
Differentiating Nanomaghemite and Nanomagnetite and Discussing Their Importance in Arsenic and Lead Removal from Contaminated Effluents: A Critical Review



Abstract
:1. Introduction
2. Synthesis Methods of Magnetic NPs
2.1. Co-Precipitation Method
2.2. Thermal Decomposition Method
2.3. Bulk Effects in Fe3O4 and γ-Fe2O3
3. Discussion about Main Differences Based on Physical Techniques
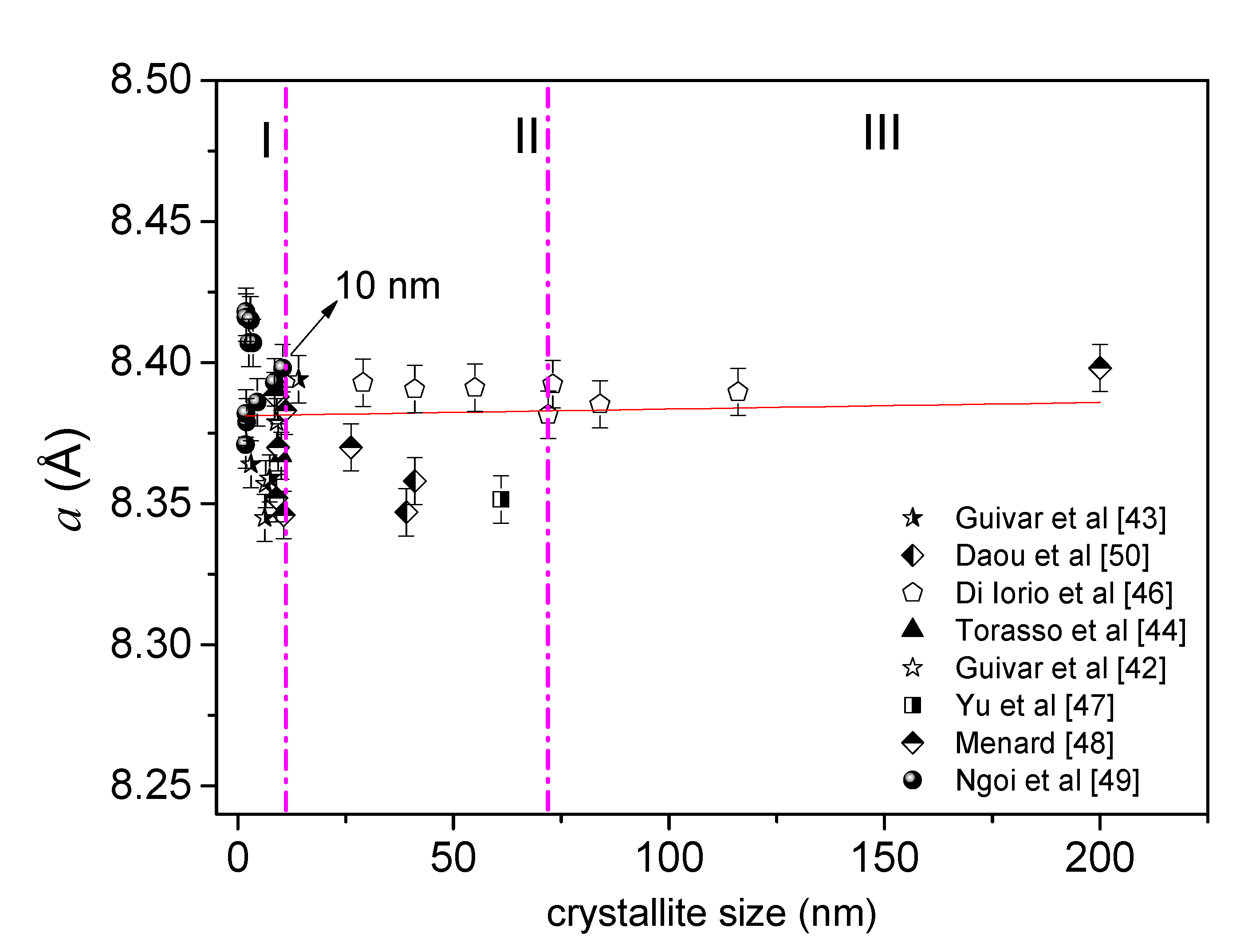
3.1. Can the XRD Technique Allow to Differentiate a Nanomagnetite or Nanomaghemite?
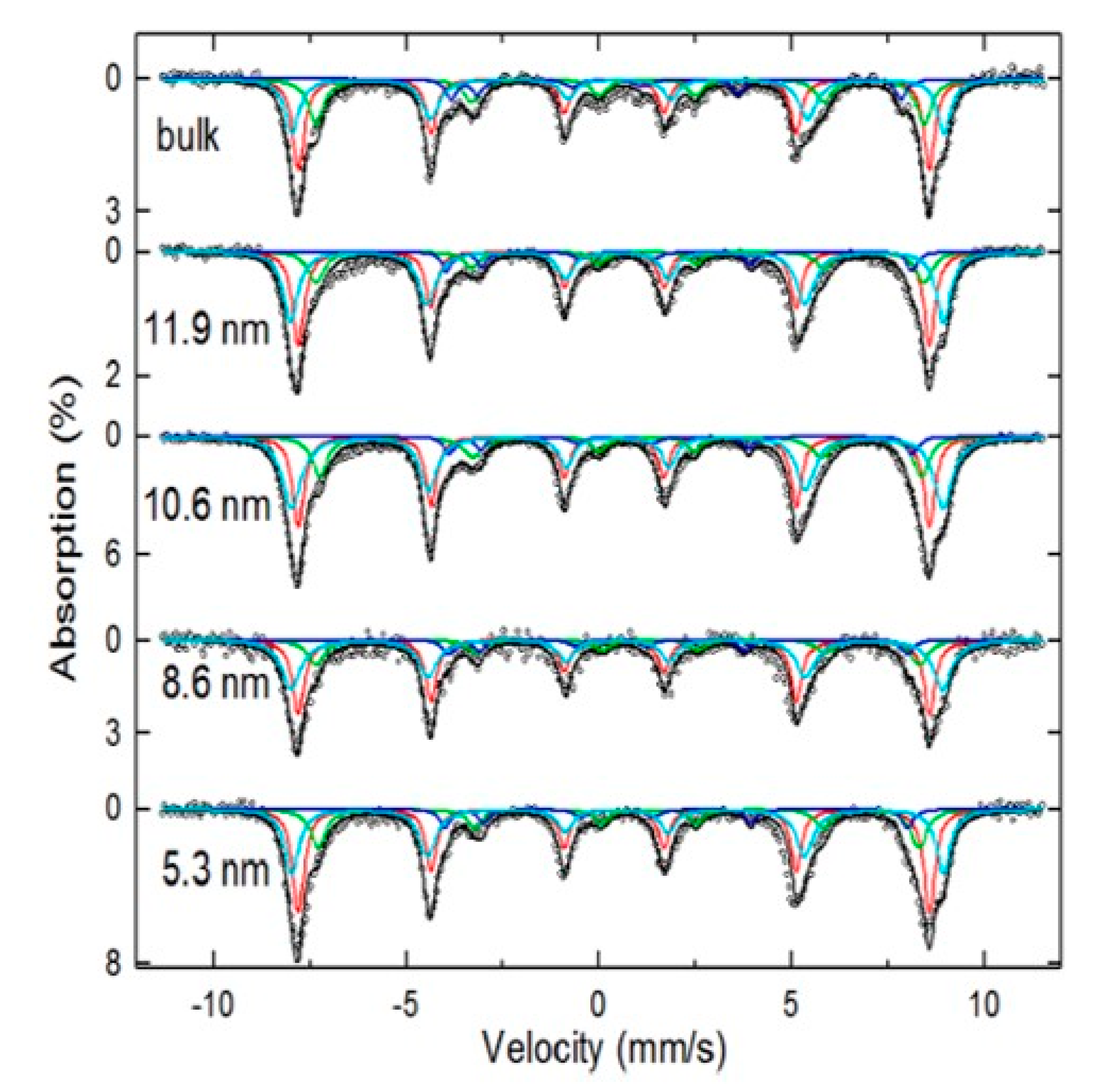
3.2. Mössbauer Technique as the Main Tool of Differentiation
3.3. High Resolution XPS and Synchrotron Radiation Techniques
4. In-Detail Discussion of the Adsorbent Properties
4.1. As Adsorption Experiments
4.1.1. Individual As Adsorptive Properties
4.1.2. Effect of pH in the Independent Removal of As(III) and As(V)
4.1.3. As Adsorption Mechanism and Adsorption Isotherm Models
4.1.4. Effect of Organic Pollutants on the As Simultaneous Uptake
4.1.5. Effect of the Coexisting Anions Cl−, NO3−, and SO42− on the As Adsorption
4.1.6. Influence of PO43− on the As Adsorption
4.1.7. Effect of the Coexisting of Metal Ions on the As Removal
4.2. Pb(II) Adsorption Experiments
4.2.1. pH and Adsorption Mechanism of Pb(II)
4.2.2. Effect of Initial Concentration on the Uptake of Pb(II)
4.2.3. Effect of Dosage on Pb(II) Removal
4.2.4. Temperature Dependence of the Pb(II) Removal
4.2.5. Simultaneous Removal of Divalent Metal Ions
4.2.6. Simultaneous Pb(II) and Organic Pollutants Adsorption
4.2.7. Removal of Pb(II) and Organic Compounds
4.2.8. Pb(II) Isotherm Models
4.2.9. Simultaneous Adsorption in Real Waters with Transition Metal-like Ions
4.3. Physicochemical Properties of Nano-Fe3O4 and Nano-γ-Fe2O3 Influencing As and Pb(II) Adsorption
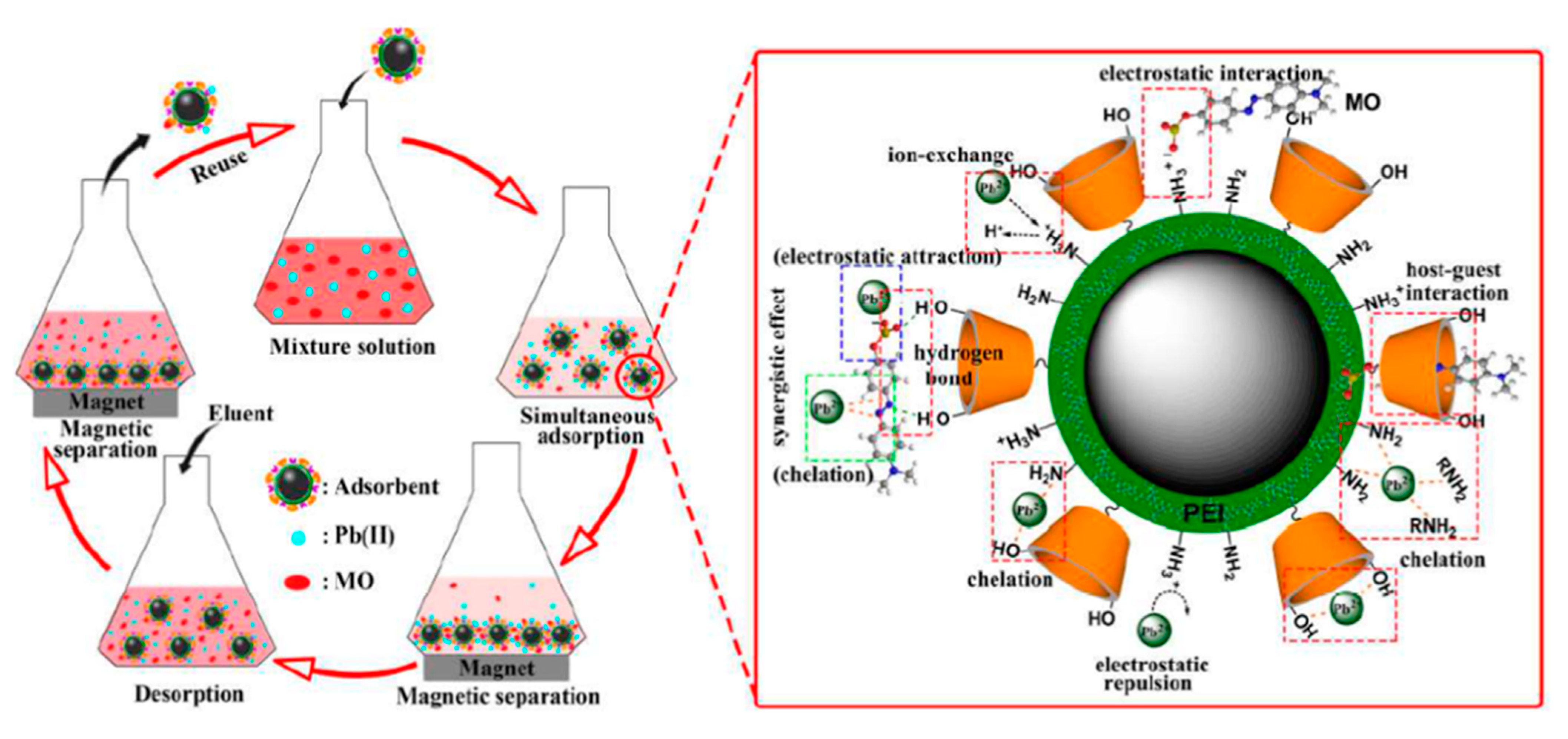
4.4. Regeneration and Reuse of Magnetic Nanoadsorbents
4.5. Cost Evaluation
5. Conclusions
6. Future Perspectives
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, 2nd ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Württemberg, Germany, 2003. [Google Scholar]
- Oh, S.J.; Cook, D.C.; Townsend, H.E. Characterization of iron oxides commonly formed as corrosion products on steel. Hyperfine Interact. 1998, 112, 59–66. [Google Scholar] [CrossRef]
- Nedkov, I.; Merodiiska, T.; Slavov, L.; Vandenberghe, R.E.; Kusano, Y.; Takada, J. Surface Oxidation, size, and shape of nano-sized magnetite obtained by co-precipitation. J. Magn. Magn. Mater. 2006, 300, 358–467. [Google Scholar] [CrossRef]
- Sun, X.; Huls, N.F.; Sigdel, A.; Sun, S. Tuning Exchange Bias in Core/Shell FeO/Fe3O4 nanoparticles. Nano Lett. 2012, 12, 246–251. [Google Scholar] [CrossRef]
- De Carvalho, J.F.; De Medeiros, S.N.; Morales, M.A.; Dantas, A.L.; Carriço, A.S. Synthesis of magnetite nanoparticles by high energy ball milling. Appl. Surf. Sci. 2013, 275, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Roth, H.-C.; Schwaminger, S.P.; Schindler, M.; Wagner, F.E.; Berensmeier, S. Influencing factors in the CO-precipitation process of superparamagnetic iron oxide nanoparticles: A model-based study. J. Magn. Magn. Mater. 2015, 377, 81–89. [Google Scholar] [CrossRef]
- Hah, H.Y.; Gray, S.; Johnson, C.E.; Johnson, J.A.; Kolesnichenko, V.; Kucheryavy, P.; Goloverda, G. Mössbauer spectroscopy of superparamagnetic Fe3O4 nanoparticles. J. Magn. Magn. Mater. 2021, 539, 168382. [Google Scholar] [CrossRef]
- Lavorato, G.C.; Rubert, A.A.; Xing, Y.; Das, R.; Robles, J.; Litterst, F.J.; Baggio-Saitovitch, E.; Phan, M.-H.; Srikanth, H.; Vericat, C.; et al. Shell-mediated control of surface chemistry of highly stoichiometric nanomagnetite nanoparticles. Nanoscale 2020, 12, 13626–13636. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Mazumder, N.; Mondal, S.; Panigrahi, K.; Banerjee, A.; Das, D.; Sarkar, S.; Roy, D.; Chattopadhyay, K.K. Size-modulation of functionalized Fe3O4: Nanoscopic customization to devise resolute piezoelectric nanocomposites. Dalton Trans. 2020, 49, 7872–7890. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.; Angappane, S.; Park, J.; An, K.; Hyeon, T.; Park, J.G. Exchange bias behavior of monodisperse Fe3O4/γ-Fe2O3 core/shell nanoparticles. Curr. Appl. Phys. 2012, 12, 808–811. [Google Scholar] [CrossRef]
- Perez, G.; Romero, M.P.; Saitovitch, E.B.; Litterst, F.J.; Araujo, J.F.D.F.; Bell, D.C.; Solorzano, G. Alkali concentration effects on the composition, morphology, and magnetic properties of magnetite, maghemite and iron oxyhydroxide nanoparticles. Solid State Sci. 2020, 106, 106295. [Google Scholar] [CrossRef]
- Da Costa, G.M.; Blanco-Andujar, C.; De Grave, E.; Pankhurst, Q.A. Magnetic Nanoparticles for in vivo use: A critical assessment of their composition. J. Phys. Chem. B 2014, 118, 11738–11746. [Google Scholar] [CrossRef]
- Tuček, J.; Zboril, R.; Petridis, D. Maghemite nanoparticles by view of Mössbauer spectroscopy. J. Nanosci. Nanotechnol. 2006, 6, 926–947. [Google Scholar] [CrossRef] [PubMed]
- Greneche, J.-M. The contribution of 57Fe Mossbauer spectrometry to investigate magnetic nanomaterials. In Mössbauer Spectrocopy, 1st ed.; Yoshida, Y., Langouche, G., Eds.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Lata, S.; Samadder, S.R. Removal of arsenic from water using nanoadsorbents and challenges: A review. J. Environ. Manag. 2016, 166, 387–406. [Google Scholar] [CrossRef]
- Hao, L.; Liu, M.; Wang, N.; Li, G. A critical review on arsenic removal from water using iron-based adsorbents. RSC Adv. 2018, 8, 39545–39560. [Google Scholar] [CrossRef]
- Bundschuh, J.; Schneider, J.; Alam, M.A.; Niazi, N.K.; Herath, I.; Parvez, F.; Tomaszewska, B.; Guilherme, L.R.G.; Maity, J.P.; López, D.L.; et al. Seven potential sources of arsenic pollution in Latin America and their environmental and health impacts. Sci. Total Environ. 2021, 780, 146274. [Google Scholar] [CrossRef]
- Habuda-Stanić, M.; Nujić, M. Arsenic removal by nanoparticles: A review. Environ. Sci. Pollut. Res. 2015, 22, 8094–8123. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Arsenic, Fact Sheet N° 372; WHO: Geneva, Switzerland, 2012. [Google Scholar]
- Bissen, M.; Frimmel, F.H. Arsenic—A review. Part II: Oxidation of arsenic and its removal in water treatment. Acta Hydrochim. Hydrobiol. 2003, 31, 97–107. [Google Scholar] [CrossRef]
- Dutta, S.; Manna, K.; Srivastava, S.K.; Gupta, A.K.; Yadav, M.K. Hollow polyalinine microsphere/Fe3O4 nanocomposite as an effective adsorbent for removal of arsenic from water. Sci. Rep. 2020, 10, 4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.-H.; Chuang, Y.-H.; Chen, T.-Y.; Tian, Y.; Li, H.; Wang, M.-K.; Zhang, W. Mechanism of arsenic adsorption on magnetite nanoparticles from water: Thermodynamic and spectroscopic studies. Environ. Sci. Technol. 2015, 49, 7726–7734. [Google Scholar] [CrossRef] [PubMed]
- Massart, R. Preparation of aqueous magnetic liquids in alkaline and acidic media. IEEE Trans. Magn. 1981, 17, 1247–1248. [Google Scholar] [CrossRef]
- Jolivet, J.P.; Froidefond, C.; Pottier, A.; Chanéac, C.; Cassaignon, S.; Tronc, E.; Euzenb, P. Size tailoring of oxide nanoparticles by precipitation in aqueous medium. A semi-quantitative modelling. J. Mater. Chem. 2004, 14, 3281–3288. [Google Scholar] [CrossRef]
- Goya, G.F.; Grazú, V.; Ibarra, M.R. Magnetic nanoparticles for cancer therapy. Curr. Nanosci. 2008, 4, 1–16. [Google Scholar] [CrossRef]
- Randrianantoandro, N.; Mercier, A.M.; Hervieu, M.; Grenèche, J.M. Direct phase transformation from hematite to maghemite during high energy ball milling. Mater. Lett. 2001, 47, 150–158. [Google Scholar] [CrossRef]
- Lux, H. Iron (II) hydroxide. In Handbook of Preparative Inorganic Chemistry, 2nd ed.; Brauer, G., Ed.; Academic Press: New York, NY, USA, 1965; Volume 2, pp. 1498–1499. [Google Scholar]
- Jolivet, J.P.; Chanéac, C.; Prené, P.; Vayssières, L.; Tronc, E. Wet Chemistry of Spinel Iron oxide Particles. J. Phys. IV 1997, 7, C1-573–C1-576. [Google Scholar] [CrossRef] [Green Version]
- Gokon, N.; Shimada, A.; Haneko, H.; Tamura, Y.; Ito, K.; Ohara, T. Magnetic coagulation and reaction rate for the aqueous ferrite formation reaction. J. Magn. Magn. Mater. 2002, 238, 47–55. [Google Scholar] [CrossRef]
- Ramos-Guivar, J.A.; López, E.O.; Greneche, J.-M.; Litterst, F.J.; Passamani, E.C. Effect of EDTA organic coating on the spin canting behavior of maghemite nanoparticles for lead (II) adsorption. Appl. Surf. Sci. 2021, 538, 148021. [Google Scholar] [CrossRef]
- Ramos Guivar, J.A.; Passamani, E.C.; Litterst, F.J. Superspinglass state in functionalized zeolite 5A-maghemite nanoparticles. AIP Adv. 2021, 11, 035223. [Google Scholar] [CrossRef]
- Liu, S.; Yu, B.; Wang, S.; Shen, Y.; Cong, H. Preparation, surface functionalization and application of Fe3O4 magnetic nanoparticles. Adv. Colloid Interface Sci. 2020, 281, 102–165. [Google Scholar] [CrossRef]
- Sun, S.; Zeng, H. Size-Controlled Synthesis of Magnetite Nanoparticles. J. Am. Chem. Soc. 2002, 124, 8204–8205. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Godoy, N.; Agredo-Diaz, D.G.; Garzón-Posada, A.O.; Parra Vargas, C.A.; Landínez Téllez, D.A.; Roa-Rojas, J. A facile method to produce magnetic nanoparticles and its influence on their magnetic and physical properties. Mater. Lett. 2021, 293, 129700. [Google Scholar] [CrossRef]
- Gorski, C.A.; Scherer, M.M. Determination of nanoparticulate magnetite stoichiometry by Mössbauer spectroscopy, acidic dissolution, and powder X-ray diffraction: A critical review. Am. Mineral. 2010, 95, 1017–1026. [Google Scholar] [CrossRef]
- Tronc, E.; Fiorani, D.; Noguès, M.; Testa, A.M.; Lucari, F.; D’Orazio, F.; Greneche, J.M.; Wernsdorfer, W.; Gálvez, N.; Chanéac, C.; et al. Surface effects in noninteracting and interacting γ-Fe2O3 nanoparticles. J. Magn. Magn. Mater. 2003, 262, 6–14. [Google Scholar] [CrossRef]
- Mørup, S.; Topsøe, H. Mossbauer studies of thermal excitations in magnetically ordered microcrystals. Appl. Phys. 1976, 11, 63–66. [Google Scholar] [CrossRef]
- Mørup, S.; Hansen, B.R. Uniform magnetic excitations in nanoparticles. Phys. Rev. B 2005, 72, 024418. [Google Scholar] [CrossRef] [Green Version]
- Millan, A.; Urtizberea, A.; Silva, N.J.O.; Palacio, F.; Amaral, V.S.; Snoeck, E.; Serin, V. Surface effects in maghemite nanoparticles. J. Magn. Magn. Mater. 2007, 312, L5–L9. [Google Scholar] [CrossRef]
- Shokrollahi, H. A review of the magnetic properties, synthesis methods and applications of maghemite. J. Magn. Magn. Mater. 2017, 426, 74–81. [Google Scholar] [CrossRef]
- Guivar, J.A.R.; Sanches, E.A.; Bruns, F.; Sadrollahi, E.; Morales, M.A.; López, E.O.; Litterst, F.J. Vacancy ordered γ-Fe2O3 nanoparticles functionalized with nanohydroxyapatite: XRD, FTIR, XPS and Mossbauer studies. Appl. Surf. Sci. 2016, 389, 721–734. [Google Scholar] [CrossRef]
- Guivar, J.A.R.; Bustamante, A.G.; Gonzalez, J.C.; Sanches, E.A.; Morales, M.A.; Raez, J.M.; López-Muñoz, M.J.; Arencibia, A. Adsorption of arsenite and arsenate by binary and ternary magnetic nanocomposites with high iron oxide content. Appl. Surf. Sci. 2018, 454, 87–100. [Google Scholar] [CrossRef]
- Guivar, J.A.R.; Sadrollahi, E.D.; Menzel, D.; Ramos Fernandes, E.G.; López, E.O.; Torres, M.M.; Arsuaga, J.M.; Arencibia, A.; Litterst, F.J. Magnetic, structural and surface properties of functionalized maghemite nanoparticles for copper and lead adsorption. RSC Adv. 2017, 7, 28763–28779. [Google Scholar] [CrossRef] [Green Version]
- Torasso, N.; Vergara-Rubio, A.; Rivas-Rojas, P.; Huck-Iriart, C.; Larrañaga, A.; Fernández-Cirelli, A.; Cerveny, S.; Goyanes, S. Enhancing arsenic adsorption via excellent dispersion of iron oxide nanoparticles inside poly(vinyl alcohol) nanofibers. J. Environ. Chem. Eng. 2020, 9, 104664. [Google Scholar] [CrossRef]
- Frison, R.; Cernuto, G.; Cervellino, A.; Zaharko, O.; Colonna, G.M.; Guagliardi, A.; Masciocchi, N. Magnetite–Maghemite Nanoparticles in the 5–15 nm Range: Correlating the Core–Shell Composition and the Surface Structure to the Magnetic Properties. A Total Scattering Study. Chem. Mater. 2013, 25, 4820–4827. [Google Scholar] [CrossRef]
- Di Lorio, E.; Colombo, C.; Cheng, Z.; Capitani, G.; Mele, D.; Ventruti, G.; Angelico, R. Characterization of magnetite nanoparticles synthetized from Fe(II)/nitrate solutions for arsenic removal from water. J. Environ. Chem. Eng. 2019, 7, 102986. [Google Scholar] [CrossRef]
- Yu, X.; Tong, S.; Ge, M.; Zuo, J.; Cao, C.; Song, W. One-step synthesis of magnetic composites of cellulose@iron oxide nanoparticles for arsenic removal. J. Mater. Chem. A 2013, 1, 959–965. [Google Scholar] [CrossRef]
- Menard, M.C.; Marschilok, A.C.; Takeuchi, K.J.; Takeuchi, E.S. Variation in the iron oxidation states of magnetite nanocrystals as a function of crystallite size: The impact on electrochemical capacity. Electrochim. Acta 2013, 94, 320–326. [Google Scholar] [CrossRef]
- Ngoi, K.H.; Wong, J.C.; Chiu, W.S.; Chia, C.H.; Jin, K.S.; Kim, H.-J.; Kim, H.-C.; Ree, M. Morphological structure details, size distributions and magnetic properties of iron oxide nanoparticles. J. Ind. Eng. Chem. 2021, 95, 37–50. [Google Scholar] [CrossRef]
- Daou, T.J.; Greneche, J.-M.; Lee, S.-J.; Lee, S.; Lefevre, C.; Bégin-Colin, S.; Pourroy, G. Spin canting of maghemite studied by NMR and in-field Mössbauer spectrometry. J. Phys. Chem. C 2010, 114, 8794–8799. [Google Scholar] [CrossRef]
- Schwaminger, S.P.; Syhr, C.; Berensmeier, S. Controlled Synthesis of Magnetic Iron Oxide Nanoparticles: Magnetite or Maghemite? Crystals 2020, 10, 214. [Google Scholar] [CrossRef] [Green Version]
- González-Alonso, D.; Espeso, J.I.; Gavilán, H.; Zeng, L.; Díaz, M.T.F.; Subías, G.; de Pedro, I.; Fernández, J.R.; Bender, P.; Barquín, L.F.; et al. Identifying the presence of magnetite in an ensemble of iron-oxide nanoparticles: A comparative neutron diffraction study between bulk and nanoscale. Nanoscale Adv. 2021, 3, 3491–3496. [Google Scholar] [CrossRef]
- Iyengar, S.J.; Joy, M.; Ghosh, C.K.; Dey, S.; Kotnala, R.K.; Ghosh, S. Magnetic, X-ray and Mossbauer studies on magnetite/maghemite core-shell nanostructures fabricated through an aqueous route. RSC. Adv. 2014, 4, 64919–64929. [Google Scholar] [CrossRef] [Green Version]
- Dormann, J.L.; D’Orazio, F.; Lucari, F.; Tronc, E.; Prené, P.; Jolivet, J.P.; Fiorani, D.; Cherkaoui, R.; Noguès, M. Thermal variation of the relaxation time of the magnetic moment of γ-Fe2O3 nanoparticles with interparticle interaction of various strengths. Phys. Rev. B 1996, 53, 14291–14297. [Google Scholar] [CrossRef]
- Wilson, D.; Langell, M.A. XPS analysis of oleylamine/oleic acid capped Fe3O4 nanoparticles as a function of temperature. Appl. Surf. Sci. 2014, 303, 6–13. [Google Scholar] [CrossRef]
- Jiménez-Villacorta, F.; Prieto, C.; Huttel, Y.; Telling, N.D.; Van Der Laan, G. X-ray magnetic circular dichroism study of the blocking process in nanostructured iron-iron oxide core-shell systems. Phys. Rev. B 2011, 84, 172404. [Google Scholar] [CrossRef] [Green Version]
- Bonanni, V.; Basini, M.; Peddis, D.; Lascialfari, A.; Rossi, G.; Torelli, P. X-ray magnetic circular dichroism discloses surface spins correlation in maghemite hollow nanoparticles. Appl. Phys. Lett. 2018, 112, 022404. [Google Scholar] [CrossRef]
- Siddiqui, S.I.; Singh, P.N.; Tara, N.; Pal, S.; Chaudhry, S.A.; Sinha, I. Arsenic removal from water by starch functionalized maghemite nano-adsorbents: Thermodynamics and kinetics investigations. Colloids Interface Sci. Commun. 2020, 36, 100263. [Google Scholar] [CrossRef]
- Navarathna, C.M.; Karunanyake, A.G.; Gunatilake, S.R.; Pittman, C.U., Jr.; Perez, F.; Mohan, D.; Mlsna, T.E. Removal of Arsenic(III) from water using magnetite precipitated onto Douglas fir biochar. J. Environ. Manag. 2019, 250, 109429. [Google Scholar] [CrossRef]
- Pizarro, C.; Escudey, M.; Caroca, E.; Pavez, C.; Zúñiga, G.E. Evaluation of zeolite, nanomagnetite and nanomagnetite-zeolite composite materials as arsenic (V) adsorbents in hydroponic tomato cultures. Sci. Total Environ. 2021, 751, 141623. [Google Scholar] [CrossRef]
- Hernández-Flores, H.; Pariona, N.; Herrera-Trejo, M.; Hdz-García, H.M.; Mtz-Enriquez, A.I. Concrete/maghemite nanocomposites as novel adsorbents for arsenic removal. J. Mol. Struct. 2018, 1171, 9–16. [Google Scholar] [CrossRef]
- Das, T.K.; Sakthivel, T.S.; Jeyaranjan, A.; Seal, S.; Bezbaruah, A.N. Ultra-high arsenic adsorption by graphene oxide iron nanohybrid: Removal mechanisms and potential applications. Chemosphere 2020, 253, 126702. [Google Scholar] [CrossRef]
- Raval, N.P.; Kumar, M. Geogenic arsenic removal through core–shell based functionalized nanoparticles: Groundwater in-situ treatment perspective in the post-COVID anthropocene. J. Hazard. Mater. 2021, 402, 123466. [Google Scholar] [CrossRef]
- Su, H.; Ye, Z.; Hmidi, N. High-performance iron oxide–graphene oxide nanocomposite adsorbents for arsenic removal. Colloids Surf. A Physicochem. Eng. Asp. 2017, 522, 161–172. [Google Scholar] [CrossRef]
- Zeng, H.; Zhai, L.; Qiao, T.; Yu, Y.; Zhang, J.; Li, D. Efficient removal of As(V) from aqueous media by magnetic nanoparticles prepared with Iron-containing water treatment residuals. Sci. Rep. 2020, 10, 9335. [Google Scholar] [CrossRef] [PubMed]
- Das, T.K.; Bezbaruah, A.N. Comparative study of arsenic removal by iron-based nanomaterials: Potential candidates for field applications. Sci. Total Environ. 2021, 10, 142914. [Google Scholar] [CrossRef] [PubMed]
- Leus, K.; Folens, K.; Nicomel, N.R.; Perez, J.P.H.; Filippousi, M.; Meledina, M.; Dîrtu, M.M.; Turner, S.; Van Tendeloo, G.; Garcia, Y.; et al. Removal of arsenic and mercury species from water by covalent triazine framework encapsulated γ-Fe2O3 nanoparticles. J. Hazard. Mater. 2018, 353, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.I.; Chaudhry, S.A. Nanohybrid composite Fe2O3-ZrO2/BC for inhibiting the growth of bacteria and adsorptive removal of arsenic and dyes from water. J. Clean. Prod. 2019, 223, 849–868. [Google Scholar] [CrossRef]
- Alchouron, J.; Navarathna, C.; Chludil, H.D.; Dewage, N.B.; Perez, F.; Hassan, E.B.; Pittman, C.U., Jr.; Vega, A.S.; Mlsna, T.E. Assessing South American Guadua chacoensis bamboo biochar and Fe3O4nanoparticle dispersed analogues for aqueous arsenic(V) remediation. Sci. Total Environ. 2020, 706, 135943. [Google Scholar] [CrossRef]
- Maziarz, P.; Matusik, J.; Leiviskä, T.; Strączek, T.; Kapusta, C.; Woch, W.M.; Tokarz, W.; Górniak, K. Toward highly effective and easily separable halloysite-containing adsorbents: The effect of iron oxide particles impregnation and new insight into As(V) removal mechanisms. Sep. Purif. Technol. 2019, 210, 390–401. [Google Scholar] [CrossRef]
- Ajith, N.; Swain, K.K. Study on the performance and interaction of different synthetic iron oxides for arsenic uptake using 76As radiotracer. Appl. Radiat. Isot. 2019, 153, 108807. [Google Scholar] [CrossRef]
- Alchouron, J.; Navarathna, C.; Rodrigo, P.M.; Snyder, A.; Chludil, H.D.; Vega, A.S.; Bosi, G.; Perez, F.; Mohan, D.; Pittman, C.U., Jr.; et al. Household arsenic contaminated water treatment employing iron oxide/ bamboo biochar composite: An approach to technology transfer. J. Colloid Interface Sci. 2021, 587, 767–779. [Google Scholar] [CrossRef]
- Nisticò, R.; Celi, L.R.; Prevot, A.B.; Carlos, L.; Magnacca, G.; Zanzo, E.; Martin, M. Sustainable magnet-responsive nanomaterials for the removal of arsenic from contaminated water. J. Hazard. Mater. 2018, 342, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Nikić, J.; Tubić, A.; Watson, M.; Maletić, S.; Šolić, M.; Majkić, T.; Agbaba, J. Arsenic Removal from Water by Green Synthesized Magnetic Nanoparticles. Water 2019, 11, 2520. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Kong, L.; Huang, X.; Liu, M.; Li, L. Removal of arsenic(V) from aqueous solutions using sulfur-doped Fe3O4 nanoparticles. RSC. Adv. 2018, 8, 40804–40812. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Zhai, L.; Qiao, T.; Zhang, J.; Li, D. Removal of As(V) by a core-shell magnetic nanoparticles synthesized with iron-containing water treatment residuals. Colloids Surf. A Physicochem. Eng. Asp. 2021, 627, 127074. [Google Scholar] [CrossRef]
- Zeng, H.; Zhai, L.; Zhang, J.; Li, D. As(V) adsorption by a novel core-shell magnetic nanoparticles prepared with Iron-containing water treatment residuals. Sci. Total Environ. 2021, 753, 142002. [Google Scholar] [CrossRef]
- Feng, L.; Cao, M.; Ma, X.; Zhu, Y.; Hu, C. Superparamagnetic high-surface-area Fe3O4 nanoparticles as adsorbents for arsenic removal. J. Hazard. Mater. 2012, 217–218, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Jing, C.; Zhang, J.; Liu, T.; Yang, S.; Wang, W. Magnetic Fe3O4@CuO nanocomposite assembled on graphene oxide sheets for the enhanced removal of arsenic(III/V) from water. Appl. Surf. Sci. 2019, 466, 746–756. [Google Scholar] [CrossRef]
- An, B.; Liang, Q.; Zhao, D. Removal of arsenic(V) from spent ion exchange brine using a new class of starch-bridged magnetite nanoparticles. Water Res. 2011, 45, 1961–1972. [Google Scholar] [CrossRef] [PubMed]
- Tuutijärvi, T.; Lu, J.; Sillanpää, M.; Chen, G. As(V) adsorption on maghemite nanoparticles. J. Hazard. Mater. 2009, 166, 1415–1420. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Kim, W.; Suh, C.Y.; Shin, D.; Ko, K.S.; Ha, K. Magnetic iron oxide nanoparticles prepared by electrical wire explosion for arsenic removal. Powder Technol. 2013, 246, 572–574. [Google Scholar] [CrossRef]
- Lin, Y.F.; Chen, J.L. Synthesis of mesoporous maghemite (γ-Fe2O3) nanostructures with enhanced arsenic removal efficiency. RSC Adv. 2013, 3, 15344–15349. [Google Scholar] [CrossRef]
- Cui, J.; Jin, Q.; Li, Y.; Li, F. The oxidation and removal of As(III) from soil using a novel magnetic nanocomposite derived-biomass wastes. Environ. Sci. Nano 2019, 6, 478–488. [Google Scholar] [CrossRef]
- He, R.; Peng, Z.; Lyu, H.; Huang, H.; Nan, Q.; Tang, J. Synthesis and characterization of an iron-impregnated biochar for aqueous arsenic removal. Sci. Total Environ. 2018, 612, 1177–1186. [Google Scholar] [CrossRef]
- Deng, M.; Wu, X.; Zhu, A.; Zhang, Q.; Liu, Q. Well-dispersed TiO2 nanoparticles anchored on Fe3O4 magnetic nanosheets for efficient arsenic removal. J. Environ. Manag. 2019, 237, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.F.; Chen, J.L.; Xu, C.Y.; Chung, T.W. One-pot synthesis of paramagnetic iron(III) hydroxide nanoplates and ferrimagnetic magnetite nanoparticles for the removal of arsenic ions. Chem. Eng. Sci. 2014, 250, 409–415. [Google Scholar] [CrossRef]
- Akin, I.; Arslan, G.; Tor, A.; Ersoz, M.; Cengeloglu, Y. Arsenic(V) removal from underground water by magnetic nanoparticles synthesized from waste red mud. J. Hazard. Mater. 2012, 235–236, 62–68. [Google Scholar] [CrossRef]
- Siddiqui, S.I.; Naushad, M.; Chaudhry, S.A. Promising prospects of nanomaterials for arsenic water remediation: A comprehensive review. Process Saf. Environ. Prot. 2019, 126, 60–97. [Google Scholar] [CrossRef]
- Narouei, F.H.; Andreescu, D.; Andreescu, S. Rapid characterization of arsenic adsorption on single magnetite nanoparticles by collisions at microelectrodes. Environ. Sci. Nano 2020, 7, 1999–2009. [Google Scholar] [CrossRef]
- Paul, B.; Parashar, V.; Mishra, A. Graphene in the Fe3O4 nano-composite switching the negative influence of humic acid coating into an enhancing effect in the removal of arsenic from water. Environ. Sci. Water Res. Technol. 2015, 1, 77–83. [Google Scholar] [CrossRef]
- Rashid, M.; Sterbinsky, G.E.; Gracia Pinilla, M.A.; Cai, Y.; O’Shea, K.E. Kinetic and Mechanistic Evaluation of Inorganic Arsenic Species Adsorption onto Humic Acid Grafted Magnetite Nanoparticles. J. Phys. Chem. C 2018, 122, 13540–13547. [Google Scholar] [CrossRef]
- Taleb, K.; Markovski, J.; Veličković, Z.; Rusmirović, J.; Rančić, M.; Pavlović, V.; Marinković, A. Arsenic removal by magnetite-loaded amino modified nano/microcellulose adsorbents: Effect of functionalization and media size. Arab. J. Chem. 2019, 12, 4675–4693. [Google Scholar] [CrossRef] [Green Version]
- Darezereshki, E.; Khodadadi Darban, A.; Abdollahy, M.; Jamshidi-Zanjani, A. Influence of heavy metals on the adsorption of arsenate by magnetite nanoparticles: Kinetics and thermodynamic. Environ. Nanotechnol. Monit. Manag. 2018, 10, 51–62. [Google Scholar] [CrossRef]
- Lung, I.; Stan, M.; Opris, O.; Soran, M.L.; Senila, M.; Stefan, M. Removal of lead(II), cadmium(II), and arsenic(III) from aqueous solution using magnetite nanoparticles prepared by green synthesis with Box–Behnken design. Anal. Lett. 2018, 51, 2517–2531. [Google Scholar] [CrossRef]
- Yoon, Y.; Zheng, M.; Ahn, Y.T.; Park, W.K.; Yang, W.S.; Kang, J.W. Synthesis of magnetite/non-oxidative graphene composites and their application for arsenic removal. Sep. Purif. Technol. 2017, 178, 40–48. [Google Scholar] [CrossRef]
- Kango, S.; Kumar, R. Magnetite nanoparticles coated sand for arsenic removal from drinking water. Environ. Earth Sci. 2016, 75, 381. [Google Scholar] [CrossRef]
- Tripathy, M.; Hota, G. Maghemite and graphene oxide embedded polyacrylonitrile electrospun nanofiber matrix for remediation of arsenate ions. ACS Appl. Polym. Mater. 2020, 2, 604–617. [Google Scholar] [CrossRef]
- Chiew, H.; Sampson, M.L.; Huch, S.; Ken, S.; Bostick, B.C. Effect of Groundwater Iron and Phosphate on the Efficacy of Arsenic Removal by Iron-Amended BioSand Filters. Environ. Sci. Technol. 2009, 43, 6295–6300. [Google Scholar] [CrossRef]
- Markovski, J.S.; Dokić, V.; Milosavljević, M.; Mitrić, M.; Perić-Grujić, A.A.; Onjia, A.E.; Marinković, A.D. Ultrasonic assisted arsenate adsorption on solvothermally synthesized calcite modified by goethite, α-MnO2 and goethite/α-MnO2. Ultrason. Sonochem. 2014, 21, 790–801. [Google Scholar] [CrossRef]
- Markovski, J.S.; Marković, D.D.; Dokić, V.R.; Mitrić, M.; Ristić, M.D.; Onjia, A.E.; Marinković, A.D. Arsenate adsorption on waste eggshell modified by goethite, α-MnO2 and goethite/α-MnO2. Chem. Eng. J. 2014, 237, 430–442. [Google Scholar] [CrossRef]
- Issa, N.B.; Rajaković-Ognjanović, V.N.; Marinković, A.D.; Rajaković, L.V. Separation and determination of arsenic species in water by selective exchange and hybrid resins. Anal. Chim. Acta 2011, 706, 191–198. [Google Scholar] [CrossRef]
- Tombácz, E.; Tóth, I.Y.; Nesztor, D.; Illés, E.; Hajdú, A.; Szekeres, M.; Vékás, L. Adsorption of organic acids on magnetite nanoparticles, pH-dependent colloidal stability and salt tolerance. Colloids Surf. A Physicochem. Eng. Asp. 2013, 435, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Foo, K.Y.; Hameed, B.H. Insights into the modeling of adsorption isotherm systems. Chem. Eng. J. 2010, 156, 2–10. [Google Scholar] [CrossRef]
- Goldberg, S.; Johnston, C.T. Mechanisms of arsenic adsorption on amorphous oxides evaluated using macroscopic measurements, vibrational spectroscopy, and surface complexation modeling. J. Colloid Interface Sci. 2001, 234, 204–216. [Google Scholar] [CrossRef]
- Bagbi, Y.; Sarswat, A.; Mohan, D.; Pandey, A.; Solanki, P.R. Lead (Pb2+) adsorption by monodispersed magnetite nanoparticles: Surface analysis and effects of solution chemistry. J. Environ. Chem. Eng. 2016, 4, 4237–4247. [Google Scholar] [CrossRef]
- Rajput, S.; Pittman, C.U., Jr.; Mohan, D. Synthesis of Magnetic Magnetite Nanoparticles (Fe3O4) and applications for lead removal (Pb2 +) and chromium (Cr6 +) of the water. J. Colloid Interface Sci. 2015, 468, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Asthana, A.; Singh, A.K.; Prasad, S.; Susan, M.A.B.H. Novel glycine-functionalized magnetic nanoparticles entrapped calcium alginate beads for effective removal of lead. Microchem. J. 2017, 130, 168–178. [Google Scholar] [CrossRef]
- Bagbi, Y.; Sarswat, A.; Mohan, D.; Pandey, A.; Solanki, P.R. Lead and chromium adsorption from water using L-cysteine functionalized magnetite (Fe3O4) nanoparticles. Sci. Rep. 2017, 7, 7672. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Tan, X.; Tan, L.; Zhang, H.; Liu, H.; Fang, M.; Hayat, T.; Wang, X. Magnetic porous polymers prepared through high internal phase emulsions for efficient removal of Pb2+ and Cd2 +. ACS Sustain. Chem. Eng. 2018, 6, 5206–5213. [Google Scholar] [CrossRef]
- Wang, N.; Yang, D.; Wang, X.; Yu, S.; Wang, H.; Wen, T.; Song, G.; Yu, Z.; Wang, X. Highly efficient removal of Pb(II) and Cu(II) using hollow Fe3O4@PDA nanoparticles with excellent applicability and reusability. Inorg. Chem. Front. 2018, 5, 2174–2182. [Google Scholar] [CrossRef]
- Hosseinzadeh, M.; Seyyed Ebrahimi, S.A.; Raygan, S.; Masoudpanah, S.M. Removal of cadmium and lead ions from aqueous solution by nanocrystalline magnetite through mechanochemical activation. J. Ultrafine Grained Nanostruct. Mater. 2016, 49, 72–79. [Google Scholar]
- Nejad, S.B.; Mohammadi, A. Epoxy-triazinetrione-functionalized magnetic nanoparticles as an efficient magnetic nanoadsorbent for the removal of malachite green and Pb (II) from aqueous solutions. J. Chem. Eng. Data 2020, 65, 2731–2742. [Google Scholar] [CrossRef]
- Chen, B.; Chen, S.; Zhao, H.; Liu, Y.; Long, F.; Pan, X. A versatile β-cyclodextrin and polyethyleneimine bi-functionalized magnetic nanoadsorbent for simultaneous capture of methyl orange and Pb(II) from complex wastewater. Chemosphere 2019, 216, 605–616. [Google Scholar] [CrossRef]
- Jia, C.; Zhao, J.; Lei, L.; Kang, X.; Lu, R.; Chen, C.; Li, S.; Zhao, Y.; Yang, Q.; Chen, Z. Novel magnetically separable anhydride-functionalized Fe3O4@SiO2@PEI-NTDA nanoparticles as effective adsorbents: Synthesis, stability and recyclable adsorption performance for heavy metal ions. RSC Adv. 2019, 9, 9533–9545. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Wu, K.; Gao, Y.; Liu, L.; Zhu, X.; Li, X.; Zhang, F. Efficient Removal of Zn(II), Pb(II), and Cd(II) in Waste Water Based on Magnetic Graphitic Carbon Nitride Materials with Enhanced Adsorption Capacity. J. Chem. Eng. Data 2018, 63, 3902–3912. [Google Scholar] [CrossRef]
- Chen, K.; He, J.; Li, Y.; Cai, X.; Zhang, K.; Liu, T.; Hu, Y.; Lin, D.; Kong, L.; Liu, J. Removal of cadmium and lead ions from water by sulfonated magnetic nanoparticle adsorbents. J. Colloid Interface Sci. 2017, 494, 307–316. [Google Scholar] [CrossRef]
- Ge, F.; Li, M.; Ye, H.; Zhao, B. Effective removal of heavy metal ions Cd2+, Zn2+, Pb2+, Cu2+ from aqueous solution by polymer-modified magnetic nanoparticles. J. Hazard. Mater. 2012, 211–212, 366–372. [Google Scholar] [CrossRef]
- Zhang, C.; Sui, J.; Li, J.; Tang, Y.; Cai, W. Efficient removal of heavy metal ions by thiol-functionalized superparamagnetic carbon nanotubes. Chem. Eng. J. 2012, 210, 45–52. [Google Scholar] [CrossRef]
- Bi, J.; Huang, X.; Wang, J.; Wang, T.; Wu, H.; Yang, J.; Lu, H.; Hao, H. Oil-phase cyclic magnetic adsorption to synthesize Fe3O4@C@TiO2-nanotube composites for simultaneous removal of Pb(II) and rhodamine B. Chem. Eng. J. 2019, 366, 50–61. [Google Scholar] [CrossRef]
- Behbahani, E.S.; Dashtian, K.; Ghaedi, M. Fe3O4-FeMoS4: Promise magnetite LDH-based adsorbent for simultaneous removal of Pb(II), Cd(II), and Cu(II) heavy metal ions. J. Hazard. Mater. 2021, 410, 124560. [Google Scholar] [CrossRef]
- Li, G.; Zhao, Z.; Liu, J.; Jiang, G. 2011. Effective heavy metal removal from aqueous systems by thiol functionalized magnetic mesoporous silica. J. Hazard. Mater. 2011, 192, 277–283. [Google Scholar]
- Wang, Y.; Hu, L.; Zhang, G.; Yan, T.; Yan, L.; Wei, Q.; Du, B. Removal of Pb(II) and methylene blue from aqueous solution by magnetic hydroxyapatite-immobilized oxidized multi-walled carbon nanotubes. J. Colloid Interface Sci. 2017, 494, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Fato, F.P.; Li, D.W.; Zhao, L.J.; Qiu, K.; Long, Y.T. Simultaneous removal of multiple heavy metal ions from river water using ultrafine mesoporous magnetite nanoparticles. ACS Omega 2019, 4, 7543–7549. [Google Scholar] [CrossRef] [Green Version]
- Pena, M.; Meng, X.; Korfiatis, G.P.; Jing, C. Adsorption mechanism of arsenic on nanocrystalline titanium dioxide. Environ. Sci. Technol. 2006, 40, 1257–1262. [Google Scholar] [CrossRef]
- Carabante, I.; Grahn, M.; Holmgren, A.; Kumpiene, J.; Hedlund, J. Adsorption of As (V) on iron oxide nanoparticle films studied by in situ ATR-FTIR spectroscopy. Colloids Surf. A Physicochem. Eng. Asp. 2009, 346, 106–113. [Google Scholar] [CrossRef]
- Ogholbeyg, A.B.; Kianvash, A.; Hajalilou, A.; Abouzari-Lotf, E.; Zarebkohan, A. Cytotoxicity characteristics of green assisted-synthesized superparamagnetic maghemite (γ-Fe2O3) nanoparticles. J. Mater. Sci. Mater. Electron. 2018, 29, 12135–12143. [Google Scholar] [CrossRef]
- Hui, B.H.; Salimi, M.N. Production of iron oxide nanoparticles by co-precipitation method with optimization studies of processing temperature, pH and stirring rate. In IOP Conference Series: Materials Science and Engineering; IOP Publishing: Bristol, UK, 2020; Volume 743, p. 012036. [Google Scholar]
- Nazari, M.; Ghasemi, N.; Maddah, H.; Motlagh, M.M. Synthesis and characterization of maghemite nanopowders by chemical precipitation method. J. Nanostruct. Chem. 2014, 4, 99. [Google Scholar] [CrossRef] [Green Version]
- Mikutta, C.; Kretzschmar, R. Spectroscopic evidence for ternary complex formation between arsenate and ferric iron complexes of humic substances. Environ. Sci. Technol. 2011, 45, 9550–9557. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, S.; Rafiee, Z.; Dashtian, K. Fe3O4-based melamine-rich covalent organic polymer for simultaneous removal of auramine O and rhodamine B. J. Chem. Eng. Data 2020, 65, 696–705. [Google Scholar] [CrossRef]
- Xue, S.; Xiao, Y.; Wang, G.; Fan, J.; Wan, K.; He, Q.; Gao, M.; Miao, Z. Adsorption of heavy metals in water by modifying Fe3O4 nanoparticles with oxidized humic acid. Colloids Surf. A Physicochem. Eng. Asp. 2021, 616, 126333. [Google Scholar] [CrossRef]
- Ramos-Guivar, J.A.; Taipe, K.; Schettino, M.A., Jr.; Silva, E.; Morales Torres, M.A.; Passamani, E.C.; Litterst, F.J. Improved removal capacity and equilibrium time of maghemite nanoparticles growth in zeolite type 5A for Pb(II) adsorption. Nanomaterials 2020, 10, 1668. [Google Scholar] [CrossRef]
- Baig, S.A.; Sheng, T.; Hu, Y.; Xu, J.; Xu, X. Arsenic removal from natural water using low-cost granulated adsorbents: A review. Clean Soil Air Water 2015, 43, 13–26. [Google Scholar] [CrossRef]
- Augusto, P.A.; Castelo-Grande, T.; Vargas, D.; Pascual, A.; Hernández, L.; Estevez, A.M.; Barbosa, D. Upscale Design, Process Development, and Economic Analysis of Industrial Plants for Nanomagnetic Particle Production for Environmental and Biomedical Use. Materials 2020, 13, 2477. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
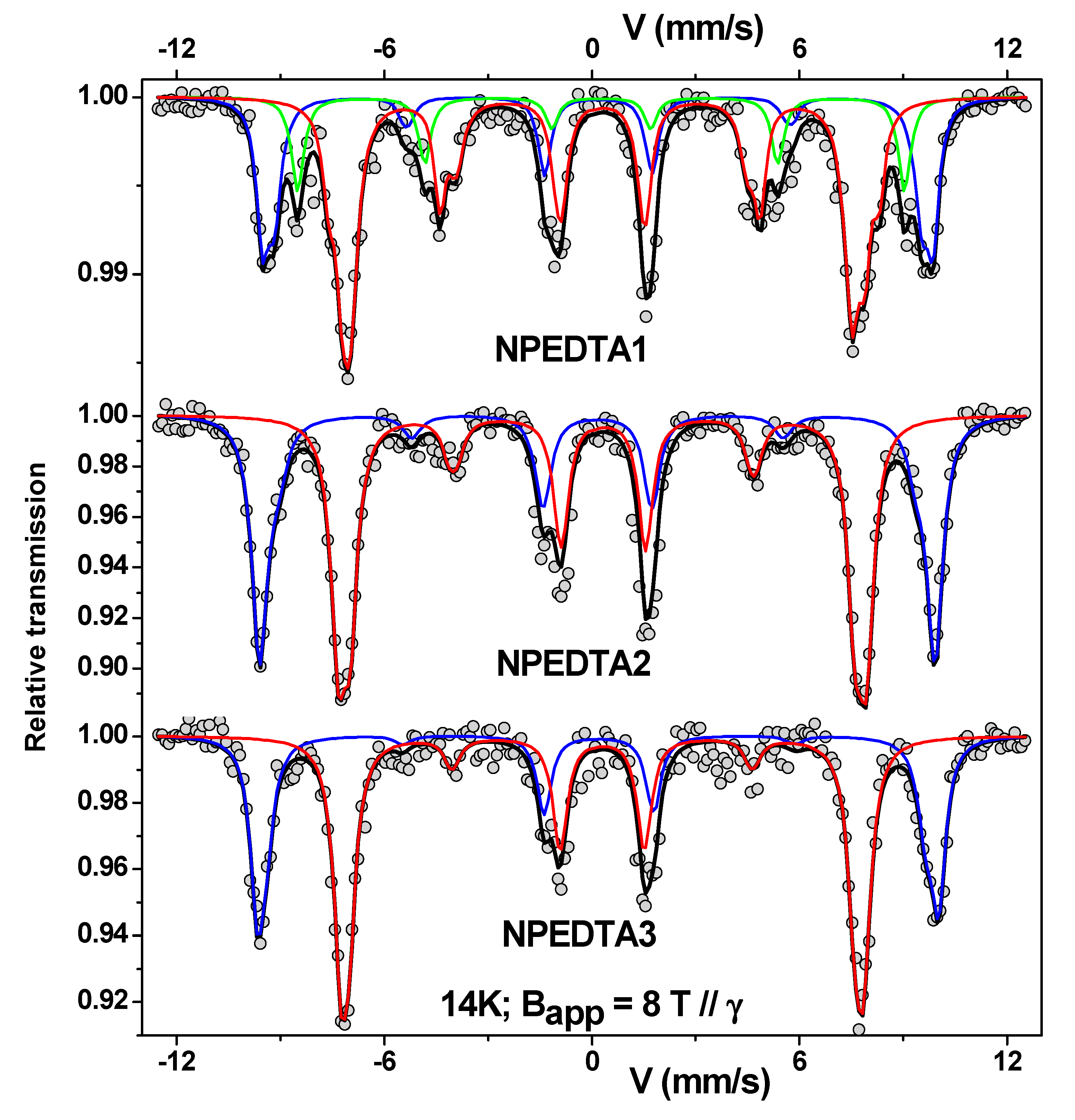
| Sample | Site | ± 0.02 | ± 0.02 | <Beff> (T) ± 0.5 | (°) ± 5 | <Bhf> (T) ± 0.5 | F (%) ± 1 | e (nm) ± 0.05 |
|---|---|---|---|---|---|---|---|---|
| NPEDTA1 | A | 0.18 | −0.01 | 58.8 | 26 | 51.8 | 28 | 0.38 |
| B | 0.29 | 0.01 | 46.2 | 42 | 52.4 | 57 | 0.89 | |
| C | 0.27 | −0.00 | 54.2 | 56 | 50.2 | 15 | 1.37 | |
| NPEDTA2 | A | 0.36 | −0.00 | 59.6 | 21 | 52.2 | 43 | 0.24 |
| B | 0.52 | 0.02 | 46.2 | 28 | 53.3 | 57 | 0.42 | |
| NPEDTA3 | A | 0.37 | −0.04 | 60.2 | 14 | 52.7 | 40 | 0.10 |
| B | 0.52 | 0.00 | 46.0 | 22 | 53.5 | 60 | 0.24 |
| Bulk γ-Fe2O3 | Nano-γ-Fe2O3 | Bulk Fe3O4, nano-Fe3O4 |
|---|---|---|
| At 14 K, it has perfect asymmetric sextets. | At RT, the sextets collapse to a doublet or singlet-superparamagnetic-like regime (size < 10 nm). At 14 K, the in-field Mössbauer measurements reveal two or three magnetic components depending on particle size. Broadenings can still be significant due to overbarrier fluctuations of smaller particles. | Bulk stoichiometric Fe3O4 depicts two characteristic sextets at RT, while the nano-Fe3O4 presents a collapse spectrum to a doublet or singlet. At 6 K, the spectrum is fitted with three components of tetrahedral Fe3+, octahedral Fe3+, and octahedral Fe2+ [7]. |
| Static hyperfine magnetic fields. | At RT, fluctuating hyperfine magnetic fields are presented. At 14 K, superparamagnetic relaxation is negligible and two defined sextets are observed. | Hyperfine magnetic fields at RT [1,7]: Isomer shifts at RT K [1,7]: |
| Hyperfine magnetic fields at 14 K [30]: | At RT, the appearance of the complex shapes with mixed components that depend on the particle size, anisotropy energies, blocking temperature distributions, and magnetic interactions are observed. At 14 K, if sizes are smaller than 10 nm, strong spin canting behavior occurs, and the hyperfine parameters slightly differ. For sizes bigger than 10 nm, the hyperfine parameters are equal to the bulk expected ones. | perfine magnetic fields at 140 K [35]: bulk Fe3O4 Hyperfine magnetic fields at 140 K [35]: 21 nm Fe3O4 Hyperfine magnetic fields at RT [7]: 5.3 nm Fe3O4 Isomer shifts at RT [1,7]: |
| Isomer shifts at 14 K [30]: | For fittings an average <> for each site must be considered. RAA for site A (37.5%) and site B (62.5%). | They showed lines due to Fe2+ at about −3.0 and −0.5 mm/s. Not observed in resolved spectrum of nano-γ-Fe2O3 at 14 K. |
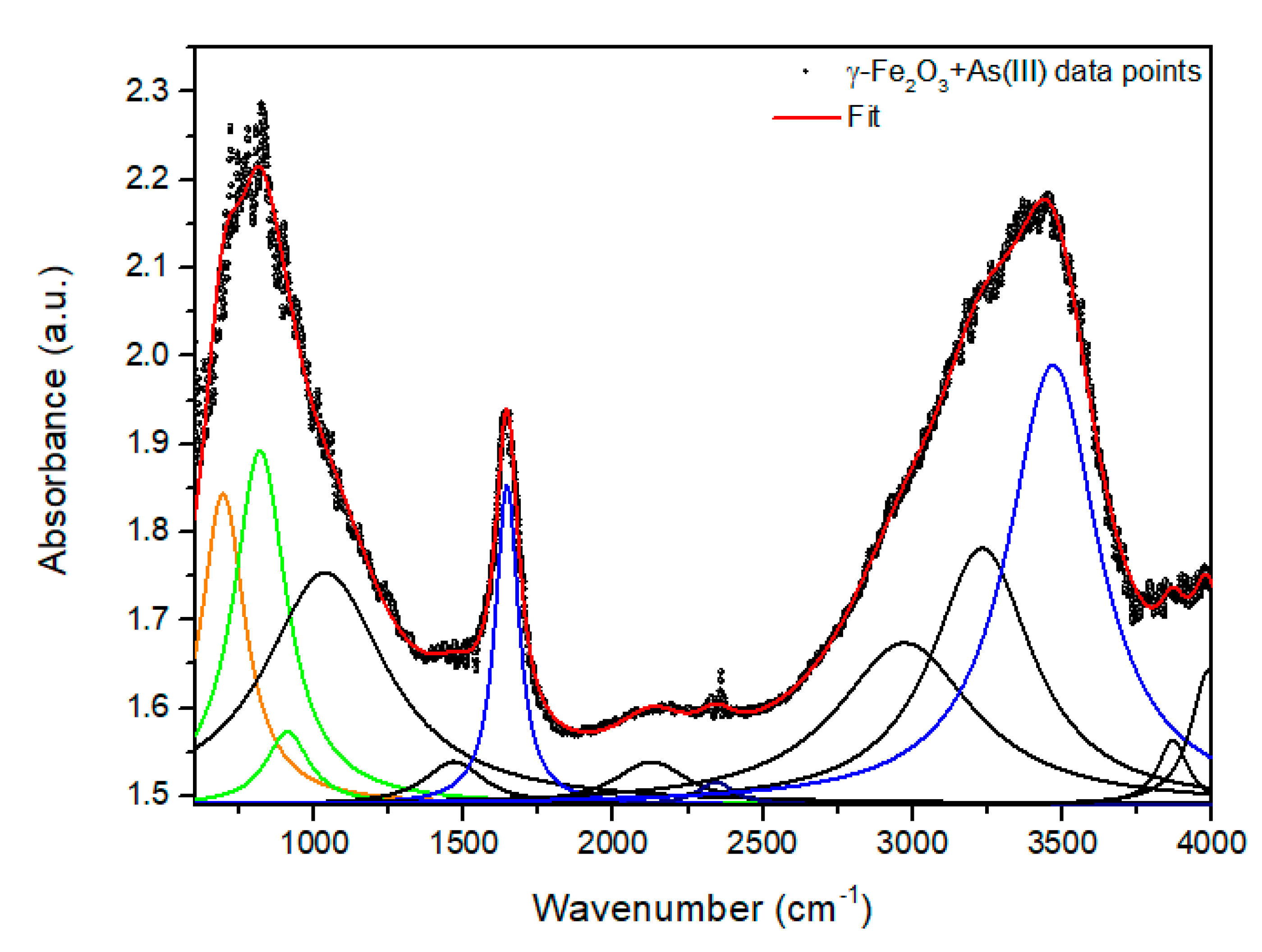
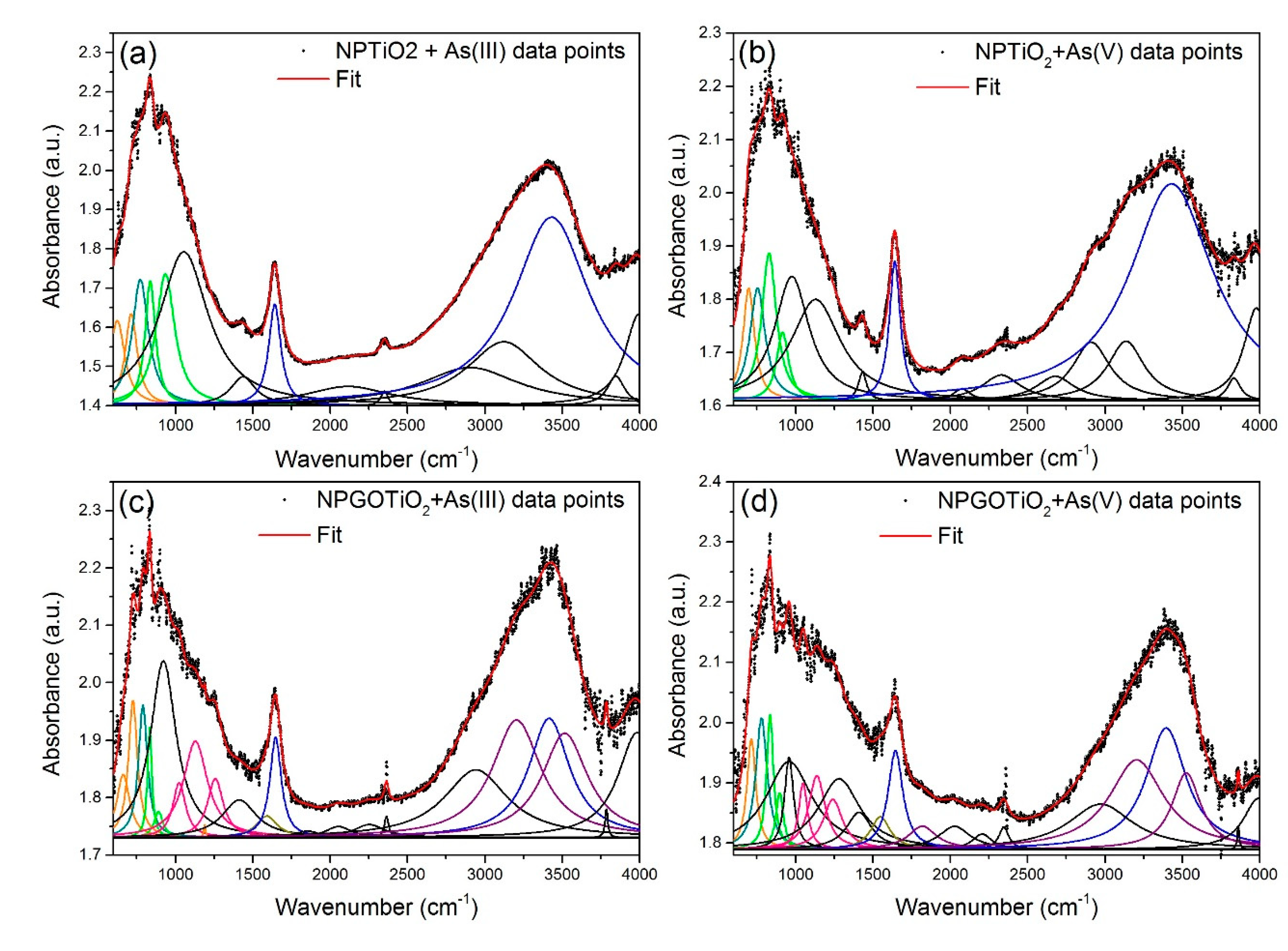
| Samples and Chemical Groups | γ-Fe2O3 + As (III) | NPTiO2 + As (III) | NPTiO2 + As (V) | NPGOTiO2 + As (III) | NPGOTiO2 + As (V) | |
|---|---|---|---|---|---|---|
| Fe-O | 694 | 624.7 | 697 | 662.7 | 714 | |
| Ti-O | - | 773 | 754 | 789 | 779 | |
| As-O | 818.9 | 837.9 | 828.9 | 834 | 834.9 | |
| C-O (alcoxy) | - | - | - | 1024 | 1048 | |
| C-O (epoxy) | - | - | - | 1129 | 1137 | |
| C-O (carboxy) | - | - | - | 1257 | 1240 | |
| C=C | - | - | - | 1591 | 1548 | |
| H2O: | O-H | 1644 | 1641 | 1641 | 1647 | 1645 |
| O-H | 3469 | 3431 | 3430 | 3414 | 3395 | |
| C-OH | - | - | - | 3205 | 3204 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos-Guivar, J.A.; Flores-Cano, D.A.; Caetano Passamani, E. Differentiating Nanomaghemite and Nanomagnetite and Discussing Their Importance in Arsenic and Lead Removal from Contaminated Effluents: A Critical Review. Nanomaterials 2021, 11, 2310. https://doi.org/10.3390/nano11092310
Ramos-Guivar JA, Flores-Cano DA, Caetano Passamani E. Differentiating Nanomaghemite and Nanomagnetite and Discussing Their Importance in Arsenic and Lead Removal from Contaminated Effluents: A Critical Review. Nanomaterials. 2021; 11(9):2310. https://doi.org/10.3390/nano11092310
Chicago/Turabian StyleRamos-Guivar, Juan A., Diego A. Flores-Cano, and Edson Caetano Passamani. 2021. "Differentiating Nanomaghemite and Nanomagnetite and Discussing Their Importance in Arsenic and Lead Removal from Contaminated Effluents: A Critical Review" Nanomaterials 11, no. 9: 2310. https://doi.org/10.3390/nano11092310