The Mechanism of Action of Cyclophosphamide and Its Consequences for the Development of a New Generation of Oxazaphosphorine Cytostatics

Institute of Biochemistry II, Goethe University Frankfurt Medical School, 60590 Frankfurt, Germany
Sci. Pharm. 2020, 88(4), 42; https://doi.org/10.3390/scipharm88040042
Submission received: 7 August 2020
/
Revised: 11 September 2020
/
Accepted: 15 September 2020
/
Published: 28 September 2020
Abstract
:Although cyclophosphamide (CP) has been used successfully in the clinic for over 50 years, it has so far not been possible to elucidate the mechanism of action and to use it for improvement. This was not possible because the basis of the mechanism of action of CP, which was found by lucky coincidence, is apoptosis, the discovery of which was honored with the Nobel Prize only in 2002. Another reason was that results from cell culture experiments were used to elucidate the mechanism of action, ignoring the fact that in vivo metabolism differs from in vitro conditions. In vitro, toxic acrolein is formed during the formation of the cytotoxic metabolite phosphoreamidemustard (PAM), whereas in vivo proapoptotic hydroxypropanal (HPA) is formed. The CP metabolites formed in sequence 4-hydroxycyclophosphamide (OHCP) are the main cause of toxicity, aldophosphamide (ALDO) is the pharmacologically active metabolite and HPA amplifies the cytotoxic apoptosis initiated by DNA alkylation by PAM. It is shown that toxicity is drastically reduced but anti-tumor activity strongly increased by the formation of ALDO bypassing OHCP. Furthermore, it is shown that the anti-tumor activity against advanced solid P388 tumors that grow on CD2F1 mice is increased by orders of magnitude if DNA damage caused by a modified PAM is poorly repairable.
1. Introduction
The development of cyclophosphamide (CP) was based on the idea of enzymatically converting a non-toxic transport form into the active form at the site of action. This idea was already realized in the drug “Honvan” (diethylstilbestrol diphosphate) used in the treatment of prostate cancer. Honvan is an ineffective prodrug that is split into the active form diethylstilbestrol in prostate tissue by acid phosphatases. This principle should be imitated when developing a cytostatic. The first and only chemotherapeutic agent available at that time was nitrogen mustard originally produced as chemical warfare agent. The aim was to convert the highly toxic nitrogen mustard into a non-toxic form by incorporating it into a phosphoric acid amide bond from which it should be released in the tumor tissue by phosphoamidase; it was believed at the time that the enzyme phosphoamidase, which is specific for catalyzing the hydrolysis of phosphorus-nitrogen bonds, was present in increased activity in the tumor tissue [1,2].
This concept turned out to be wrong because the assumption of increased phosphoamidase activity in tumor cells was not correct. However, the result of the substances synthesized according to this premise was CP one of the most successful chemotherapy drugs for tumor therapy. It was quickly recognized that CP is an inactive prodrug that is hydroxylated by cyt. P450 in the liver to 4-hydroxycyclophosphamide (OHCP). All attempts in the last 50 years to improve CP, which was found by serendipity failed because the real mechanism of action of CP was unknown. An example of the failed attempt to improve oxazaphosphorine cytostatics (OX) is the development of Glufosfamide (β-D-glucose-isophosphoreamide mustard), which has been synthesized on the basis of the rationale that cancer cells have an increased uptake of glucose. Glufosfamide was developed with the intention of enriching the alkylating moiety of Ifosfamide—that is isophosphoreamide mustard—in tumor cells [3]. The hoped-for increase in effectiveness did not materialize because the second component, the triggering of apoptosis by the OX metabolite 3-hydroxypropanal (HPA), was not taken into account due to ignorance of the true mechanism of action. Only after the discovery of apoptosis and the enzymatic cleavage of the CP metabolite aldophosphamide (ALDO) with the formation of the proapoptotic CP metabolite 3-hydroxypropanal (HPA) was it possible to develop a conclusive concept for the mechanism of action of CP. The aim of this review is to show how, in knowledge of the mechanism of action and the main causes of toxicity, a new generation of OX can be developed. In animal experiments, model compounds of this new OX are by orders of magnitude more effective than the ones currently used in the clinic.
2. Summary on Materials and Methods
For the synthesis of aldophosphamide-thiazolidine (N,N-2-(2-chloroethyl)-phosphoric acid-diamide-2-(2′-thiazolidinyle-4′-carbonic acid-)-ethylester, NSC-613060), see [4]. Aldophosphamide perhydrothiazine (N,N-2-(2-chloroethyl)-phosphoric acid-diamide-2-(2′-1′,3′-thiazinyle-4′-carbonic acid-)-ethylester was synthesized according to Dausmann [5]. The aldophosphamide-perhydrothiaznie derivatives IAP (N- (2-chloroethyl)- N′-(2-chloroethyl)-phosphoric acid-amide-2-(2′-1′,3′-thiazinyle-4′-carbonic acid-)-ethylester, and SUM-IAP (N- (2-chloroethyl)- N′-(mesylethyl)-phosphoric acid-amide-2-(2′-1′,3′-thiazinyle-4′-carbonic acid-)-ethylester, were synthesized according to Zimmermann et al. [6]. 4-S-ethanol-sulfido-cyclophosphamide (N,N-2-(2-chloroethyl)-amino)-N-2-oxo-4-ethanesulfido-1,3,2-oxazaphospho- rinane) was synthesized as described in [7].
Determination of CP and the CP metabolites OHCP, ALDO, 4-ketocyclophosphamide (KCP) and carboxyphosphamide (CARB) was carried out as described in [8]. For HPLC determination and incubation experiments with rat serum ultrafiltrate and rat serum, see [9].
Animals
Female inbred NMRI mice from the Ivanovas Breeding Institute, Kisslegg, Germany; female CD2F1 mice from Zentralinstitut für Versuchstierkunde, Hannover, Germany. NMRI mice and CD2F1 mice were housed under the following conditions: room temperature 18–22 °C, dark and light periods of 12 h, free access to laboratory chow (Altromin 1324; Altromin GmbH, Lage, Germany) and water. Animals were randomly divided into experimental groups.
Male nu/nu mice were provided by Prof. Fortmeyer University Hospital Frankfurt am Main. For details on breeding and husbandry, see: Fortmeyer H.P. Schriftenreihe Versuchstierkunde Nr.8; Paul Patrey Verlag Berlin, Hamburg 1981 [10].
Antitumor activity was assayed in female CD2F1 mice. 106 P388 mice leukemia cells were transplanted subcutaneously under the skin of the flank. Therapy was started one day after tumor transplantation or 7 days after tumertransplantation, when tumor area was approximately 0.5 cm2. For the determination of tumor size, see [8], for the procedure of toxicity tests, see [4], and, for the detrmination of leukocytes, see [11].
3. Metabolism of Cyclophosphamide
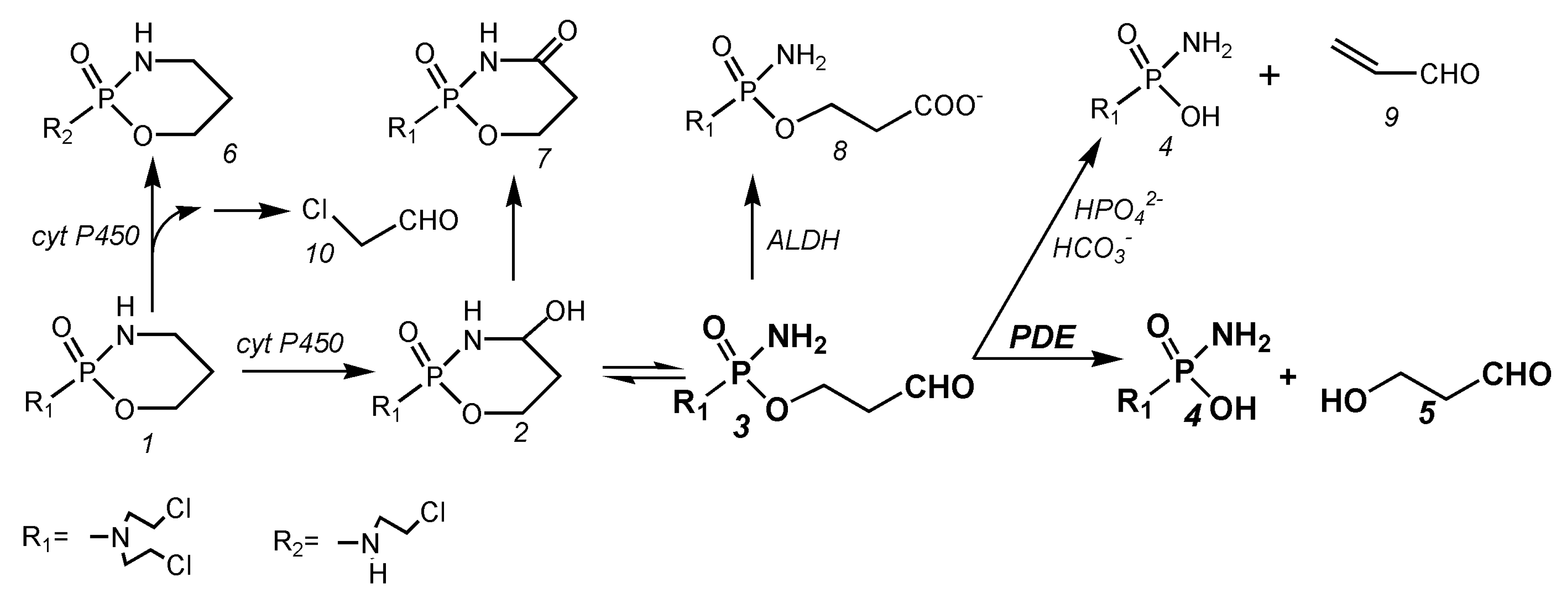
After the administration of CP in humans and animals, the main metabolic pathways of CP are as follows (Figure 1):
After the hydroxylation of CP (1 Figure 1) by hepatic P450 enzymes, the resulting OHCP (2 Figure 1) forms an equilibrium with his tautomeric aldehyde ALDO (3 Figure 1). OHCP and ALDO are not cytotoxic. In vivo the tautomers OHCP and ALDO are irreversibly inactivated by liver enzymes. OHCP is converted by enzymatic oxidation into the non-cytotoxic KCP (7 Figure 1), and ALDO is detoxified by aldehyde dehydrogenases into the cytotoxically inactive CARB (8 Figure 1)
In cell culture experiments the part of ALDO that has not been detoxified to CARB is decomposed by β elimination of acrolein (9 Figure 1) to PAM (4 Figure 1) This reaction is catalyzed by secondary phosphate ions and to a lesser extent by bicarbonate ions [12]. In vivo, however, ALDO is enzymatically decomposed by phosphodiesterases (PDE) to HPA (5 Figure 1) and PAM [9]. PAM is the alkylating moiety which is thought to unfold cell toxicity by DNA alkylation. A small part of CP is converted to dechlorocyclophosphamide (6 Figure 1) by side-chain hydroxylation. Toxic chloroacetaldehyde (10 Figure 1) is produced as a by-product.
4. Part 1: Causes of Toxicity and Mechanism of Action of Cyclophosphamide
4.1. Balance of the Cyclophosphamide Metabolism
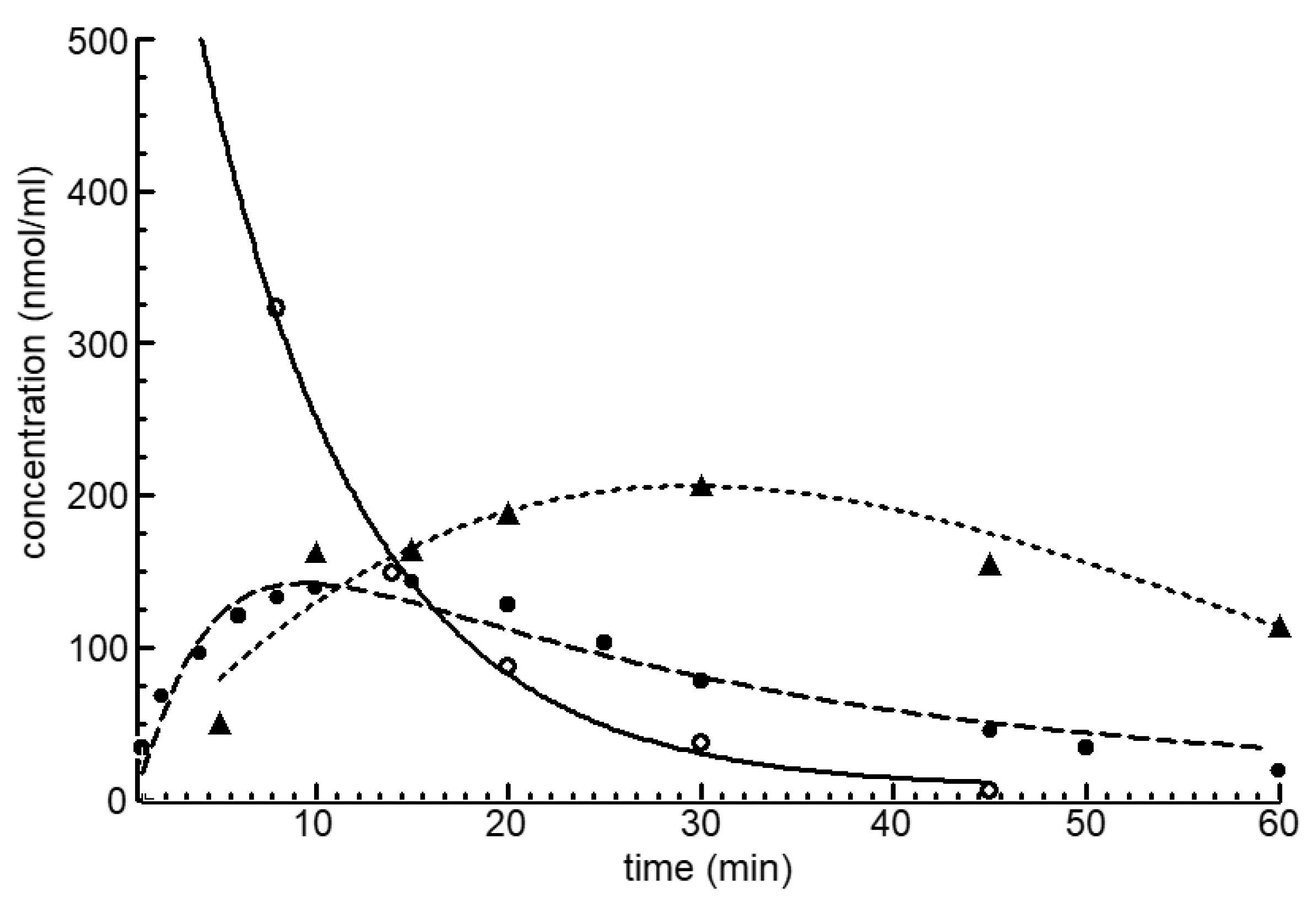
Figure 2 shows the blood level curves of CP, OHCP and of the detoxification products KCP and CARB after the intravenous injection of 100 mg/kg CP in female NMRI mice. The detoxification products KCP and CARB accumulate because they are eliminated more slowly from the blood than OHCP.
However, more important than these pharmacokinetic parameters is the question how much of the CP administered is hydroxylated in the liver by the P450 enzyme system to OHCP and how much of the OHCP formed escapes detoxification to KCP and CARB and is used for therapeutic and toxic reactions. This question was answered in separate experiments [8]. The results are summarized in Table 1.
Table 1 shows that, of 358 nmol/g corresponding to 100 mg/kg CP after intravenous administration in female NMRI mice, 92% are converted into OHCP. In total, 80% of the formed OHCP are detoxified to KCP and CARB, so that, ultimately, 20% of the CP converted into OHCP is available for protein binding, therapeutic and toxic reactions. If renal excretion is neglected, a maximum of 8% of the applied CP is converted into chloroacetaldehyde (10, Figure 1) and dechlorocyclophosphamide (6, Figure 1) by side chain hydroxylation.
This result is important for the determination of the metabolite responsible for the toxicity of CP. CP, KCP and CARB are nontoxic inert compounds, and acrolein, which is often discussed as the main cause of the toxicity of CP, can be ruled out because it is not produced in vivo [9]. Therefore, only OHCP, ALDO, HPA, PAM and chloroacetaldehyde are candidates for CP toxicity (see Figure 1). HPA is also quite out of the question as a cause of CP toxicity. It has proven to be non-toxic to eukaryotes and is used as food additive to prevent spoilage by growth of pathogens because it is active against bacterial, viruses and fungi. Neurotoxic and nephrotoxic chloroacetaldehyde cannot be avoided as long as the P450 enzyme system is involved in the metabolism of OX. Candidates for the toxicity of CP are therefore OHCP, ALDO and PAM
4.2. The Reason of CP Toxicity
Table 2 shows the results of acute toxicity experiments with OHCP and PAM in male nude mice. The experiment shows that OHCP, the LD50 value of which was calculated from these data to be 530 µmol/kg (143 mg/kg), is more toxic than PAM, the LD50 of which was calculated to be 810 µmol/kg (183 mg/kg). Because substances injected intraperitoneally must pass the liver before entering the systemic circulation only a part of a drug that is metabolized in the liver enters the systemic circulation (first pass effect). Since OHCP/ALDO is metabolized in the liver to the non-toxic KCP and CARB, the difference in toxicity between OHCP and PAM is greater than that shown by the toxicity experiment.
An indication of deviation of the real toxicity of OHCP from the measured toxicity are given from the already shown studies to the balance of the CP metabolism. The studies—Table 1—show that only a maximum of 20% of the OHCP formed after CP administration is available for toxic reactions. From this, it follows that the real toxicity of OHCP is about 3 to 5 times greater than the toxicity test indicates. As will be shown in Section 5.1, OHCP is responsible for the toxicity of the tautomeric forms OHCP/ALDO.
4.3. The Mechanism of Action of Cyclophosphamide
When OHCP is incubated in rat serum ultrafiltrate pH 7, 37 °C, a stable equilibrium between OHCP and ALDO occurs. The half-life for the decrease in concentration of the pair of tautomers OHCP/ALDO is about 23 h. If the same experiment is done with whole rat serum, then the OHCP/ALDO equilibrium also sets in, but the half-life for the decrease in concentration of OHCP/ALDO is only 20 min [9]. This faster decrease in concentration in rat serum than in the rat serum, which has been made free of proteins, indicates that the decomposition of OHCP/ALDO is enzyme catalyzed. The splitting enzyme was identified to be a phosphodiesterase (PDE) [13]. The Michaelis constant for the enzyme in human serum was determined to be 1 mM.
HPLC analysis of the serum samples with OHCP after protein precipitation and treatment with 2,4-dinitrophenylhydrazine showed time-dependent increasing concentrations of the 2,4-dinitrophenylhydrazones of 3-hydroxypropanal [9]. Based on these findings, it was concluded that the decomposition reaction of ALDO in whole rat serum is not the spontaneous β-elimination of acrolein but HPA is formed according to the reaction sequence highlighted in bold in Figure 1.
4.4. The Special Feature of Cyclophosphamide Compared to Other Alkylating Agents
When an alkylating substance acts on a cell, only a few of the possible alkylation reactions are therapeutically effective. A measure of the therapeutic effect is the therapeutic index—that is, the ratio from the amount of a therapeutic agent that causes the therapeutic effect to the amount that causes toxicity. According to Brock [14] and Brock and Hohorst [15], the therapeutic index—measured in Yoshida ascites sarcoma-bearing rats—was determined to be 120 for OHCP but only 3.5 for PAM and 2.5 for nitrogen mustard. This result indicates that DNA alkylations by PAM released from ALDO in vivo are much more efficient than DNA alkylations produced by bare PAM injected.
In 9L cells, overexpressing the anti-apoptotic Bcl-2 protein, with OHCP Schwartz and Waxman [16] could only determine a cytostatic effect but no cytotoxic apoptosis. These findings demonstrate that the mechanism of action of OHCP proceeds in two steps. First occurs DNA damage by PAM and, secondly, the damaged cell is eradicated by p53 mediated apoptosis. These findings suggest that the second step is responsible for the more than 30 fold higher therapeutic index of OHCP.
In the following, it is shown—supported by scientific literature—that HPA strongly enhances the apoptosis of cells, the DNA of which is damaged by PAM, which is the reason for the high therapeutic index of OHCP compared to PAM.
4.5. Scientific Evidence for the Enhancement of PAM-Induced Apoptosis by HPA
HPA is produced by Lactobacillus reuteri and submitted to the culture medium [17]. Experiments by Iyer [18], who investigated the effects of the supernatant of L. reuteri cultures (LR) on tumor necrosis factor activated apoptose signaling pathways in human leukemia cells, showed that reuterin inhibits the formation of the anti-apoptotic proteins Bcl-2 and Bcl-xL and the TNF-dependent NF-κB activation by inhibition of the translocation of the p65 subunit of NF-κB into the nucleus. The experiments of Iyer et al. further showed that the degradation of IκB is suppressed by a lack of ubiquitination by LR. Thus, by the inhibition of the nuclear translocation of NF-κB, apoptosis is enhanced [19]. In further experiments, Iyer et al. showed that certain branches of the MAPK signaling pathways are modified by LR. They found increased phosphorylation of the p38 and JNK proteins, indicating activation of apoptosis and cell cycle arrest. In addition, they measured the decreasing phosphorylation of ERK, which is a sign of decreased proliferation, cell division, and cell differentiation.
4.6. Reaction Scheme for the Mechanism of Action of Oxazaphosphorine Cytostatics Involving HPA
The discovery of HPA as a CP metabolite [9], the scientific work of Schwarz and Waxman [16], who have demonstrated that the function of PAM is to induce cytotoxic apoptosis by DNA alkylation, and the results published by Iyer and coworkers [18], showing that HPA is a proapoptotic aldehyde, lead to the scheme for the mechanism of action of CP and other OX shown in Figure 3.
CP is hydroxylated by P450 enzymes in the liver to OHCP, which is in equilibrium with ALDO, ALDO is the pharmacologic active metabolite. A part of ALDO is oxidized by aldehyde dehydrogenase to the non-toxic CARB (not shown). The amount of ALDO not oxidized is decomposed by PDE to the alkylating PAM and HPA. PAM damages DNA by alkylation. The alkylated DNA is either repaired immediately or—if this is not possible—the tumor suppressor protein p53 is activated, which induces cell cycle stop to give the cell time to repair the damage. If DNA repair is not possible, p53 induces apoptosis, which is—and this is special for CP and other OX—enhanced by HPA, which supports apoptosis by inhibiting the anti-apoptotic proteins Bcl-2 and Bcl-xL, and by inhibiting NFκB activation. Additional, HPA promotes apoptosis by enhancing mitogen activated protein kinase (MAPK) activity by enhancing JNK and p38 phosphorylation.
4.7. Conclusions from the Studies on the Toxicity and the Mechanism of Action of Cyclophosphamide for the Development of a New Generation of Oxazaphosphorine Cytostatics
The consequences of the studies on toxicity and the mechanism of action of OX for the development of a new generation of OX are a) to avoid the unnecessary toxicity in the formation of OHCP through the formation of the toxic by-product chloroacetaldehyde and avoidance of toxicity of OHCP itself and b) increase the efficiency of cytotoxic apoptosis initiated by DNA damage by PAM.
5. Part 2: Bypassing Toxicity and Increasing Cytotoxic Apoptosis of Oxazaphosphorine Cytostatics
5.1. Decrease in Toxicity by Bypassing the Formation of 4-Hydroxycyclophosphamide in the Formation of Aldophosphamide
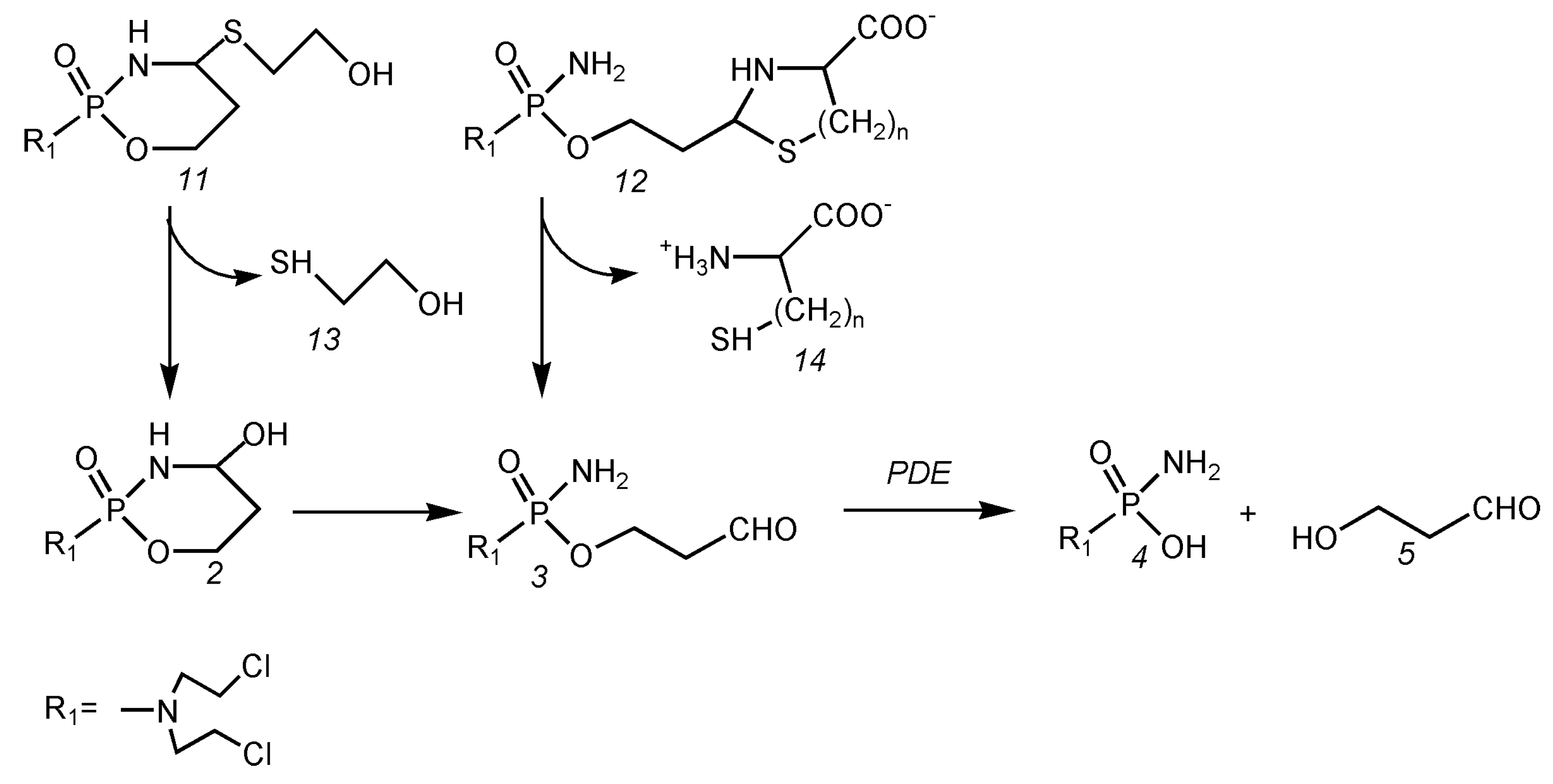
In order to determine the proportion of OHCP to the total toxicity of CP, toxicity tests were carried out with 4-(S-ethanol)-cyclophosphamide (11 Figure 4) and the thiazolidine from ALDO (12, n = 1 Figure 4) and the perhydrothiazine from ALDO (12, n = 2 Figure 4). The results are summarized in Table 3.
4-(S-ethanol)-cyclophosphamide) is 7 to 9 times more toxic than aldophosphamide-thiazolidine and aldophosphamide-perhydrothiazine, which hydrolyze directly to ALDO bypassing OHCP.
This experiment shows, impressively, that, by avoiding the formation of OHCP, the toxicity is drastically reduced.
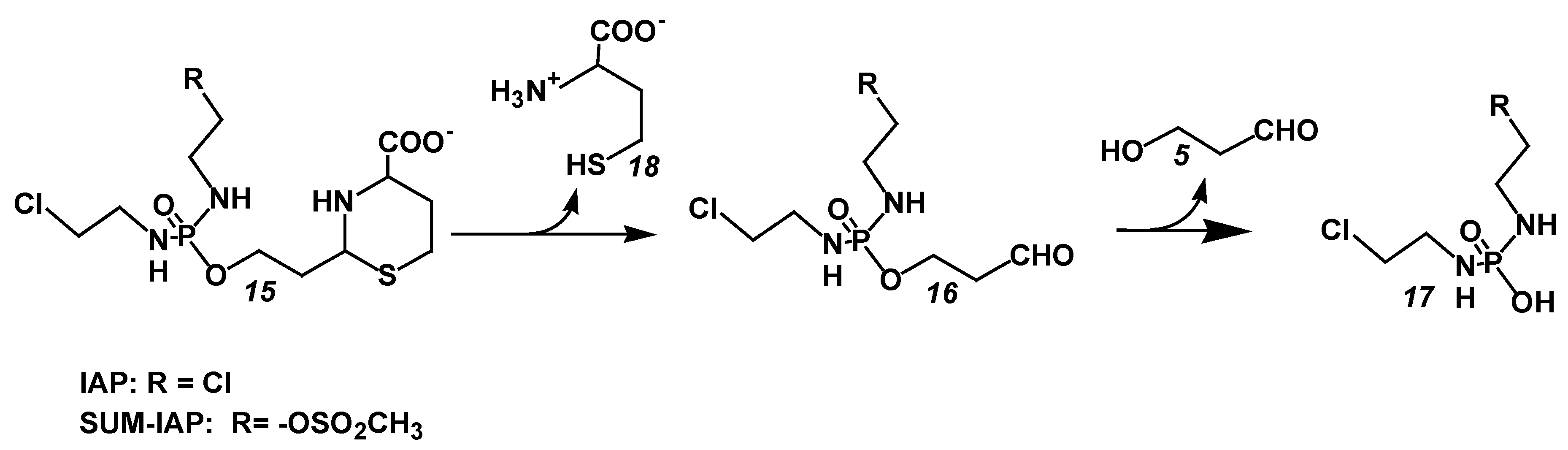
The thiazolidines and perhydrothiazines from ALDO thus meet the first requirement for the development of a new generation of OX. They are considerably less toxic than the parent substance CP. Whether they are also candidates for the chemotherapy of tumor diseases was examined by comparative therapy experiments with the clinically proven OX Ifosfamide (IF) and the perhydrothiazine from I-aldophosphamide ((IAP) 15 Figure 5). IAP hydrolyzes to I aldophosphamide (IALDO) 16 Figure 5)) and L-homocysteine (18 Figure 5). IALDO is split by PDE into I phosphoreamide mustard ((IPAM) 17 Figure 5) and HPA (5 Figure 5).
The experiment was carried out with I-aldophosphamide perhydrothiazine (IAP) because the synthesis and handling of the strongly hygroscopic and amorphous aldophosphamide thiazolidine was problematic. In contrast, the synthesis of IAP is not a problem. The substance was used as a stable, readily water-soluble lyophylizate for the following experiments.
The results of therapy experiments in P388 tumor-bearing female CD2F1 mice are summarized in Table 4.
Therapy with IAP cures 2/6 or 4/5 animals (surviving time > 100 d) while only survival time extensions are achieved with the same equitoxic dose IF. This experiment demonstrates the superiority of IAP that can be dosed higher than IF because it forms IALDO directly by bypassing the toxic OHIF and is therefore much less toxic than IF.
5.2. Increase in the Antitumor Activity of I-Aldoperhydrthiazines by Modulating the Alkylating Function
According to the proposed mechanism of action of CP and other OX, DNA damage caused by PAM or IPAM is not the cause of cell death; it merely has the function of initiating cytotoxic apoptosis. Since cellular DNA repair mechanisms repair DNA damage very efficiently and quickly, only a small part of the DNA damage caused by the alkylating metabolites trigger apoptosis. In order to increase this proportion, it is necessary to generate DNA damage that cannot be repaired or is difficult to repair. The alkylating function of all OX consists of 2 chloroethyl groups, which generate easily repairable DNA inter strand crosslinks. Therefore, most of the DNA damage caused by the currently used OX is repaired before it initiates apoptosis. According to Povirk and Shuker [20] mesyl-ethyl groups form intra strand cross-links that are poorly repaired (http://www.atdbio.com/content/16/Nucleic-acid-drug-interactions, accessed on 01 March 2018)
Figure 6 shows the results of therapy experiments with IAP and an IAP derivative—SUM-IAP—in which a chloroethyl group of IAP is substituted by a mesyl ethyl group (R = –OSO2CH3, 15 Figure 5). A small dose of IAP and SUM-IAP of 0.29 mmol/kg was administered on day 7–11 after tumor transplantation when the tumor area was approximately 0.5 cm2. The experiment with IAP, which produces easily repairable inter strand crosslinks, shows only a marginal growth delay of the tumor, whereas, in the experiment with SUM-IAP that generates poorly repairable DNA intra strand cross links, the tumor burden is reduced below detection level for 8 days before tumor cells that have become resistant grow again. In a further experiment (not shown) with only one injection of 1.32 mmol/kg IAP and SUM-IAP (521, respectively, 600 mg/kg), the result was in principle the same, only growth delay in the experiment with IAP but decrease in tumor burden below detection level in the experiment with SUM-IAP.
To compare the antitumor activity of IAP and SUM-IAP quantitatively, the tumor growth curves after therapy with IAP and SUM-IAP were evaluated using the Alexander and Mikulski back-extrapolation method [21]. From the slopes of the tumor growth curves, it can be concluded that there is a 104–105 times increase in antitumor activity when one 2-chloroethyl group in IAP is substituted by a mesyl ethyl group [11].
6. The New Generation of Oxazaphosphorine Cytostatics Are Candidates for Immunological Tumor Therapy
It is well known that CP in the lower dose range has—in addition to its antitumor activity in the higher dose range—immunomodulatory properties [22,23]. It has been shown for CP that it activates anti-tumor-driven CD8+ T-cell (CTL) responses [24], supports tumor-driven CD4+ T-cell (Th) differentiation [25] and stimulates dendritic cell maturation [26]. The fact that CP has an immune stimulating effect in low doses is due to the special sensitivity of T cell inhibiting regulatory T cells (Treg) to ALDO. Heylmann and collegues [27] compared the sensitivity of Treg, CTL and Th to Mafosfamide, which hydrolyses to OHCP within minutes. They could show that the high sensitivity of Treg compared to CTL and Th is due to induction of apoptosis and decrease in ability of Treg to repair damaged DNA. Thus, OX are antitumor substances that combine 2 mechanisms of action. These are the immediate cytotoxic apoptosis triggered by DNA alkylation and antitumor-driven CTL response. The latter, however, only with low doses.
The question arises: What influence does the type of DNA damage—repairable or irreparable—have on the immunomodulatory abilities of OX? This question is answered by an experiment summarized in Figure 7 and already published in [28]. Solid growing P388 tumor-bearing mice were treated with 666 mg/kg SUM-IAP on days 7 and 8 after tumor transplantation. This treatment causes a reduction in the tumor below detection limit from day 11 to day 22 (not shown). The tumor was then measurable again, animals also suffered from metastases and died on days 25–30 (Figure 7).
In the same experiment, but with an additional therapy cycle on days 14 and 15, no regrowth of the primary tumor and no formation of metastasis were observed. All animals survived the observation period of 100 days and can be regarded as cured, concurrent a sharp increase in no of leukocytes is measured (Figure 7). The latter is proof that the regrowth of the primary tumor, regrowth of tumor cells that have become resistant, and the formation of metastases are prevented by anti-tumor-driven immunological mechanisms.
This experiment shows, impressively, that by creating irreparable DNA alkylations, the second mechanism of action of OX, the anti-tumor-driven stimulation of the immune system, is greatly increased. With the new generation of OX, which were presented here, for the first time anti-tumor drugs are available, which can simultaneously eradicate tumor cells by cytotoxic apoptosis and by anti-tumor-driven immunological mechanisms of action.
Funding
This study was funded by the Bundesministerium für Forschung und Technologie.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Meyer, J.; Weinmann, J.P. Phosphamidase Content of Normal and Pathologic Tissues of the Oral Cavity. J. Histochem. Cytochem. 1953, 1, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomitka, K.; Takeuchi, T. On the phosphamidase reaction of tumor tissues. Gann 1955, 46, 333–334. [Google Scholar]
- Mazur, L.; Opydo-Chanek, M.; Stojak, M. Glufosfamide as a new oxazaphosphorine anticancer agent. Anti-Cancer Drugs 2011, 22, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Voelcker, G. Causes and possibilities to circumvent cyclophosphamide toxicity. Anti-Cancer Drugs 2020, 31, 617–622. [Google Scholar] [CrossRef]
- Dausman, D. Synthese von N,N-Bis-(2chlorethyl)-Phosphorsäureamid-Derivaten als Substrat für 3′-5′Exonucleasen und Untersuchungen zur Umsetzung von “aktiviertem Cyclophosphamid” mit 1,2 und 1,3 Dinukleophilen. Ph.D. Thesis, Universität Frankfurt am Main, Frankfurt, Germany, 1988. [Google Scholar]
- Zimmermann, J.; Bauer, H.H.; Hohorst, H.J.; Voelcker, G. Synthesis of I-aldofosfamide-perhydrothiazines. Arzneimittelforschung 2000, 50, 843–847. [Google Scholar] [CrossRef]
- Peter, G.; Wagner, T.; Hohorst, H.J. Studies on 4-hydroperoxycyclophosphamide (NSC-181815): A simple preparation method and its application for the synthesis of a new class of “activated” sulfur-containing cyclophosphamide (NSC-26271) derivatives. Cancer Treat. Rep. 1976, 60, 429–435. [Google Scholar]
- Voelcker, G.; Haeglsperger, R. Pharmacokinetics of cyclophosphamide and cyclophosphamide metabolites in the mouse and their influence on the therapeutic effect of “activated” cyclophosphamide (4-hydroxycyclophosphamide) (author’s transl). Arzneimittelforschung 1982, 32, 639–647. [Google Scholar]
- Voelcker, G. Enzyme Catalyzed Decomposition of 4-Hydroxycyclophosphamide. Open Conf. Proceeding J. 2017, 8, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Fortmeyer, H.P. Schriftenreihe Versuchstierkunde Nr.8; Paul Patrey Verlag: Berlin, Germany; Hamburg, Germany, 1981. [Google Scholar]
- Voelcker, G.; Pfeiffer, B.; Schnee AHohorst, H.J. Increased Antitumour Activity of mesyl-I-aldophosphamide-perhydrothiazine, in Vivo but Not in Vitro, Compared to I-aldophosphamide-perhydrothiazine. J. Cancer Res. Clin. Oncol. 2000, 126, 74–78. [Google Scholar]
- Low, J.E.; Borch, R.F.; Sladek, N.E. Conversion of 4-hydroperoxycyclophosphamide and 4-hydroxycyclophosphamide to phosphoramide mustard and acrolein mediated by bifunctional catalysis. Cancer Res. 1982, 42, 830–837. [Google Scholar]
- Hohorst, H.J.; Bielicki, L.; Voelcker, G. The Enzymatic Basis of Cyclophosphamide Specificity. Adv. Enzym. Regul. 1986, 25, 99–122. [Google Scholar] [CrossRef]
- Brock, N. Comparative pharmacologic study in vitro and in vivo with cyclophosphamide (NSC-26271), cyclophosphamide metabolites, and plain nitrogen mustard compounds. Cancer Treat. Rep. 1976, 60, 301–308. [Google Scholar] [PubMed]
- Brock, N.; Hohorst, H.J. The problem of specificity and selectivity of alkylating cytostatics: Studies on N-2-chlorethylamido-oxazaphosphorines. Z. Krebsforsch 1977, 88, 185–215. [Google Scholar] [CrossRef]
- Schwartz, P.S.; Waxman, D.J. Cyclophosphamide induces caspase 9-dependent apoptosis in 9L tumor cells. Mol. Pharmacol. 2001, 60, 1268–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleusix, V.; Lacroix, C.; Vollenweider, S.; Duboux, M.; Le Blay, G. Inhibitory activity spectrum of reuterin produced by Lactobacillus reuteri against intestinal bacteria. BMC Microbiol. 2007, 12, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, C.; Kosters, A.; Sethi, G.; Kunnumakkara, A.B.; Aggarwal, B.B.; Versalovic, J. Probiotic Lactobacillus reuteri promotes TNF-induced apoptosis in human myeloid leukemia-derived cells by modulation of NF-kappaB and MAPK signalling. Cell. Microbiol. 2008, 10, 1442–1452. [Google Scholar] [CrossRef]
- Wu, M.; Lee, H.; Bellas, R.E.; Schauer, S.L.; Arsura, M.; Katz, D.; FitzGerald, M.J.; Rothstein, T.L.; Sherr, D.H.; Sonenshein, G.E. Inhibition of NF-kappaB/Rel induces apoptosis of murine B cells. EMBO J. 1996, 2, 4682–4690. [Google Scholar] [CrossRef]
- Povirk, L.F.; Shuker, D.E. DNA damage and mutagenesis induced by nitrogen mustards. Mutat. Res. 1994, 318, 205–226. [Google Scholar] [CrossRef]
- Alexander, P.; Mikulski, Z. Differences in the Response of Leukaemia Cells in Tissue Culture to Nitrogen Mustard and to Dimethyl Myleran. Biochem. Pharmacol. 1961, 5, 275–282. [Google Scholar] [CrossRef]
- Sistigu, A.; Viaud, S.; Chaput, N.; Bracci, L.; Proietti, E.; Zitvogel, L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin. Immunopathol. 2011, 33, 369–383. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Ditsworth, D.; Catanzaro, J.M.; Sabino, G.; Furie, M.B.; Kew, R.R.; Crawford, H.C.; Zong, W.X. DNA alkylating therapy induces tumor regression through an HMGB1-mediated activation of innate immunity. J. Immunol. 2011, 186, 3517–3526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Most, R.G.; Currie, A.J.; Cleaver, A.L.; Salmons, J.; Nowak, A.K.; Mahendran, S.; Larma, I.; Prosser, A.; Robinson, B.W.; Smyth, M.J.; et al. Cyclophosphamide chemotherapy sensitizes tumor cells to TRAIL-dependent CD8 T cell-mediated immune attack resulting in suppression of tumor growth. PLoS ONE 2009, 4, e6982. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.C.; Blazar, B.R.; Mellor, A.L.; Munn, D.H.; Zhou, G. Chemotherapy rescues tumor-driven aberrant CD4+ T-cell differentiation and restores an activated polyfunctional helper phenotype. Blood 2010, 115, 2397–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharabi, A.; Laronne-Bar-On, A.; Meshorer, A.; Haran-Ghera, N. Chemoimmunotherapy reduces the progression of multiple myeloma in a mouse model. Cancer Prev. Res. 2010, 3, 1265–1276. [Google Scholar] [CrossRef] [Green Version]
- Heylmann, D.; Bauer, M.; Becker, H.; van Gool, S.; Bacher, N.; Steinbrink, K.; Kaina, B. Human CD4+CD25+ regulatory T cells are sensitive to low dose cyclophosphamide: Implications for the immune response. PLoS ONE 2013, 8, e83384. [Google Scholar] [CrossRef] [Green Version]
- Voelcker, G. Immunostimulating and cancer-reductive experimental therapy with the oxazaphosphorine cytostatic SUM-IAP. Anti-Cancer Drugs 2018, 29, 411–415. [Google Scholar] [CrossRef]
Figure 1.
Main metabolic pathways of cyclophosphamide (for details, see text). 1 cyclophosphamide (CP), 2 4-hydroxycyclophosphamide (OHCP), 3 aldophosphamide (ALDO), 4 phosphoreamidemustard (PAM), 5 hydroxypropanal (HPA), 6 dechlorocyclophosphamide, 7 4-ketocyclophosphamide (KCP), 8 carboxyphosphamide (CARB), 9 acrolein, 10 chloroacetaldehyde.
Figure 1.
Main metabolic pathways of cyclophosphamide (for details, see text). 1 cyclophosphamide (CP), 2 4-hydroxycyclophosphamide (OHCP), 3 aldophosphamide (ALDO), 4 phosphoreamidemustard (PAM), 5 hydroxypropanal (HPA), 6 dechlorocyclophosphamide, 7 4-ketocyclophosphamide (KCP), 8 carboxyphosphamide (CARB), 9 acrolein, 10 chloroacetaldehyde.

Figure 2.
Blood level curves of CP (○), OHCP (●) and detoxification products KCP plus CARB (▲) after intravenous injection of 100 mg/kg CP in female NMRI mice. CP is eliminated from the blood with a half-life of 6 min. This corresponds to the half-life of the formation of OHCP of 5 min. The detoxification products KCP and CARB accumulate because they are eliminated from the blood with elimination half-lives of 41 and 20 min more slowly than OHCP for which an elimination half-life of 16 min was calculated.
Figure 2.
Blood level curves of CP (○), OHCP (●) and detoxification products KCP plus CARB (▲) after intravenous injection of 100 mg/kg CP in female NMRI mice. CP is eliminated from the blood with a half-life of 6 min. This corresponds to the half-life of the formation of OHCP of 5 min. The detoxification products KCP and CARB accumulate because they are eliminated from the blood with elimination half-lives of 41 and 20 min more slowly than OHCP for which an elimination half-life of 16 min was calculated.

Figure 3.
Mechanism of action of oxazaphosphorine cytostatics (OX) shown here as an example for CP (for details, see text).
Figure 3.
Mechanism of action of oxazaphosphorine cytostatics (OX) shown here as an example for CP (for details, see text).

Figure 4.
Hydrolysis of 4-(S-ethanol)-cyclophosphamide (11) via OHCP and direct hydrolysis of aldophosphamide thiazolidine (12 n = 1) and aldophosphamide perhydrothiazine (12 n = 2) to aldophosphamide (3) and enzymatic cleavage of aldophosphamide to phosphoreamidemustard (4) and hydroxypropanal (5).
Figure 4.
Hydrolysis of 4-(S-ethanol)-cyclophosphamide (11) via OHCP and direct hydrolysis of aldophosphamide thiazolidine (12 n = 1) and aldophosphamide perhydrothiazine (12 n = 2) to aldophosphamide (3) and enzymatic cleavage of aldophosphamide to phosphoreamidemustard (4) and hydroxypropanal (5).

Figure 5.
Hydrolysis of I-aldophosphamide (IAP) (R = Cl) and SUM-IAP (R = –OSO2CH3 (15)) to I-aldophosphamide (16) and homocysteine (18) and the formation of I-phosporeamidemustard (17) and 3-hydroxypropanal (5).
Figure 5.
Hydrolysis of I-aldophosphamide (IAP) (R = Cl) and SUM-IAP (R = –OSO2CH3 (15)) to I-aldophosphamide (16) and homocysteine (18) and the formation of I-phosporeamidemustard (17) and 3-hydroxypropanal (5).

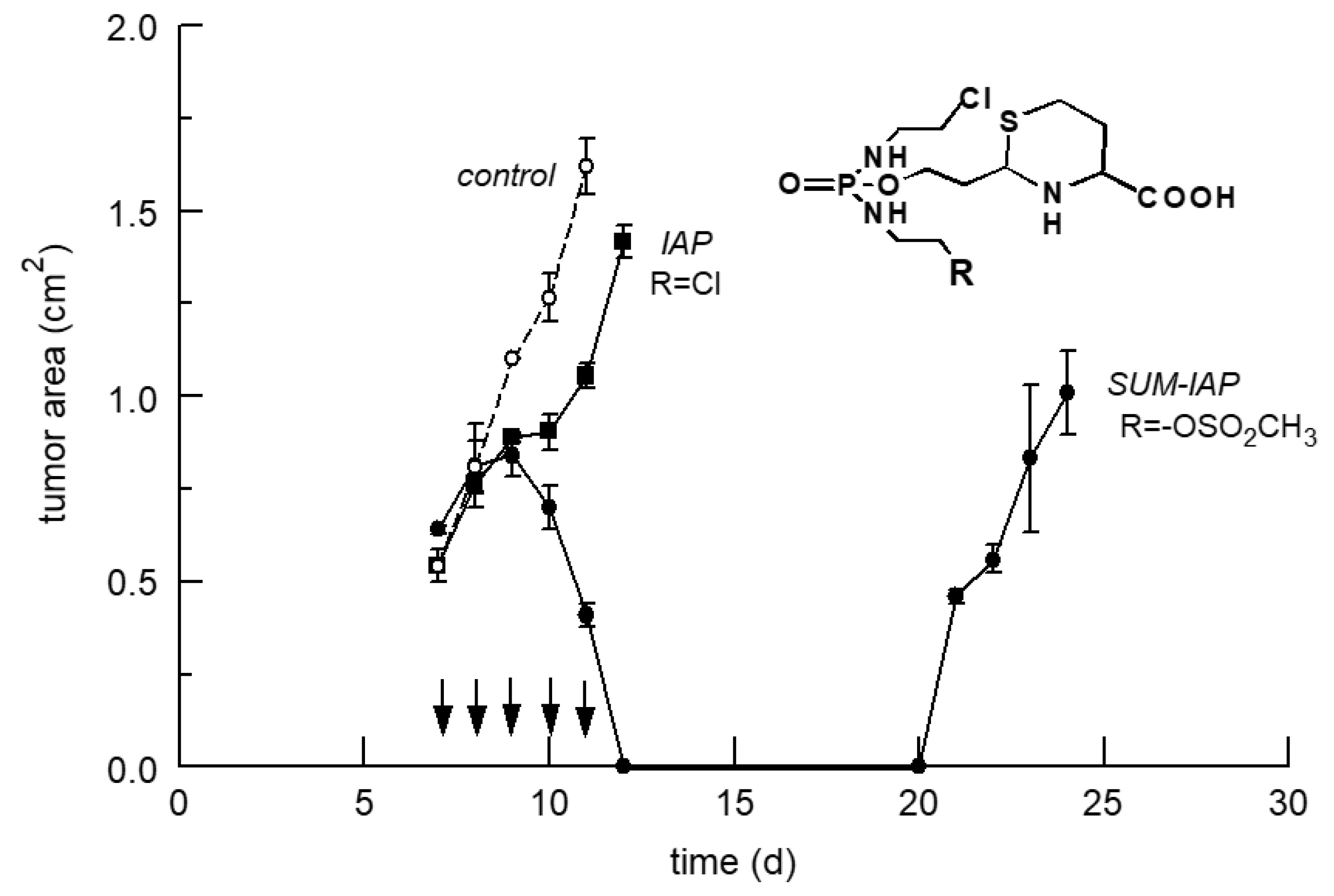
Figure 6.
Tumor growth curves of subcutaneously transplanted P388 tumors in female CD2F1 mice following therapy with 0.29 mmol IAP (115 mg/kg) (■) or SUM-IAP (133 mg/kg) (●) i.p. on day 7–11 (arrows), mean ± standard deviation from 3 animals. “Control” (○) shows the tumor growth curve of subcutaneously transplanted P388 mice leukemia cells in untreated female CD2F1 mice.
Figure 6.
Tumor growth curves of subcutaneously transplanted P388 tumors in female CD2F1 mice following therapy with 0.29 mmol IAP (115 mg/kg) (■) or SUM-IAP (133 mg/kg) (●) i.p. on day 7–11 (arrows), mean ± standard deviation from 3 animals. “Control” (○) shows the tumor growth curve of subcutaneously transplanted P388 mice leukemia cells in untreated female CD2F1 mice.

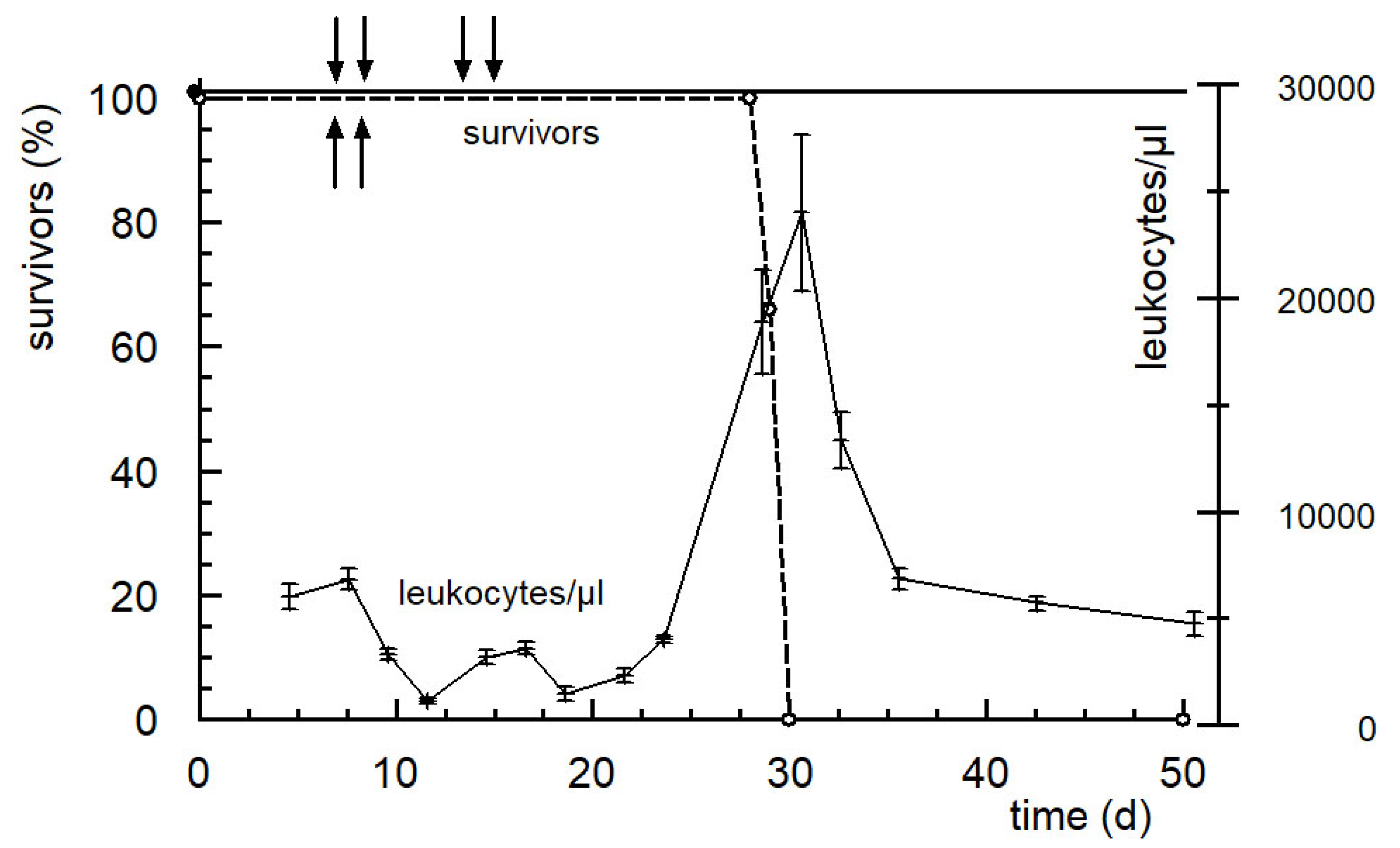
Figure 7.
Survival curves of CD2F1 mice bearing subcutaneously transplanted solid growing P388 tumors following therapy with 666 mg/kg SUM-IAP on days 7 and 8 (upward arrows, broken line, 3 animals) and therapy following 666 mg/kg SUM-IAP on days 7 and 8 and on days 14 and 15 (downward arrows, solid line, 5 animals) and the number of leucocytes following therapy on days 7.8 and 14.15 (mean values +/− SD). The median survival time of untreated tumor-bearing controls was 11–12 days.
Figure 7.
Survival curves of CD2F1 mice bearing subcutaneously transplanted solid growing P388 tumors following therapy with 666 mg/kg SUM-IAP on days 7 and 8 (upward arrows, broken line, 3 animals) and therapy following 666 mg/kg SUM-IAP on days 7 and 8 and on days 14 and 15 (downward arrows, solid line, 5 animals) and the number of leucocytes following therapy on days 7.8 and 14.15 (mean values +/− SD). The median survival time of untreated tumor-bearing controls was 11–12 days.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Balance of cyclophophamide metabolism in female NMRI mice after the intravenous administration of 100 mg/kg cyclophosphamide calculated from the mean of 5–8 female NMRI mice. The 2nd column in the % column indicates how much of the OHCP formed is oxidized to KCP and Carb.
Table 1.
Balance of cyclophophamide metabolism in female NMRI mice after the intravenous administration of 100 mg/kg cyclophosphamide calculated from the mean of 5–8 female NMRI mice. The 2nd column in the % column indicates how much of the OHCP formed is oxidized to KCP and Carb.
| Bioavailability | |||
|---|---|---|---|
| Compound | nmol/g | % | |
| cyclophosphamide | 358 | 100 | |
| OHCP + ALDO | 328 | 92 | 100 |
| KCP + CARB | 266 | 74 | 80 |
| protein binding therap., toxic. reactions | 62 | 17 | 20 |
Table 2.
Acute toxicity of OHCP and PAM in male nude mice, single intraperitoneal injection, observation period 30 days.
Table 2.
Acute toxicity of OHCP and PAM in male nude mice, single intraperitoneal injection, observation period 30 days.
| LD50 (µmol/kg) | |||||
|---|---|---|---|---|---|
| OHCP (µmol/kg) | 437 | 480 | 531 | 708 | ~530 |
| mortality | 0/7 | 1/8 | 5/10 | 10/10 | |
| PAM (µmol/kg) | 548 | 685 | 805 | 973 | ~810 |
| mortality | 0/6 | 0/6 | 11/21 | 6/6 |
Table 3.
Acute toxicity of 4-(S-ethanol)-cyclophosphamide (11 Figure 4), aldophosphamide thiazolidine (12, n = 1 Figure 4) and aldophosphamide perhydrothiazine (12, n = 2 Figure 4) in female NMRI mice, single intraperitoneal injection, observation period 30 days.
| Dosage (mmol/kg) | LD50 (mmol/kg) | ||||
|---|---|---|---|---|---|
| 4-(S-ethanol)-CP (11 Figure 4) | 0.53 | 0.64 | 0.78 | 0.94 | ~0.7 |
| mortality | 0/5 | 2/5 | 3/5 | 5/5 | |
| aldophosphamid thiazolidine (12, n = 1 Figure 4) | 4.9 | 5.9 | 8.9 | ~6 | |
| mortality | 0/5 | 3/7 | 5/5 | ||
| 4aldophosphamid perhydrothiazine (12, n = 2 Figure 4) | 4.5 | 5.5 | ~5 | ||
| mortality | 0/5 | 5/5 | |||
Table 4.
Antitumor activity of equitoxic dosages of Ifosfamide and IAP in P388 tumor-bearing mice. Therapy was started on day 1 after 106 P388 tumor cells were subcutaneously transplantated in female CD2F1 mice. Ifosfamide was administered intraperitoneally, IAP was administered subcutaneously. 1 increase in life span (ILS), 2 surviving time > 100 d.
Table 4.
Antitumor activity of equitoxic dosages of Ifosfamide and IAP in P388 tumor-bearing mice. Therapy was started on day 1 after 106 P388 tumor cells were subcutaneously transplantated in female CD2F1 mice. Ifosfamide was administered intraperitoneally, IAP was administered subcutaneously. 1 increase in life span (ILS), 2 surviving time > 100 d.
| Dosage (mg/kg) | ILS (%) 1 | Long Time Survivors 2 | |
|---|---|---|---|
| Ifosfamide | 175 d1-4 | 233 | 0/5 |
| 350 d1-2 | 289 | 0/5 | |
| IAP (R = Cl, 15 Figure 5) | 750 d1-4 | 244 | 2/6 |
| 1500 d1-2 | 400 | 4/5 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Voelcker, G. The Mechanism of Action of Cyclophosphamide and Its Consequences for the Development of a New Generation of Oxazaphosphorine Cytostatics. Sci. Pharm. 2020, 88, 42. https://doi.org/10.3390/scipharm88040042
AMA Style
Voelcker G. The Mechanism of Action of Cyclophosphamide and Its Consequences for the Development of a New Generation of Oxazaphosphorine Cytostatics. Scientia Pharmaceutica. 2020; 88(4):42. https://doi.org/10.3390/scipharm88040042
Chicago/Turabian StyleVoelcker, Georg. 2020. "The Mechanism of Action of Cyclophosphamide and Its Consequences for the Development of a New Generation of Oxazaphosphorine Cytostatics" Scientia Pharmaceutica 88, no. 4: 42. https://doi.org/10.3390/scipharm88040042