
Synthesis, Characterization, Antimicrobial Screening and Free-Radical Scavenging Activity of Some Novel Substituted Pyrazoles

Abstract
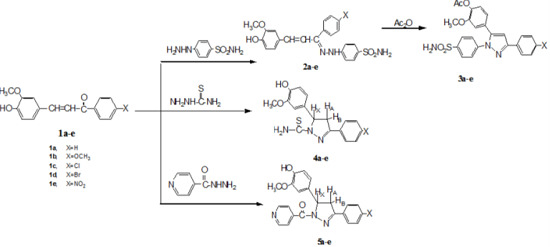
:
1. Introduction

2. Results and Discussion
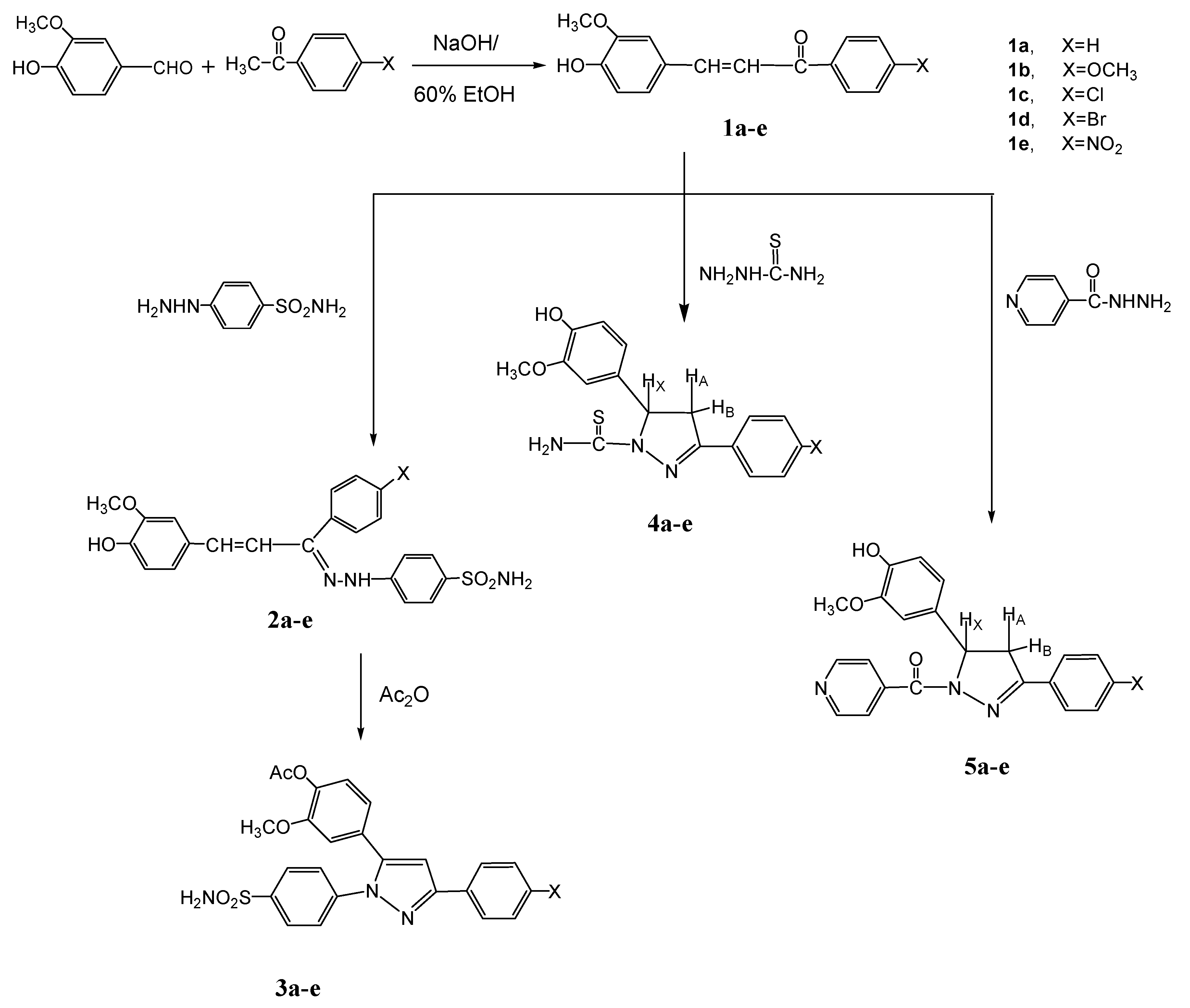
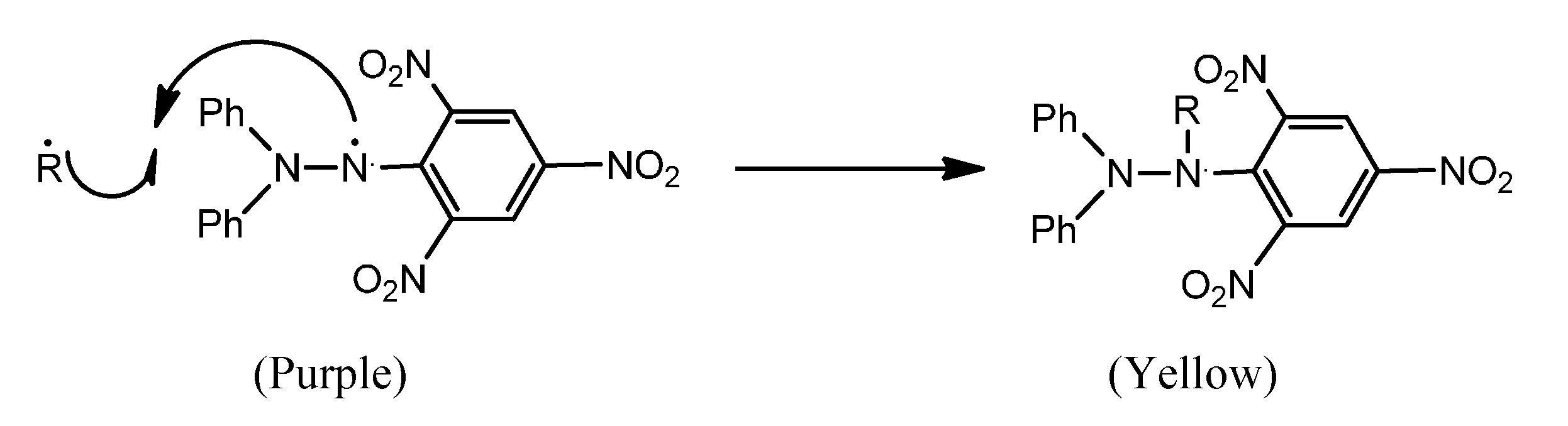
2.1. Chemistry

2.2. Pharmacological Activity
2.2.1. In Vitro Antibacterial Screening of Synthesized Compounds

| Compound No. | Zone of Inhibition (mm) | Minimum Inhibitory Concentration (MIC) mg/mL | ||
|---|---|---|---|---|
| S. aureus | C. albicans | S. aureus | C. albicans | |
| 1a | - | 15 | - | - |
| 1b | - | 15 | - | - |
| 1c | 21 | 20 | 0.1 | 0.05 |
| 1d | 15 | 18 | 0.063 | 0.063 |
| 1e | - | 15 | - | - |
| 2a | 19 | 22 | 0.063 | 0.031 |
| 2b | 18 | 25 | 0.125 | 0.031 |
| 2c | 22 | 26 | 0.05 | 0.05 |
| 2d | 18 | 20 | 0.063 | 0.125 |
| 2e | 17 | 17 | - | - |
| 4a | - | 15 | - | - |
| 4b | - | 15 | - | - |
| 4c | 21 | 24 | 0.05 | 0.05 |
| 4d | - | 15 | - | - |
| 4e | 17 | 17 | - | - |
| 5a | - | 20 | - | 0.25 |
| 5b | - | 15 | - | - |
| 5c | 17 | 20 | 0.1 | 0.05 |
| 5d | 14 | 20 | 0.12 | 0.5 |
| 5e | 12 | 15 | - | - |
| Rifampicin | 32 | - | - | - |
| Ampicillin | 30 | - | - | - |
| DMSO | - | 14 | - | - |
| Concentrations mg/mL | 1 | 0.50 | 0.25 | 0.125 | 0.063 | 0.031 | 1 | 0.50 | 0.25 | 0.125 | 0.063 | 0.031 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Microorganism Growth | S. aureus | C. albicans | ||||||||||
| 1c | − | − | * | + | + | + | − | * | + | + | + | + |
| 2c | − | − | * | + | + | + | − | − | * | + | + | + |
| 4c | − | * | + | + | + | + | − | − | − | * | + | + |
| 5c | − | − | * | + | + | + | − | − | − | * | + | + |
2.2.2. Evaluation of Antioxidant and Anti-inflammatory Activities
Antioxidant Activity (DPPH Based Free Radical Scavenging Activity)

Anti-Inflammatory Activity (Scavenging of Nitric Oxide Radical)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | Mean Absorbance ± S.D.* at Different Concentrations | Efficacy at 0.25/0.25 Vitamin C | |||
|---|---|---|---|---|---|
| 0.25 mg/mL | 0.5 mg/mL | 0.75 mg/mL | 1 mg/mL | ||
| 1c | 9.4 ± 0.5 | −65 ± 3.5 | −69 ± 5.2 | −88 ± 3.5 | 0.13 |
| 2c | 58 ± 3.2 | 60 ± 2.5 | 91 ± 4.2 | 94 ± 6.5 | 0.81 |
| 3c | 20 ± 2.4 | 12 ± 1.2 | −12 ± 1.2 | −5 ± 0.5 | 0.28 |
| 4c | 78.8 ± 1.2 | 43.5 ± 2.5 | 31.8 ± 3.1 | −69 ± 6 | 1.1 |
| 5c | 64.7 ± 4.3 | 51 ± 2.3 | 21 ± 2.5 | −7 ± 0.8 | 0.9 |
| Vitamin C | 72 ± 2.5 | 76 ± 1.8 | 89 ± 2.1 | 94 ± 2.8 | |
| Compound No. | Mean Absorbance ± S.D.* at Different Concentrations | Efficacy at 0.25/0.25 Vitamin C | |||
|---|---|---|---|---|---|
| 0.25 mg/mL | 0.5 mg/mL | 0.75 mg/mL | 1 mg/mL | ||
| 1c | 36 ± 1.3 | 38 ± 2.1 | 41 ± 4.1 | 44 ± 1.5 | 0.88 |
| 2c | 17 ± 1.7 | 19 ± 1.2 | 25 ± 1.5 | 29 ± 1.8 | 0.4 |
| 3c | 7 ± 1.1 | 14 ± 1.2 | 25 ± 1.5 | 47 ± 3.5 | 0.94 |
| 4c | 19 ± 1.9 | 36 ± 3.2 | 58 ± 2.4 | 74 ± 4.5 | 1.5 |
| 5c | 26 ± 1.9 | 34 ± 1.5 | 42 ± 2.5 | 62 ± 5.2 | 1.03 |
| Vitamin C | 50 ± 1.2 | ||||
3. Experimental Section
3.1. General Information
3.1.1. General Procedure for the Preparation of 1a–e
3.1.2. General Procedure for the Preparation of 2a–e
3.1.3. General Procedure for the Preparation of 3a–e
3.1.4. General Procedure for the Preparation of 4a–e
3.1.5. General Procedure for the Preparation of 5a–e
3.2. Determination of Antimicrobial Activity
3.3. DPPH Based Free Radical Scavenging Activity
3.4. Nitric Oxide Radical Scavenging Activity
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Taylor, E.C.; Patel, H.H. Synthesis of Pyrazolo [3,4-d] Pyrimidine Analogues of the potentagent. N-{4-[2-(2-amino-4(3H)-oxo-7H-pyrrolo[2,3-d]Pyrimidin-5-yl) ethyl]benzoyl}-l-Glutamic acid (LY231514). Tetrahedron 1992, 48, 8089–8100. [Google Scholar]
- Song, H.; Liu, Y.; Xiong, L.; Li, Y.; Yang, N.; Wang, Q. Design, Synthesis and Insecticidal Activity of Novel Pyrazole Derivatives Containing α-hydroxymethyl-N-Benzylcarboxamide, α-Chloromethyl-N-Benzyl Carboxamide, and 4,5-Dihydrooxazole moieties. J. Agric. Food Chem. 2012, 60, 1470–1479. [Google Scholar] [CrossRef] [PubMed]
- Sharshira, E.M.; Hamada, N.M.M. Synthesis and Antimicrobial Evaluation of Some Pyrazole Derivatives. Molecules 2012, 17, 4962–4971. [Google Scholar] [CrossRef] [PubMed]
- Rashad, A.E.; Shamroukh, A.H.; Hegab, M.I.; Awad, H.M. Synthesis of Some Biologically Active Pyrazoles and C-Nucleosides. Acta Chim. Slov. 2005, 52, 429–434. [Google Scholar]
- Rashad, A.E.; Hegab, M.I.; Abdel-Megeid, R.E.; Micky, J.A.; Abdel-Megeid, F.M.E. Synthesis and Antiviral Evaluation of Some New Pyrazole and Fused Pyrazolo Pyrimidine Derivatives. Bioorg. Med. Chem. 2008, 16, 7102–7106. [Google Scholar] [CrossRef] [PubMed]
- Bhat, B.A.; Dhar, K.L.; Saxena, A.K.; Shanmugavel, M.; Qazi, G.N. Synthesis and Biological Evaluation of Chalcones and Their Derived Pyrazoles as Potential Cytotoxic Agents. Bioorg. Med. Chem. Lett. 2005, 15, 3177–3180. [Google Scholar] [CrossRef] [PubMed]
- Horrocks, P.; Pickard, M.R.; Parekh, H.H.; Patel, S.P.; Pathak, R.B. Synthesis and Biological Evaluation of 3-(4-Chlorophenyl)-4-Substituted Pyrazole Derivatives. Org. Biomol. Chem. 2013, 11, 4891–4898. [Google Scholar] [CrossRef] [PubMed]
- Kalirajan, R.; Sivakumar, S.U.; Jubie, S.; Gowramma, B.; Suresh, B. Synthesis and Biological Evaluation of Some Heterocyclic Derivatives of Chalcones. Int. J. ChemTech Res. 2009, 1, 27–34. [Google Scholar]
- Holla, B.S.; Akberali, P.M.; Shivanada, M.K. Studies on Arylfuran Derivative: Part X. Synthesis and Antibacterial Properties of Arylfuryl-Δ2-Pyrazolines. Farmaco 2000, 55, 256–263. [Google Scholar]
- Maggio, B.; Daidone, G.; Raffa, D.; Plescia, S.; Mantione, L.; Cutuli, V.M.C.; Mangano, N.G.; Caruso, A. Synthesis and Pharmacological Study of ethyl 1-methyl-5-(substituted-3,4-dihydro-4-oxoquinazolin-3-yl)-1H-pyrazole-4-acetates. Eur. J. Med. Chem. 2001, 36, 737–742. [Google Scholar] [CrossRef]
- Clinton, R.O.; Manson, A.J.; Stonner, F.W.; Beyler, A.L.; Potts, G.O.; Arnold, A. Steroidal [3,2-c] pyrazoles. J. Am. Chem. Soc. 1959, 81, 1513–1514. [Google Scholar] [CrossRef]
- Kalirajan, R.; Palanivelu, M.; Rajamanickam, V.; Vinothapooshan, G.; Andarajagopal, K. Synthesis and Biological Evaluation of Some Heterocyclic Derivatives of Chalcones. Int. J. Chem. Sci. 2007, 5, 73–80. [Google Scholar]
- Urmila, G.; Vineeta, S.; Vineeta, K.; Sanjana, C. Synthesis and Antifungal Activity of New Fluorine Containing 4-(Substituted Phenylazo) Pyrazoles and Isoxazoles. Indian J. Heterocycl. Chem. 2005, 14, 265–266. [Google Scholar]
- Patel, C.K.; Rami, C.S.; Panigrahi, B.; Patel, C.N. Synthesis and Biological Evaluation of (4-Substituted Benzylidene)-3-Methyl-1-(Substituted Phenyl Sulfonyl and Substituted Benzoyl)-1H-Pyrazol-5(4H)-one As Anti-inflammatory Agent. J. Chem. Pharm. Res. 2010, 2, 73–78. [Google Scholar]
- Yang, J.F.; Cao, H.; Liu, H.; Li, B.Q.; Ma, Y.M. Synthesis and Bioactivity of Novel Bis-heterocyclic Compounds Containing Pyrazole and Oxadiazoline. J. Chin. Chem. Soc. 2011, 58, 369–375. [Google Scholar] [CrossRef]
- MallikarjunaRao, R.; Sreeramulu, J.; Ravindranath, L.K.; NagarajaReddy, G.; Hanumanthurayudu, K.; Nageswara Reddy, G.; Jayaraju, A.; Madhusudhan, P. Synthesis and biological screening of some Pyridine and Pyrrole Derivatives of Pyrazolo [3,4-c] pyrazoles. J. Chem. Pharm. Res. 2012, 4, 272–278. [Google Scholar]
- Mohareb, R.M.; El-Sayed, N.N.E.; Abdelaziz, M.A. Uses of Cyanoacetylhydrazine in Heterocyclic Synthesis: Novel Synthesis of Pyrazole Derivatives with Anti-tumor Activities. Molecules 2012, 17, 8449–8463. [Google Scholar]
- Kumar, K.A.; Jayaroopa, P. Pyrazoles: Synthetic Strategies and Their Pharmaceutical Applications-An Overview. Int. J. PharmTech Res. 2013, 5, 1473–1486. [Google Scholar]
- Piste, P.B. Facile Synthesis and Antimicrobial Screening of Pyrazole Derivatives. World. J. Pharm. Res. 2014, 3, 735–742. [Google Scholar]
- Kamal, A.; Shaik, A.B.; Polepalli, S.; Reddy, V.S.; Kumar, G.B.; Gupta, S.; Krishna, K.V.S.R.; Nagabhushana, A.; Mishra, R.K.; Jain, N. Pyrazole-Oxadiazole Conjugates: Synthesis, Antiproliferative Activity and Inhibition of Tubulin Polymerization. Org. Biomol. Chem. 2014, 12, 7993–8007. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, A.; Bari, S.S.; Bhalla, J. Synthesis of Novel Pyrazolylmethylene-Pyrimidine Heterocycles: Potential Synthons for Hybrid β-Lactams. Can. Chem. Trans. 2015, 3, 72–84. [Google Scholar]
- Abdelhamid, A.O.; Zohdi, H.F.; Sallam, M.M.M.; Ahmed, N.A. Reactions with Hydra- zonoyl Halides. 31. Synthesis of Some New Pyrrolidino [3,4-c] pyrazolines, Pyrazoles, and Pyrazolo [3,4-d] pyridazines. Molecules 2000, 5, 967–973. [Google Scholar] [CrossRef]
- Nakamichi, N.; Kawashita, Y.; Hayashi, M. Oxidative Aromatization of 1,3,5-Trisubstituted pyrazolines and Hantzsch 1,4-dihydropyridines by Pd/C in acetic acid. Org. Lett. 2002, 4, 3955–3957. [Google Scholar] [CrossRef] [PubMed]
- El-Emary, T.I. Synthesis of Newly Substituted Pyrazoles and Substituted Pyrazolo [3,4-b] pyridines Based on 5-Amino-3-methyl-1-phenyl pyrazole. J. Chin. Chem. Soc. 2007, 54, 507–518. [Google Scholar] [CrossRef]
- Hamada, N.M. Synthesis and Spectral Studies of Some Novel Pyrazole Derivatives from Chalcones Precursors. Heterocycl. Commun. 2009, 15, 327–334. [Google Scholar] [CrossRef]
- Hu, H.; Ge, C.; Ding, L.; Zhang, A. Synthesis of Novel 1-[(2,6-Dichloro-4-trifluoro-methyl)phenyl]-3-aryl-1H-pyrazole-4-carbaldehydes. Molecules 2010, 15, 7472–7481. [Google Scholar] [CrossRef] [PubMed]
- Sharshira, E.M.; Hamada, N.M.M. Synthesis and in Vitro Antimicrobial Activity of Some Pyrazolyl-1-carboxamide Derivatives. Molecules 2011, 16, 7736–7745. [Google Scholar] [CrossRef] [PubMed]
- Ghorab, M.M.; Ragab, F.A.; Heiba, H.I.; Agha, H.M. Synthesis of Some Novel Sulfonamides Containing Biologically Active Alkanoic Acid, Acetamide, Thiazole, and Pyrrole Moieties of Expected Antitumor and Radiosensitizing Activities. J. Basic Appl. Chem. 2011, 1, 8–14. [Google Scholar]
- Fadda, A.A.; Abdel-Latif, E.; El-Mekawy, R.E. Synthesis of Some New Aryl Azo Thiophene and Arylazopyrazole Derivatives as Antitumor Agents. Pharmacol. Pharm. 2012, 3, 148–157. [Google Scholar] [CrossRef]
- Ghorab, M.M.; El-Gazzar, M.G.; Alsaid, M.S. Synthesis, Characterization and Anti-Breast Cancer Activity of New 4-Aminoantipyrine-Based Heterocycles. Int. J. Mol. Sci. 2014, 15, 7539–7553. [Google Scholar] [CrossRef] [PubMed]
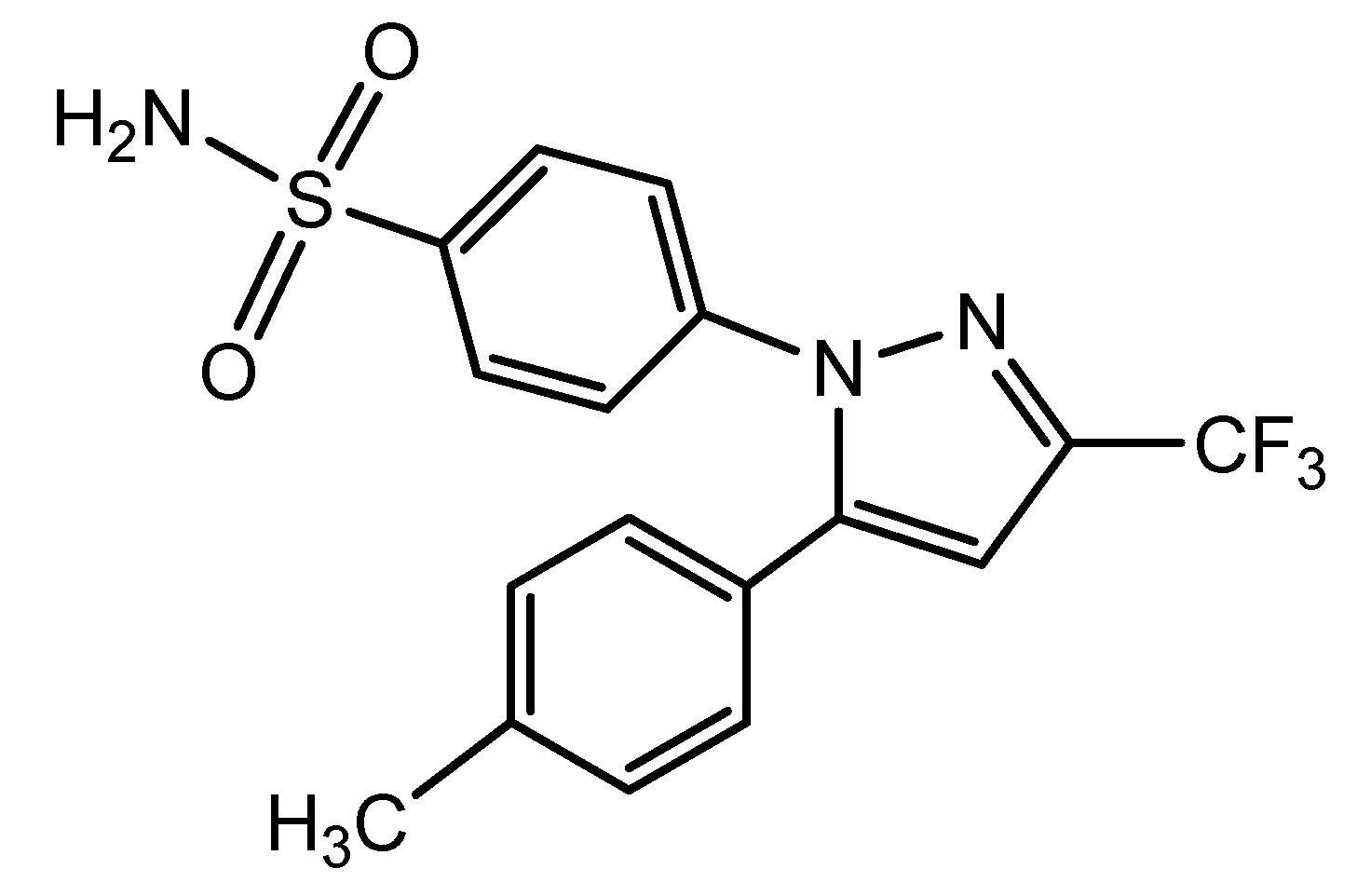
- Penning, T.D.; Talley, J.J.; Bertenshawm, S.R.; Carter, J.S.; Collins, P.W.; Docter, S.; Graneto, M.J.; Lee, L.F.; Malecha, J.W.; Miyashiro, J.M.; et al. Synthesis and Biological Evaluation of the 1,5-Diarylpyrazole Class of Cyclooxygenase-2 Inhibitors: Identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazole-1-yl]benzenesulfonamide. J. Med. Chem. 1997, 40, 1347–1365. [Google Scholar] [CrossRef] [PubMed]
- Korgaokar, S.S.; Patil, P.H.; Shah, M.J.; Parekh, H.H. Studies on Pyrazolines: Preparation and Antimicrobial Activity of 3-(3-(p-Chlorophenylsulphonamidophenyl)-5Aryl-1H/Acetyl Pyrazolines. Ind. J. Pharm. Sci. 1996, 58, 222–225. [Google Scholar]
- Amir, M.; Kumar, H.; Khan, S.A. Synthesis and Pharmacological Evaluation of Pyrazoline Derivatives as New Anti-Inflammatory and Analgesic Agents. Bioorg. Med. Chem. Lett. 2008, 18, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.; Rajendraprasad, Y. Synthesis and analgesic studies of some new 2-pyrazolines. J. Chem. 2012, 9, 1810–1815. [Google Scholar] [CrossRef]
- Palaska, E.; Erol, D.; Demirdamar, R. Synthesis and antidepressant activities of some 1,3,5-triphenyl-2-pyrazolines. Eur. J. Med. Chem. 1996, 31, 43–47. [Google Scholar] [CrossRef]
- Palaska, E.; Aytemir, M.; Uzbay, I.T.; Erol, D. Synthesis and Antidepressant Activities of some 3,5-Diphenyl-2-Pyrazolines. Eur. J. Med. Chem. 2001, 36, 539–543. [Google Scholar] [CrossRef]
- Gok, S.; Demet, M.M.; Özdemir, A.; Turan-Zitouni, G. Evaluation of Antidepressant-Like Effect of 2-Pyrazoline Derivatives. Med. Chem. Res. 2010, 19, 94–101. [Google Scholar]
- Dmytro, H.; Borys, Z.; Olexandr, V.; Lucjusz, Z.; Andrzej, G.; Roman, L. Synthesis of Novel Thiazolone-Based Compounds Containing Pyrazoline Moiety and Evaluation of Their Anticancer Activity. Eur. J. Med. Chem. 2009, 44, 1396–1404. [Google Scholar]
- Raman, K.; Pandey, B.R.; Barthwal, J.P.; Parmar, S.S. Antiinflammatory and Antiproteolytic Properties of 1,3-Disubstituted-5-(2-arylindol-3-yl)-Δ2-pyrazolines. Eur. J. Med. Chem. Chim. Ther. 1980, 15, 567–569. [Google Scholar]
- Mui, M.S.; Siew, B.N.; Buss, A.D.; Crasta, S.C.; Kah, L.G.; Sue, K.L. Synthesis of N-1 Acidic Functionality Affording Analogues With Enhanced Antiviral Activity Against HIV. Bioorg. Med. Chem. Lett. 2002, 12, 679–699. [Google Scholar]
- Turan-Zitounim, G.; Chevallet, P.; Kiliç, F.S.; Erol, K. Synthesis of Some Thiazolyl-Pyrazoline Derivatives and Preliminary Investigation of Their Hypotensive Activity. Eur. J. Med. Chem. 2000, 35, 635–641. [Google Scholar] [CrossRef]
- Ahmad, A.; Husain, A.; Khan, S.A.; Mujeeb, M.; Bhandari, A. Synthesis, Antimicrobial and Antitubercular Activities of Some Novel Pyrazoline Derivatives. J. Saudi Chem. Soc. 2014. [Google Scholar] [CrossRef]
- Soni, N.; Pande, K.; Kalsi, R.; Gupta, T.K.; Parmar, S.S.; Barthwal, J.P. Inhibition of Rat Brain Monoamine Oxidase and Succinic Dehydrogenase by Anticonvulsant Pyrazolines. Res. Commun. Mol. Pathol. Pharm. 1987, 56, 129–132. [Google Scholar]
- Rairord, L.C.; Gundy, G.V. Condensation of Vanillin Substitution Products with Aceto- phenone. J. Am. Chem. Soc. 1932, 54, 1191–1193. [Google Scholar] [CrossRef]
- Modzelewska, A.; Pettit, C.; Achanta, G.; Davidson, N.E.; Huang, P.; Khan, S.R. Anticancer activities of novel chalcone and bis-chalcone derivatives. Bioorg. Med. Chem. 2006, 14, 3491–3495. [Google Scholar] [CrossRef] [PubMed]
- Ansari, F.L.; Nazir, S.; Noureen, H.; Miraza, B. Combinatorial Synthesis and Antibacterial Evaluation of an Indexed Chalcone Library. Chem. Biodivers. 2005, 2, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Sadhu, S.K.; Okuyama, E.; Fujimoto, H.; Ishibashi, M. Separation of Leucas Aspera, a Medicinal Plant of Bangladesh, Guided by Prostaglandin Inhibitory and Antioxidant Activities. Chem. Pharm. Bull. 2003, 51, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Brand-Willams, W.; Cuvelier, M.E.; Berset, C. Use of a Free Radical Method to Evaluate Antioxidant Activity. Lebensm. Wiss. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Espin, J.C.; Soler-Rivas, C.; Wichers, H.J. Characterization of Total Free Radical Scavenger Capacity of Vegetable Oils and Oil Fraction Using 2,2-Diphenyl-1-picrylhydrazyl Radical. J. Agric. Food. Chem. 2000, 48, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Yu, L. Free Radical Scavenging Properties of Conjugated Linoleic Acids. J. Agric. Food Chem. 2001, 49, 3452–3456. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, E.R. Oxidation and Antioxidants in Fat and Oil Processing. J. Am. Oil Chem. Soc. 1978, 55, 809–841. [Google Scholar] [CrossRef]
- Hossain, M.M.; Shaha, S.K.; Foysal Aziz, F. Antioxidant Potential Study of Some Synthesized N-Heterocycles. Bangladesh Med. Res. Counc. Bull. 2009, 35, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Herbert, V. Prooxidant Effects of Antioxidant Vitamins. J. Nutr. 1996, 126, 1197S–1200S. [Google Scholar] [PubMed]
- Fukumotoand, L.R.; Mazza, G. Assessing Antioxidant and Prooxidant Activities of Phenolic Compounds. J. Agric. Food Chem. 2000, 48, 3597–3604. [Google Scholar] [CrossRef]
- Hagerman, A.E.; Riedl, K.M.; Jones, G.A.; Sovik, K.N.; Ritchard, N.T.; Hartzfeld, P.W. High Molecular Weight Plant Polyphenolics (tannins) as Biological Antioxidants. J. Agric. Food Chem. 1998, 46, 1887–1892. [Google Scholar] [CrossRef]
- Nabavi, S.M.; Ebrahimzadeh, M.A.; Nabavi, S.F.; Hamidinia, A.; Bekhradnia, A.R. Determination of Antioxidant Activity, Phenol and Flavonoids Content of Parrotia Persica Mey. Pharmacol. Online 2008, 2, 560–567. [Google Scholar]
- Nabavi, S.M.; Ebrahimzadeh, M.A.; Nabavi, S.F.; Jafari, M. Free Radical Scavenging Activity and Antioxidant Capacity of Eryngium Caucasicum Trautv and Froripia Subpinnata. Pharmacol. Online 2008, 3, 19–25. [Google Scholar]
- Gulati, K.; Ray, A.; Masood, A.; Vijayan, V.K. Involvement of Nitric Oxide (NO) in the Regulation of Stress Susceptibility and Adaptation in Rats. Ind. J. Exp. Biol. 2006, 44, 809–815. [Google Scholar]
- Krishnakumar, K.; Bhat, A.R.; Umaa, K.; Chandrasekharan, A.K. Synthesis and Valuation of Mannich Bases of Certain Novel Nitro Hydroxy 1,2-Pyrazolines. Anc. Sci. Life 2004, 24, 103–108. [Google Scholar] [PubMed]
- Mounika, S. Synthesis Characterization and Evaluation of Some Pyrazoline Derivatives. Int. J. Innov. Pharm. Sci. Res. 2013, 1, 235–251. [Google Scholar]
- Sharma, N.; Mohanakrishnanb, D.; Sharma, U.K.; Kumar, R.; Richa Sinha, A.K.; Sahal, D. Design, Economical Synthesis and Antiplasmodial Evaluation of Vanillin Derived Allylated Chalcones and Their Marked Synergism with Artemisinin against Chloroquine Resistant Strains of Plasmodium falciparum. Eur. J. Med. Chem. 2014, 79, 350–368. [Google Scholar] [CrossRef] [PubMed]
- Marcocci, L.; Maguire, J.; Droy-Lefaix, M.T.; Packer, L. The Nitric Oxide Scavenging Properties of Ginkgo Biloba Extract EGB 761. Biochem. Biophys. Res. Commun. 1994, 201, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Sreejayan, N.; Rao, M.N.A. Nitric Oxide Scavenging by Curcuminoids. J. Pharm. Pharmacol. 1997, 49, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Samples Availability: Samples of the compounds 1a–e, 3c,d, 4b–d and 5c,d are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamada, N.M.M.; Abdo, N.Y.M. Synthesis, Characterization, Antimicrobial Screening and Free-Radical Scavenging Activity of Some Novel Substituted Pyrazoles. Molecules 2015, 20, 10468-10486. https://doi.org/10.3390/molecules200610468
Hamada NMM, Abdo NYM. Synthesis, Characterization, Antimicrobial Screening and Free-Radical Scavenging Activity of Some Novel Substituted Pyrazoles. Molecules. 2015; 20(6):10468-10486. https://doi.org/10.3390/molecules200610468
Chicago/Turabian StyleHamada, Nagwa Mohamed Mahrous, and Nadia Yousef Megally Abdo. 2015. "Synthesis, Characterization, Antimicrobial Screening and Free-Radical Scavenging Activity of Some Novel Substituted Pyrazoles" Molecules 20, no. 6: 10468-10486. https://doi.org/10.3390/molecules200610468