacteria may play an important role in the induction of senescence in CD4+ T lymphocytes. |
| Differential activation of p38 MAPK signaling during periodontitis. | The phospho-p38 MAPK intensity score in immunostained tissues was positively correlated with clinical periodontal parameters of the disease linked to inflammation and bone loss, implying that p38 MAPK activation is one of the main signaling pathways involved in human periodontal inflammation and its severity. Inhibition of p38 MAPK activation in preclinical models of periodontitis prevented bone loss. | [84,85,86] | There could be a relation between the activation of the p38 MAPK signaling pathway in senescent CD4+ T lymphocytes and SASP production. |
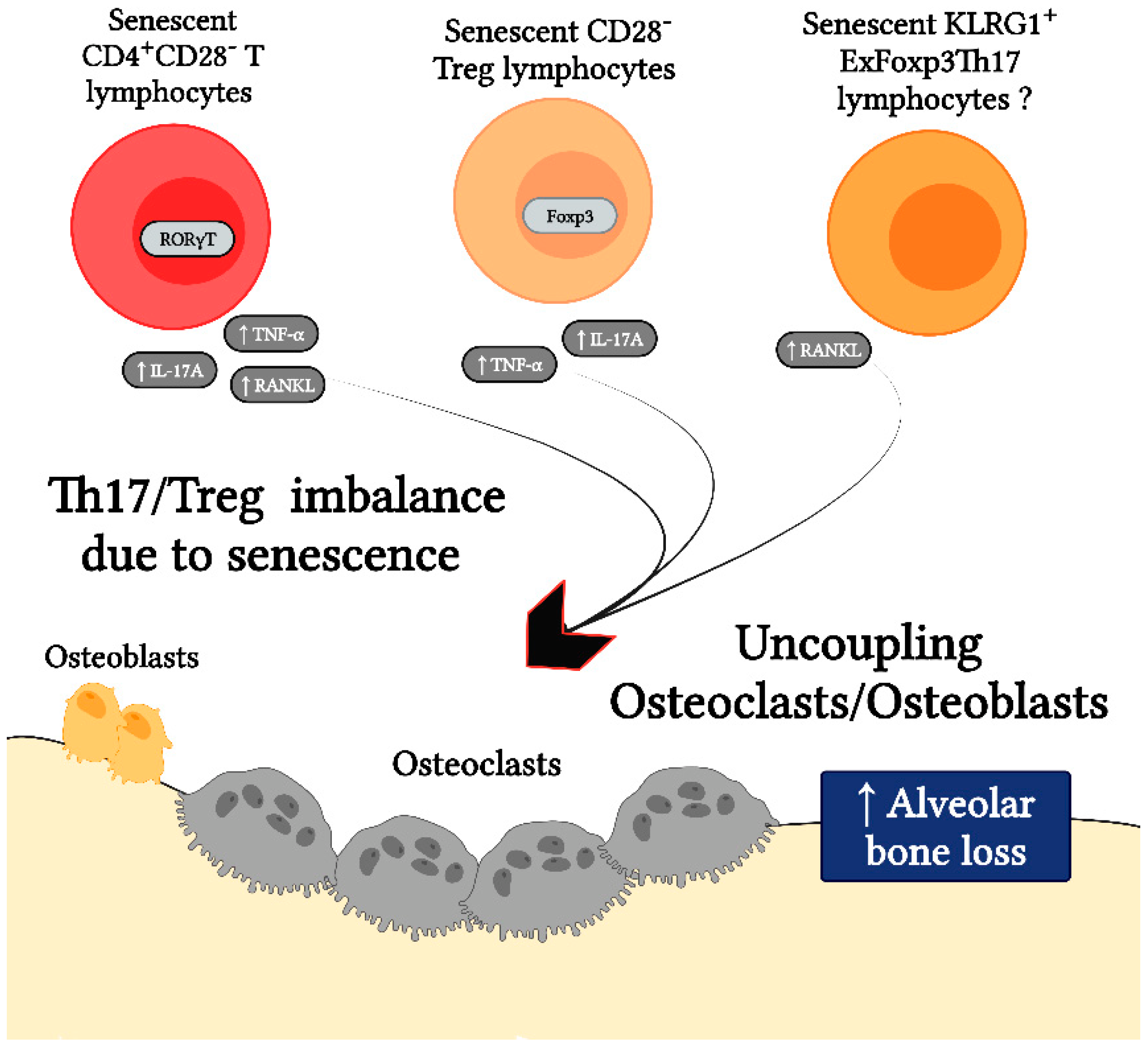
| Senescent CD4+ T lymphocytes exhibit a Th17-biased secretory profile. | Senescent CD4+CD28− T lymphocytes show a preferential polarization towards the Th17 phenotype, with the increased expression of RORγt. In addition, CD4+ T lymphocytes from elderly subjects show a Th17-biased cytokine production profile, due to the defects in autophagy and mitochondrial bioenergetics, which in turn are associated with redox imbalance and activation of the Th17 master regulator STAT-3 to bind to IL-17A promoters. | [73,87] | The senescence of CD4+ T lymphocytes during periodontitis may favor the Th17 lymphocyte polarization. |
| Senescent CD4+ T lymphocytes are a common feature of chronic osteolytic pathologies. | Senescent CD4+CD28− T lymphocytes are present in chronic osteolytic pathologies such as rheumatoid arthritis, osteopenia, osteoporosis, osteomyelitis, and during the loss of orthopedic bone implants due to infectious causes. In these contexts, senescent CD4+CD28− T lymphocytes show a greater osteoclastogenic capacity due to a higher production of RANKL and TNF-α, as compared with their non-senescent counterparts. | [74,77,78,79] | Senescent CD4+ T lymphocytes may be directly linked to alveolar bone loss due to the increased production of pro-osteoclastogenic mediators. |
| Senescent Tregs show impaired suppressor function and increased production of pro-inflammatory profile cytokines. | A novel subset of senescent CD28− Treg is described, which insufficiently suppressed the proliferation of effector T lymphocytes and produced a pro-inflammatory cytokine pattern. | [44] | Senescent Tregs in peridontitis may be related to an imbalance between their regulatory and effector functions. |
| During experimental periodontitis, exFoxp3Th17 KLRG1+ lymphocytes are generated. | During experimental periodontitis, Foxp3+ T lymphocytes are converted into exFoxp3Th17 cells, expressing KLRG1. KLRG1 is a hallmark of cellular senescence in T lymphocytes. Thus, exFoxp3Th17 cells play a key role in the pathogenesis of periodontitis by expressing high amounts of IL-17A and RANKL and showing a potent osteoclastogenic capacity in vivo. | [71] | Senescent Tregs appear to be generated in the context of periodontitis and play a key role during the alveolar bone resorption due to the polarization bias towards the Th17 phenotype. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Osuna, L.; Sierra-Cristancho, A.; Cafferata, E.A.; Melgar-Rodríguez, S.; Rojas, C.; Carvajal, P.; Cortez, C.; Vernal, R. Senescent CD4+CD28− T Lymphocytes as a Potential Driver of Th17/Treg Imbalance and Alveolar Bone Resorption during Periodontitis. Int. J. Mol. Sci. 2022, 23, 2543. https://doi.org/10.3390/ijms23052543
González-Osuna L, Sierra-Cristancho A, Cafferata EA, Melgar-Rodríguez S, Rojas C, Carvajal P, Cortez C, Vernal R. Senescent CD4+CD28− T Lymphocytes as a Potential Driver of Th17/Treg Imbalance and Alveolar Bone Resorption during Periodontitis. International Journal of Molecular Sciences. 2022; 23(5):2543. https://doi.org/10.3390/ijms23052543
Chicago/Turabian StyleGonzález-Osuna, Luis, Alfredo Sierra-Cristancho, Emilio A. Cafferata, Samanta Melgar-Rodríguez, Carolina Rojas, Paola Carvajal, Cristian Cortez, and Rolando Vernal. 2022. "Senescent CD4+CD28− T Lymphocytes as a Potential Driver of Th17/Treg Imbalance and Alveolar Bone Resorption during Periodontitis" International Journal of Molecular Sciences 23, no. 5: 2543. https://doi.org/10.3390/ijms23052543