Sequence Analysis of Egyptian Foot-and-Mouth Disease Virus Field and Vaccine Strains: Intertypic Recombination and Evidence for Accidental Release of Virulent Virus

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Field Samples and Inactivated Vaccine
2.2. FTA Card Storage Experiment
2.3. RNA Extraction
2.3.1. Liquid Samples
2.3.2. FTA Cards
2.4. FMDV Real-Time RT-PCR
2.5. Virus Isolation
2.6. Viral Genome Sequencing
2.6.1. VP1-Coding Region
2.6.2. Full-Length Viral Genome
2.6.3. Vaccine Composition Analysis
2.6.4. Nucleotide Sequence Alignments and Phylogenetic Analysis
3. Results and Discussion
3.1. Animal Samples
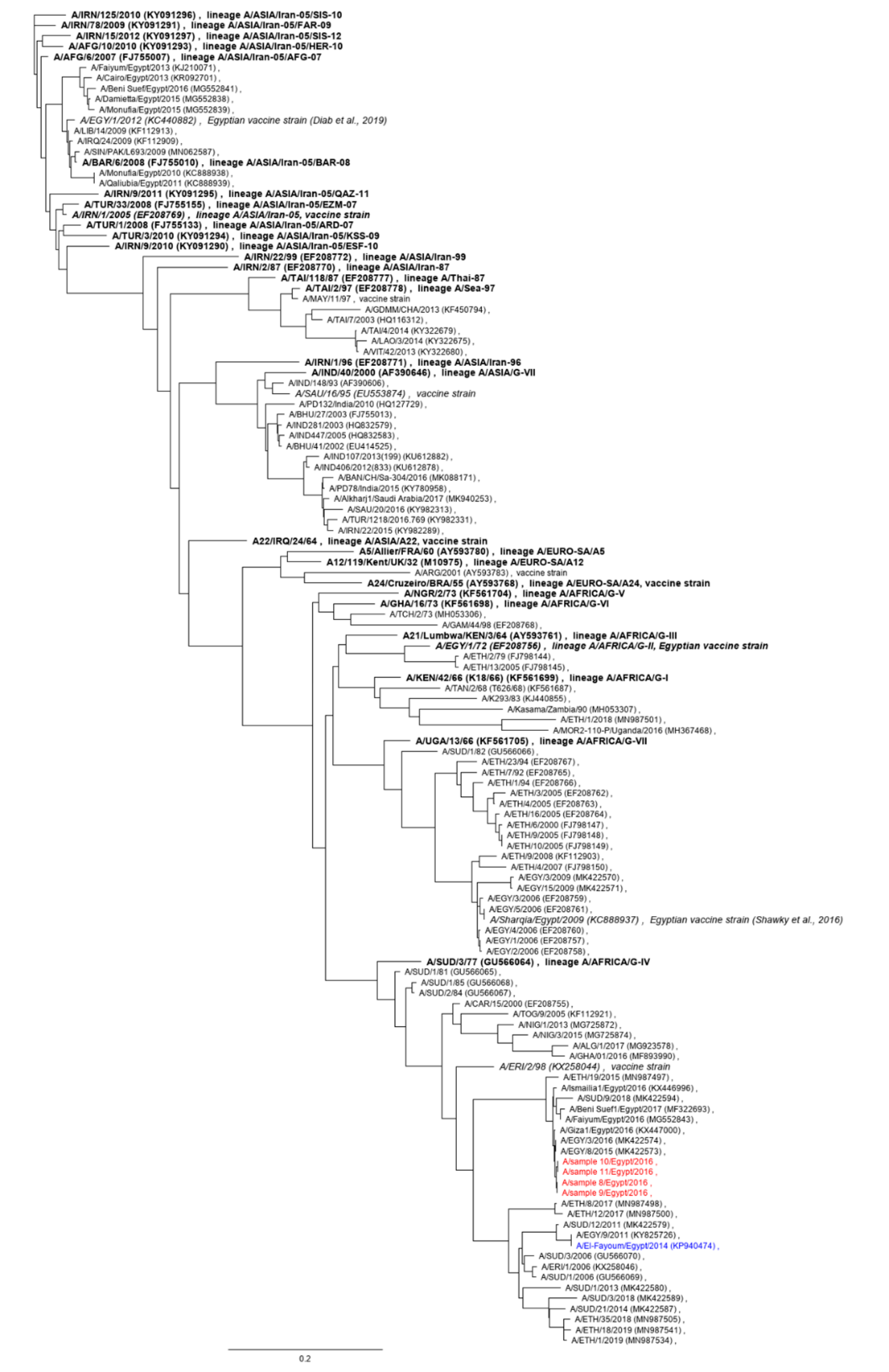
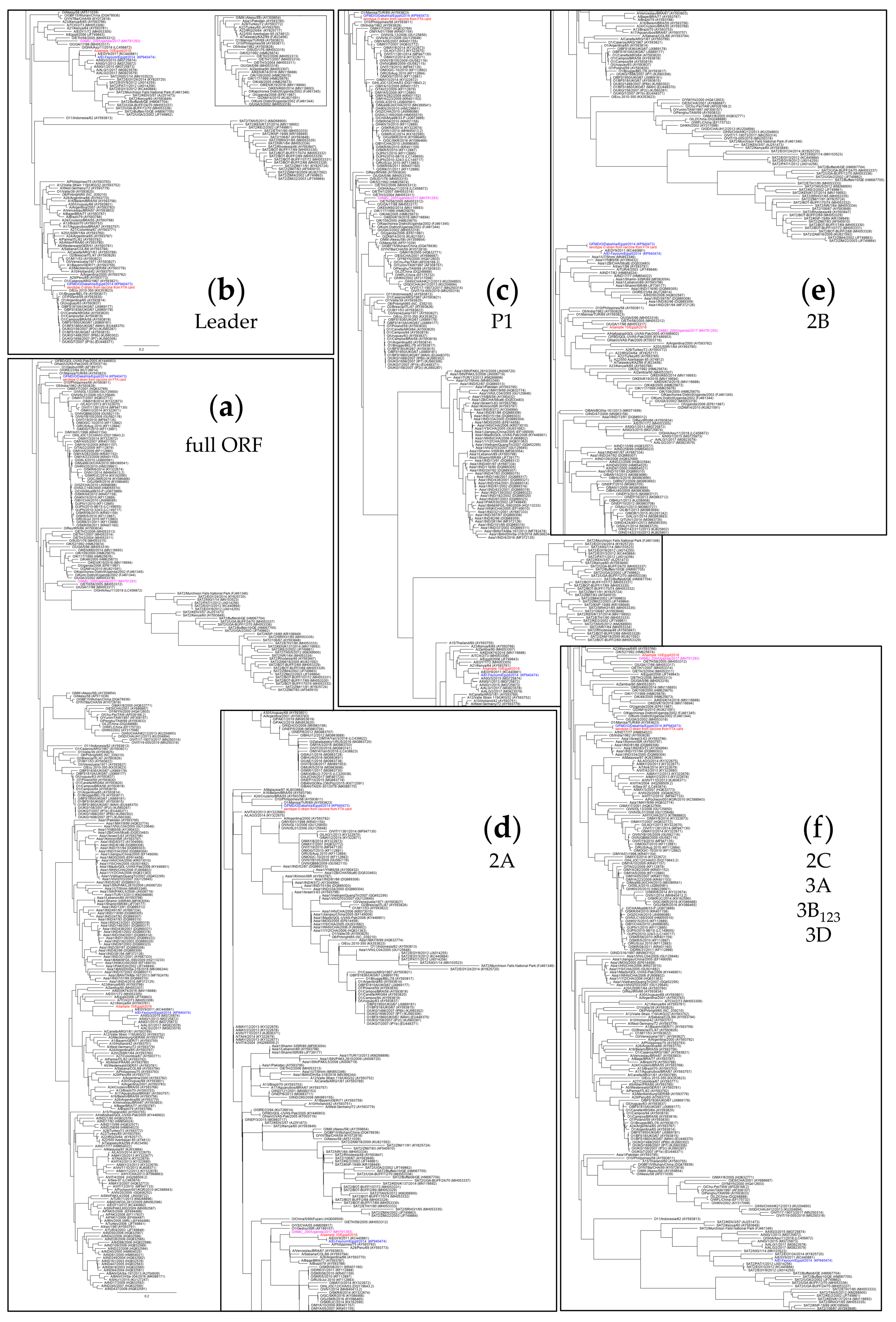
3.2. Full-Length Sequence of Serotype A Isolate from 2016
3.3. FTA Card Storage Experiment
3.4. Composition Analysis of 2019 Inactivated FMDV Vaccine
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grubman, M.J.; Baxt, B. Foot-and-mouth disease. Clin. Microbiol. Rev. 2004, 17, 465–493. [Google Scholar] [CrossRef] [Green Version]
- Brito, B.P.; Rodriguez, L.L.; Hammond, J.M.; Pinto, J.; Perez, A.M. Review of the Global Distribution of Foot-and-Mouth Disease Virus from 2007 to 2014. Transbound. Emerg. Dis. 2017, 64, 316–332. [Google Scholar] [CrossRef]
- Carrillo, C.; Tulman, E.R.; Delhon, G.; Lu, Z.; Carreno, A.; Vagnozzi, A.; Kutish, G.F.; Rock, D.L. Comparative genomics of foot-and-mouth disease virus. J. Virol. 2005, 79, 6487–6504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arzt, J.; Juleff, N.; Zhang, Z.; Rodriguez, L.L. The pathogenesis of foot-and-mouth disease I: Viral pathways in cattle. Transbound. Emerg. Dis. 2011, 58, 291–304. [Google Scholar] [CrossRef]
- Diaz-San Segundo, F.; Medina, G.N.; Stenfeldt, C.; Arzt, J.; de Los Santos, T. Foot-and-mouth disease vaccines. Vet. Microbiol. 2017, 206, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Jamal, S.M.; Nazem Shirazi, M.H.; Ozyoruk, F.; Parlak, U.; Normann, P.; Belsham, G.J. Evidence for multiple recombination events within foot-and-mouth disease viruses circulating in West Eurasia. Transbound. Emerg. Dis. 2020, 67, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Pezzoni, G.; Bregoli, A.; Grazioli, S.; Barbieri, I.; Madani, H.; Omani, A.; Sadaoui, H.; Bouayed, N.; Wadsworth, J.; Bachanek-Bankowska, K.; et al. Foot-and-mouth disease outbreaks due to an exotic virus serotype A lineage (A/AFRICA/G-IV) in Algeria in 2017. Transbound. Emerg. Dis. 2019, 66, 7–13. [Google Scholar] [CrossRef]
- Forth, L.F.; Höper, D.; Beer, M.; Eschbaumer, M. High-Resolution Composition Analysis of an Inactivated Polyvalent Foot-and-Mouth Disease Vaccine. Pathogens 2020, 9, 63. [Google Scholar] [CrossRef] [Green Version]
- Callahan, J.D.; Brown, F.; Osorio, F.A.; Sur, J.H.; Kramer, E.; Long, G.W.; Lubroth, J.; Ellis, S.J.; Shoulars, K.S.; Gaffney, K.L.; et al. Use of a portable real-time reverse transcriptase-polymerase chain reaction assay for rapid detection of foot-and-mouth disease virus. J. Am. Vet. Med. Assoc. 2002, 220, 1636–1642. [Google Scholar] [CrossRef]
- Dill, V.; Eschbaumer, M. Reliable detection, sequencing, and transfection of foot-and-mouth disease virus RNA from badly preserved vesicular epithelium. J. Vet. Diagn. Investig. 2019, 31, 778–782. [Google Scholar] [CrossRef]
- LaRocco, M.; Krug, P.W.; Kramer, E.; Ahmed, Z.; Pacheco, J.M.; Duque, H.; Baxt, B.; Rodriguez, L.L. A continuous bovine kidney cell line constitutively expressing bovine alphavbeta6 integrin has increased susceptibility to foot-and-mouth disease virus. J. Clin. Microbiol. 2013, 51, 1714–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dill, V.; Beer, M.; Hoffmann, B. Simple, quick and cost-efficient: A universal RT-PCR and sequencing strategy for genomic characterisation of foot-and-mouth disease viruses. J. Virol. Methods 2017. [Google Scholar] [CrossRef] [PubMed]
- Chrzastek, K.; Lee, D.H.; Smith, D.; Sharma, P.; Suarez, D.L.; Pantin-Jackwood, M.; Kapczynski, D.R. Use of Sequence-Independent, Single-Primer-Amplification (SISPA) for rapid detection, identification, and characterization of avian RNA viruses. Virology 2017, 509, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Scheuch, M.; Höper, D.; Beer, M. RIEMS: A software pipeline for sensitive and comprehensive taxonomic classification of reads from metagenomics datasets. BMC Bioinform. 2015, 16, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Dill, V.; Eschbaumer, M. Cell culture propagation of foot-and-mouth disease virus: Adaptive amino acid substitutions in structural proteins and their functional implications. Virus Genes 2020, 56, 1–15. [Google Scholar] [CrossRef] [Green Version]
- WRLFMD. Genotyping Report WRLFMD/2017/00014. Available online: https://perma.cc/5M8F-9MUS (accessed on 21 July 2020).
- WRLFMD. FMD Vaccine Matching Strain Differentiation Report WRLFMD/2016/00024. Available online: https://perma.cc/T468-FUD4 (accessed on 21 July 2020).
- WRLFMD. FMD Vaccine Matching Strain Differentiation Report WRLFMD/2017/00014. Available online: https://perma.cc/JB26-FWVQ (accessed on 21 July 2020).
- Shafik, N.G.; Darwish, D.M.; Abousenna, M.S.; Galal, M.; Ahmed, A.R.; Attya, M.; Saad, M.A.; Abdelhakim, M. Efficacy of a Commercial Local Trivalent Foot and Mouth Disease (FMD) Vaccine against Recently Isolated O-EA3. Int. J. Vet. Sci. 2019, 8, 33–35. [Google Scholar]
- MEVAC. Ruminants Vaccines. Available online: https://perma.cc/BZ4T-HZ8W (accessed on 21 July 2020).
- WRLFMD. Quarterly Report 2020 Quarter 1 (Jan–Mar). Available online: https://perma.cc/J4BF-LPH3 (accessed on 22 July 2020).
- Ludi, A.B.; Horton, D.L.; Li, Y.; Mahapatra, M.; King, D.P.; Knowles, N.J.; Russell, C.A.; Paton, D.J.; Wood, J.L.; Smith, D.J.; et al. Antigenic variation of foot-and-mouth disease virus serotype A. J. Gen. Virol. 2014, 95, 384–392. [Google Scholar] [CrossRef]
- Brehm, K.E.; Kumar, N.; Thulke, H.H.; Haas, B. High potency vaccines induce protection against heterologous challenge with foot-and-mouth disease virus. Vaccine 2008, 26, 1681–1687. [Google Scholar] [CrossRef] [PubMed]
- El-Bagoury, G.F.; Sharawi, S.S.A.; El-Nahas, E.M.; Darwish, D.M.; Saad, M.A. Evaluation of cross-protection between FMD serotypes O and A local Egyptian isolate with vaccinal strains in the local commercial and imported vaccines by challenge test. Benha Vet. Med. J. 2015, 28, 241–246. [Google Scholar] [CrossRef]
- OIE. Manual of Diagnostic Tests and Vaccines for Terrestrial Animals, Chapter 3.1.8. Foot and Mouth Disease. Available online: https://perma.cc/EG94-MX4N (accessed on 27 August 2020).
- VSVRI. Bivalent Inactivated Foot and Mouth Disease Oil Vaccine (Types O1&A). Available online: https://perma.cc/D4NQ-R3JN (accessed on 22 July 2020).
- Knowles, N.J.; Wadsworth, J.; Reid, S.M.; Swabey, K.G.; El-Kholy, A.A.; Abd El-Rahman, A.O.; Soliman, H.M.; Ebert, K.; Ferris, N.P.; Hutchings, G.H.; et al. Foot-and-mouth disease virus serotype A in Egypt. Emerg. Infect. Dis. 2007, 13, 1593–1596. [Google Scholar] [CrossRef] [PubMed]
- Shawky, S.M.; Thabet, N.S.; Orabi, S.H.; Nayel, M.A. Comparative Study on the Hemato-Biochemical and Immunological Effects of the Hexavalent FMD Vaccine Alone or in Combination with Trivalent FMD Vaccine in Cattle. J. Biosci. Med. 2016, 4, 16–26. [Google Scholar] [CrossRef] [Green Version]
- WRLFMD. Genotyping Report WRLFMD/2019/00005. Available online: https://perma.cc/V6HM-9CP9 (accessed on 22 July 2020).
- Sobhy, N.M.; Bayoumi, Y.H.; Mor, S.K.; El-Zahar, H.I.; Goyal, S.M. Outbreaks of foot and mouth disease in Egypt: Molecular epidemiology, evolution and cardiac biomarkers prognostic significance. Int. J. Vet. Sci. Med. 2018, 6, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Bachanek-Bankowska, K.; Di Nardo, A.; Wadsworth, J.; Mioulet, V.; Pezzoni, G.; Grazioli, S.; Brocchi, E.; Kafle, S.C.; Hettiarachchi, R.; Kumarawadu, P.L.; et al. Reconstructing the evolutionary history of pandemic foot-and-mouth disease viruses: The impact of recombination within the emerging O/ME-SA/Ind-2001 lineage. Sci. Rep. 2018, 8, 14693. [Google Scholar] [CrossRef] [Green Version]
- Heath, L.; van der Walt, E.; Varsani, A.; Martin, D.P. Recombination patterns in aphthoviruses mirror those found in other picornaviruses. J. Virol. 2006, 80, 11827–11832. [Google Scholar] [CrossRef] [Green Version]
- Mwiine, F.N.; Velazquez-Salinas, L.; Ahmed, Z.; Ochwo, S.; Munsey, A.; Kenney, M.; Lutwama, J.J.; Maree, F.F.; Lobel, L.; Perez, A.M.; et al. Serological and phylogenetic characterization of foot and mouth disease viruses from Uganda during cross-sectional surveillance study in cattle between 2014 and 2017. Transbound. Emerg. Dis. 2019, 66, 2011–2024. [Google Scholar] [CrossRef]
- Abdul-Hamid, N.F.; Firat-Sarac, M.; Radford, A.D.; Knowles, N.J.; King, D.P. Comparative sequence analysis of representative foot-and-mouth disease virus genomes from Southeast Asia. Virus Genes 2011, 43, 41–45. [Google Scholar] [CrossRef]
- Jamal, S.M.; Ferrari, G.; Ahmed, S.; Normann, P.; Belsham, G.J. Molecular characterization of serotype Asia-1 foot-and-mouth disease viruses in Pakistan and Afghanistan; emergence of a new genetic Group and evidence for a novel recombinant virus. Infect. Genet. Evol. 2011, 11, 2049–2062. [Google Scholar] [CrossRef]
- McCahon, D.; King, A.M.; Roe, D.S.; Slade, W.R.; Newman, J.W.; Cleary, A.M. Isolation and biochemical characterization of intertypic recombinants of foot-and-mouth disease virus. Virus Res. 1985, 3, 87–100. [Google Scholar] [CrossRef]
- Wright, C.F.; Knowles, N.J.; Di Nardo, A.; Paton, D.J.; Haydon, D.T.; King, D.P. Reconstructing the origin and transmission dynamics of the 1967-68 foot-and-mouth disease epidemic in the United Kingdom. Infect. Genet. Evol. 2013, 20, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diab, E.; Bazid, A.I.; Fawzy, M.; El-Ashmawy, W.R.; Fayed, A.A.; El-Sayed, M.M. Foot-and-mouth disease outbreaks in Egypt during 2013-2014: Molecular characterization of serotypes A, O, and SAT2. Vet. World 2019, 12, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Samuel, A.R.; Knowles, N.J.; Mackay, D.K. Genetic analysis of type O viruses responsible for epidemics of foot-and-mouth disease in North Africa. Epidemiol. Infect. 1999, 122, 529–538. [Google Scholar] [CrossRef] [PubMed]
- VSVRI. Polyvalent Inactivated Foot and Mouth Disease Oil Vaccine (Types O1,A&SAT2). Available online: https://perma.cc/A3NC-8V7Y (accessed on 22 July 2020).
- WRLFMD. Country Reports: Egypt, Samples Tested. Available online: https://perma.cc/KDK8-CQ9F (accessed on 22 July 2020).
- Knowles, N.J.; Samuel, A.R.; Davies, P.R.; Midgley, R.J.; Valarcher, J.F. Pandemic strain of foot-and-mouth disease virus serotype O. Emerg. Infect. Dis. 2005, 11, 1887–1893. [Google Scholar] [CrossRef]
- Abu-Elnaga, H.I.; Rizk, S.A.; Daoud, H.M.; Mohamed, A.A.; Mossad, W.; Gamil, M.A.; Soudy, A.F.; El-Shehawy, L.I. Comparative nucleotide sequencing of the VP1 capsid gene of recent isolates of foot-and-mouth disease virus serotype O from Egypt. Arch. Virol. 2020. [Google Scholar] [CrossRef]
- Beck, E.; Strohmaier, K. Subtyping of European foot-and-mouth disease virus strains by nucleotide sequence determination. J. Virol. 1987, 61, 1621–1629. [Google Scholar] [CrossRef] [Green Version]
- Cottam, E.M.; Wadsworth, J.; Shaw, A.E.; Rowlands, R.J.; Goatley, L.; Maan, S.; Maan, N.S.; Mertens, P.P.; Ebert, K.; Li, Y.; et al. Transmission pathways of foot-and-mouth disease virus in the United Kingdom in 2007. PLoS Pathog. 2008, 4, e1000050. [Google Scholar] [CrossRef] [Green Version]
- Carrillo, C.; Dopazo, J.; Moya, A.; Gonzalez, M.; Martinez, M.A.; Saiz, J.C.; Sobrino, F. Comparison of vaccine strains and the virus causing the 1986 foot-and-mouth disease outbreak in Spain: Epizootiological analysis. Virus Res. 1990, 15, 45–55. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Serotype | Sample Material | Origin | Year | Host | Sequence | Accession Number |
|---|---|---|---|---|---|---|---|
| Type | |||||||
| sample 8 | A | saliva | Dakahlia | 2016 | cow | 1D/VP1 | MT863264 |
| sample 9 | A | saliva | Damietta | 2016 | cow | 1D/VP1 | MT863265 |
| sample 10 | A | saliva | Damietta | 2016 | buffalo | 1D/VP1 | MT863266 |
| culture supernatant | - | - | - | whole genome | MT863268 | ||
| sample 11 | A | saliva | Dakahlia | 2016 | buffalo | 1D/VP1 | MT863267 |
| FTA card | O | inactivated vaccine | Egypt | 2019 | whole genome | MT863269 |
| Isolate | ORF | L | 1A | 1B | 1C | 1D | 2A | 2B | 2C | 3A | 3B1 | 3B2 | 3B3 | 3C | 3D |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A/El-F/2014 | 87.3 | 88.1 | 89.8 | 89.3 | 90.5 | 86.6 | 98.2 | 93.9 | 83.9 | 78.2 | 76.8 | 79.2 | 83.3 | 87.2 | 88.3 |
| O/AMU_200 | 85.9 | 76.3 | 85.1 | 72.5 | 73.9 | 64.9 | 94.4 | 94.4 | 94.9 | 91.9 | 98.6 | 87.5 | 94.4 | 95.3 | 94.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abd El Rahman, S.; Hoffmann, B.; Karam, R.; El-Beskawy, M.; Hamed, M.F.; Forth, L.F.; Höper, D.; Eschbaumer, M. Sequence Analysis of Egyptian Foot-and-Mouth Disease Virus Field and Vaccine Strains: Intertypic Recombination and Evidence for Accidental Release of Virulent Virus. Viruses 2020, 12, 990. https://doi.org/10.3390/v12090990
Abd El Rahman S, Hoffmann B, Karam R, El-Beskawy M, Hamed MF, Forth LF, Höper D, Eschbaumer M. Sequence Analysis of Egyptian Foot-and-Mouth Disease Virus Field and Vaccine Strains: Intertypic Recombination and Evidence for Accidental Release of Virulent Virus. Viruses. 2020; 12(9):990. https://doi.org/10.3390/v12090990
Chicago/Turabian StyleAbd El Rahman, Sahar, Bernd Hoffmann, Reham Karam, Mohamed El-Beskawy, Mohammed F. Hamed, Leonie F. Forth, Dirk Höper, and Michael Eschbaumer. 2020. "Sequence Analysis of Egyptian Foot-and-Mouth Disease Virus Field and Vaccine Strains: Intertypic Recombination and Evidence for Accidental Release of Virulent Virus" Viruses 12, no. 9: 990. https://doi.org/10.3390/v12090990