
In Silico, In Vitro and In Cellulo Models for Monitoring SARS-CoV-2 Spike/Human ACE2 Complex, Viral Entry and Cell Fusion

 ,
,  ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Silico Molecular Dynamics Simulation of the SARS-CoV-2 S/ACE2 Complex
2.2. Proteins and Antibodies
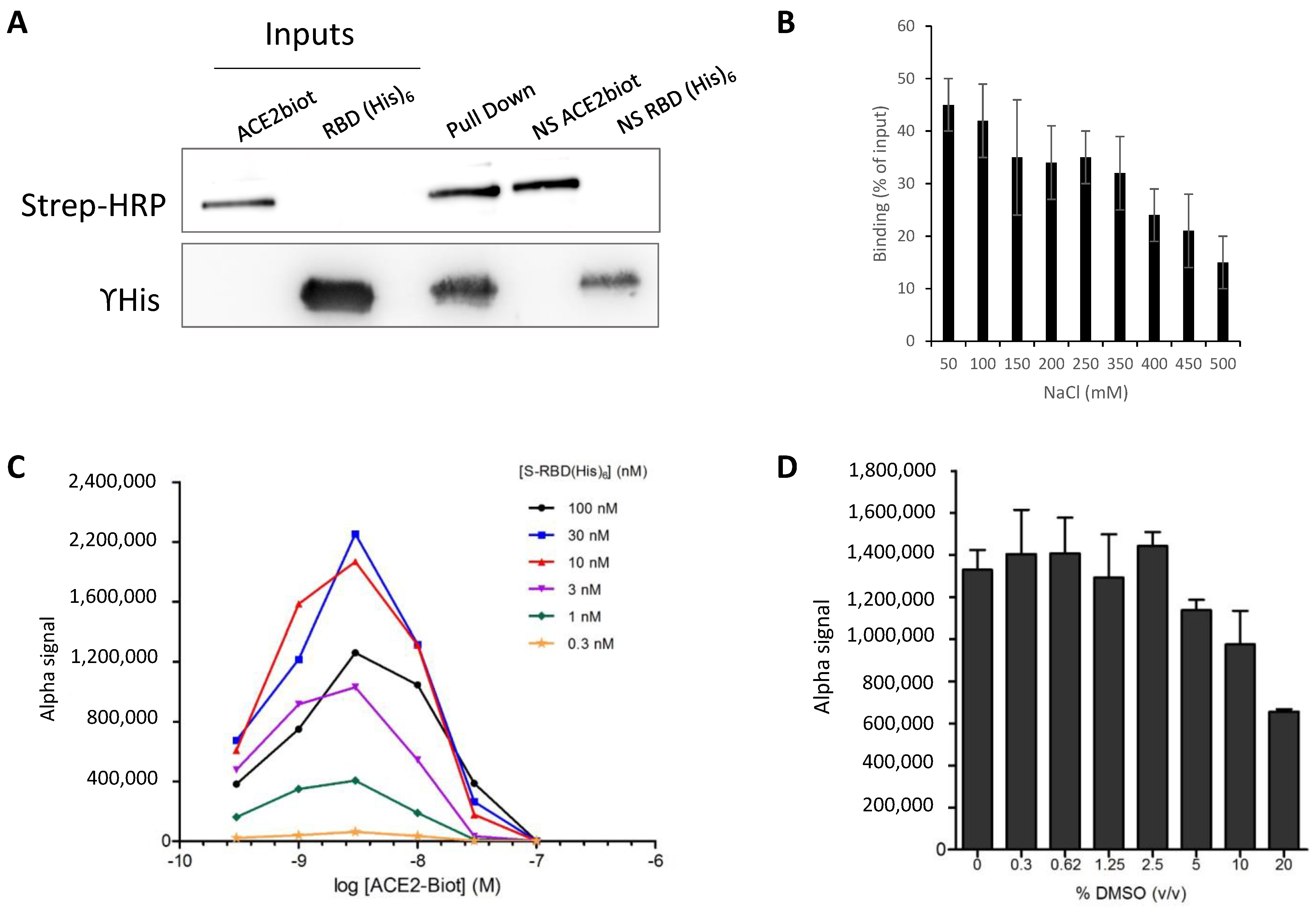
2.3. Pull Down Assay
2.4. AlphaLISA Binding Assays
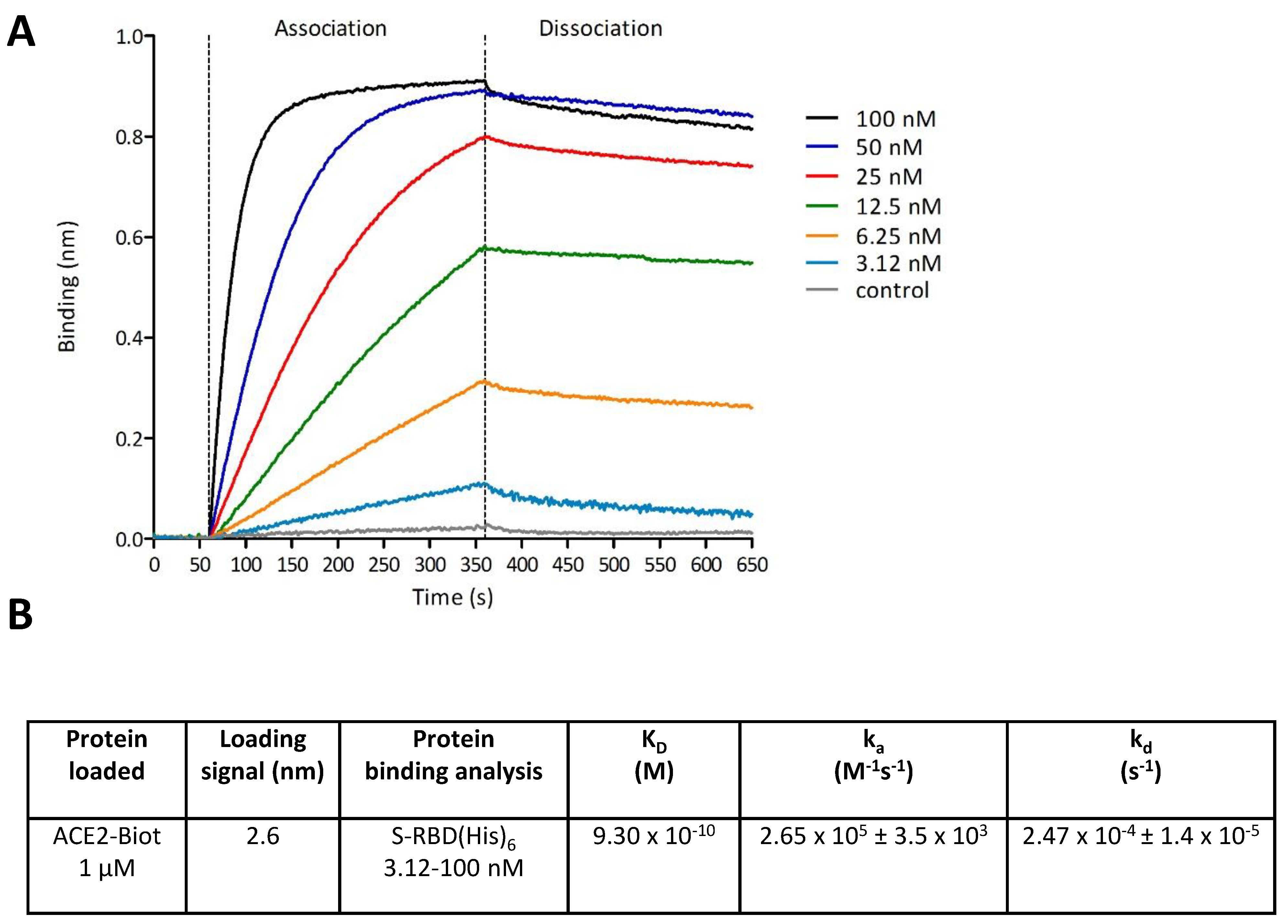
2.5. Bio-Layer Interferometry Binding Assays
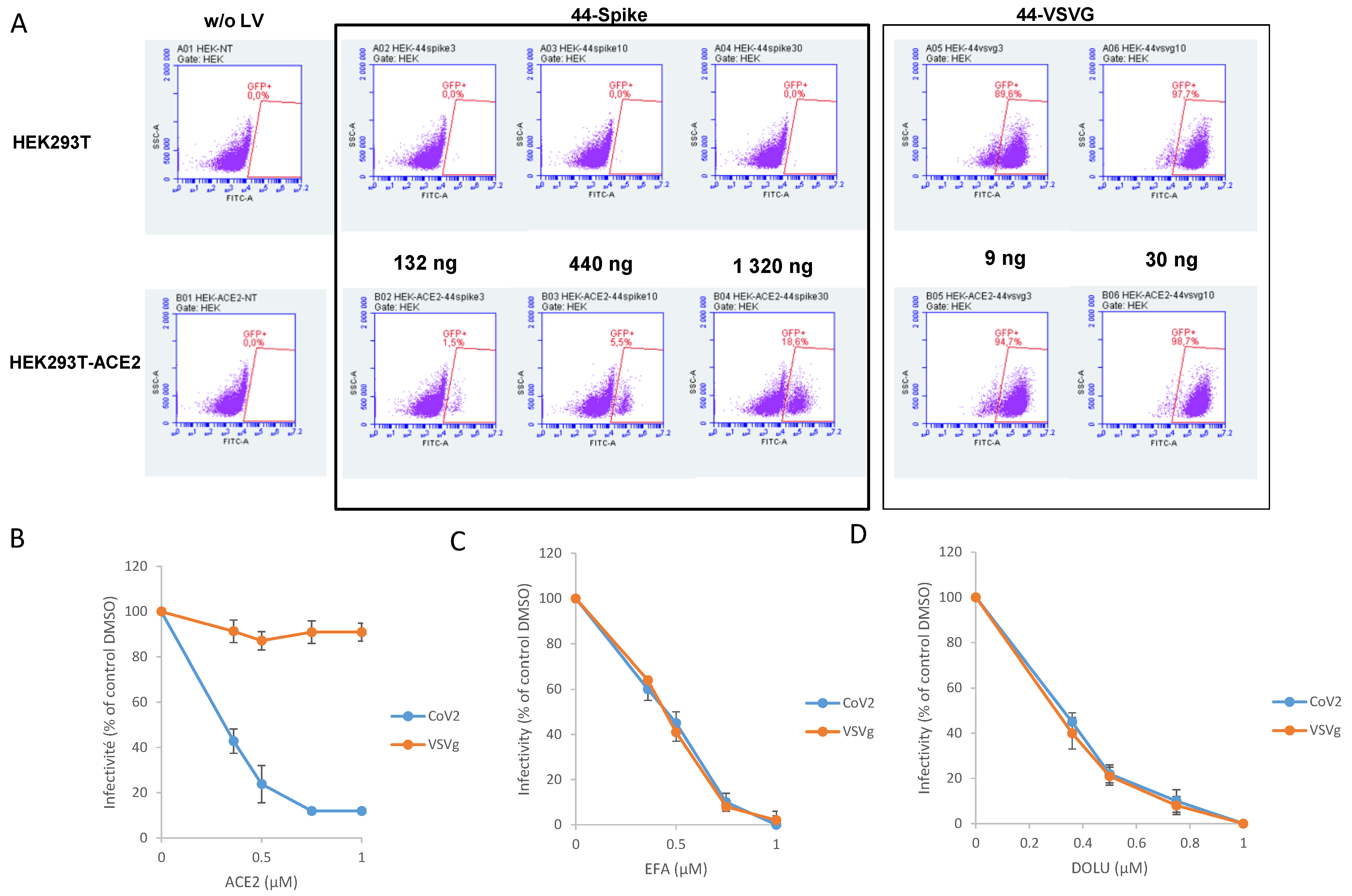
2.6. Cells and Lentiviral Vectors Production
2.7. Quantitative PCR of Proviral DNA
2.8. Cell Imaging
2.9. In Cellulo Imaging of the SARS-CoV-2 S-RBD/ACE2 Interaction
3. Results
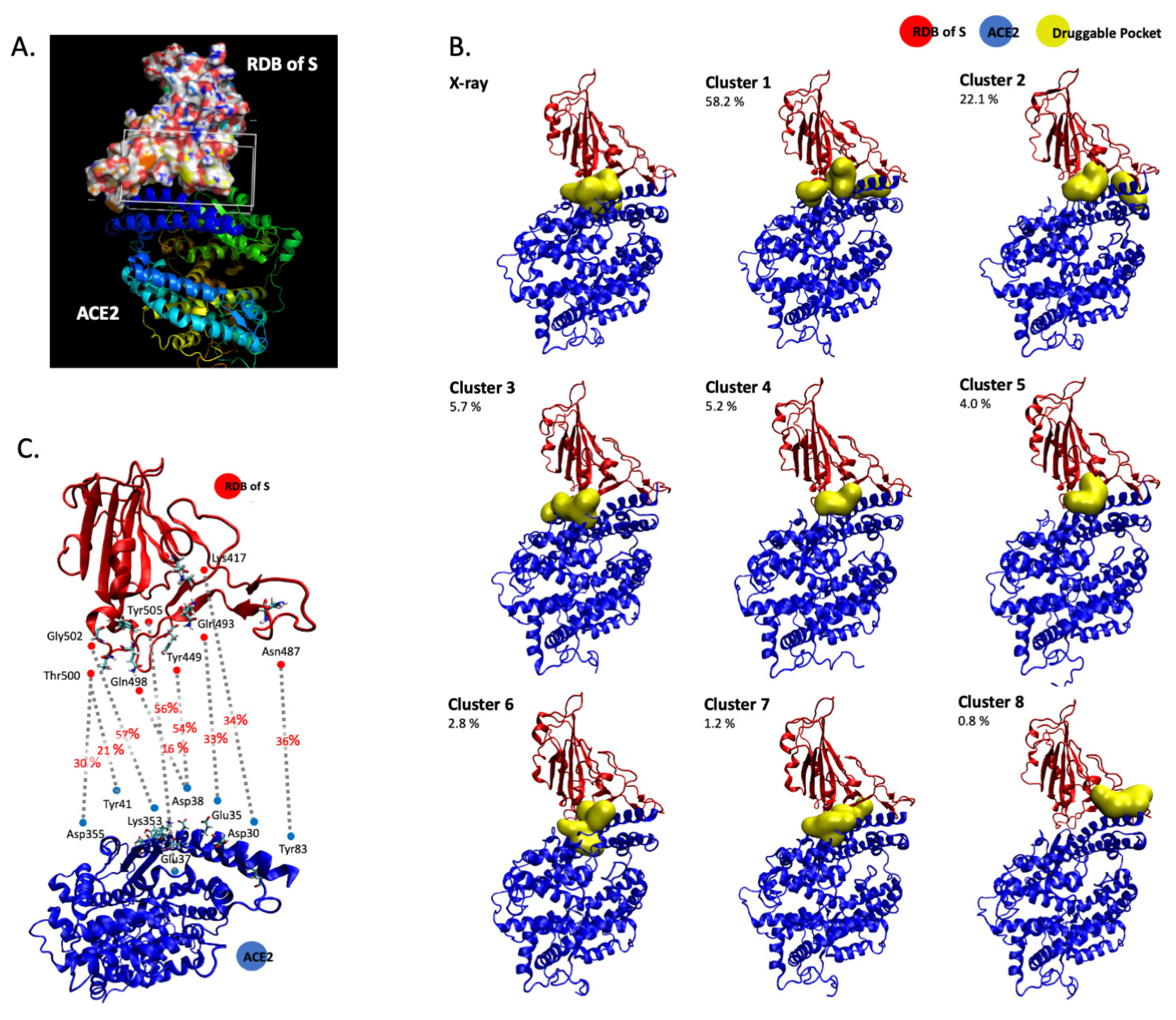
3.1. In Silico Molecular Dynamics Simulation of the SARS-CoV-2 S/ACE2 Complex
3.2. In Vitro Monitoring of the SARS-CoV-2 S-RBD/ACE2 Complex and Determination of Kinetic Constants
3.3. In Cellulo Monitoring of S/ACE2-Mediated Viral Entry
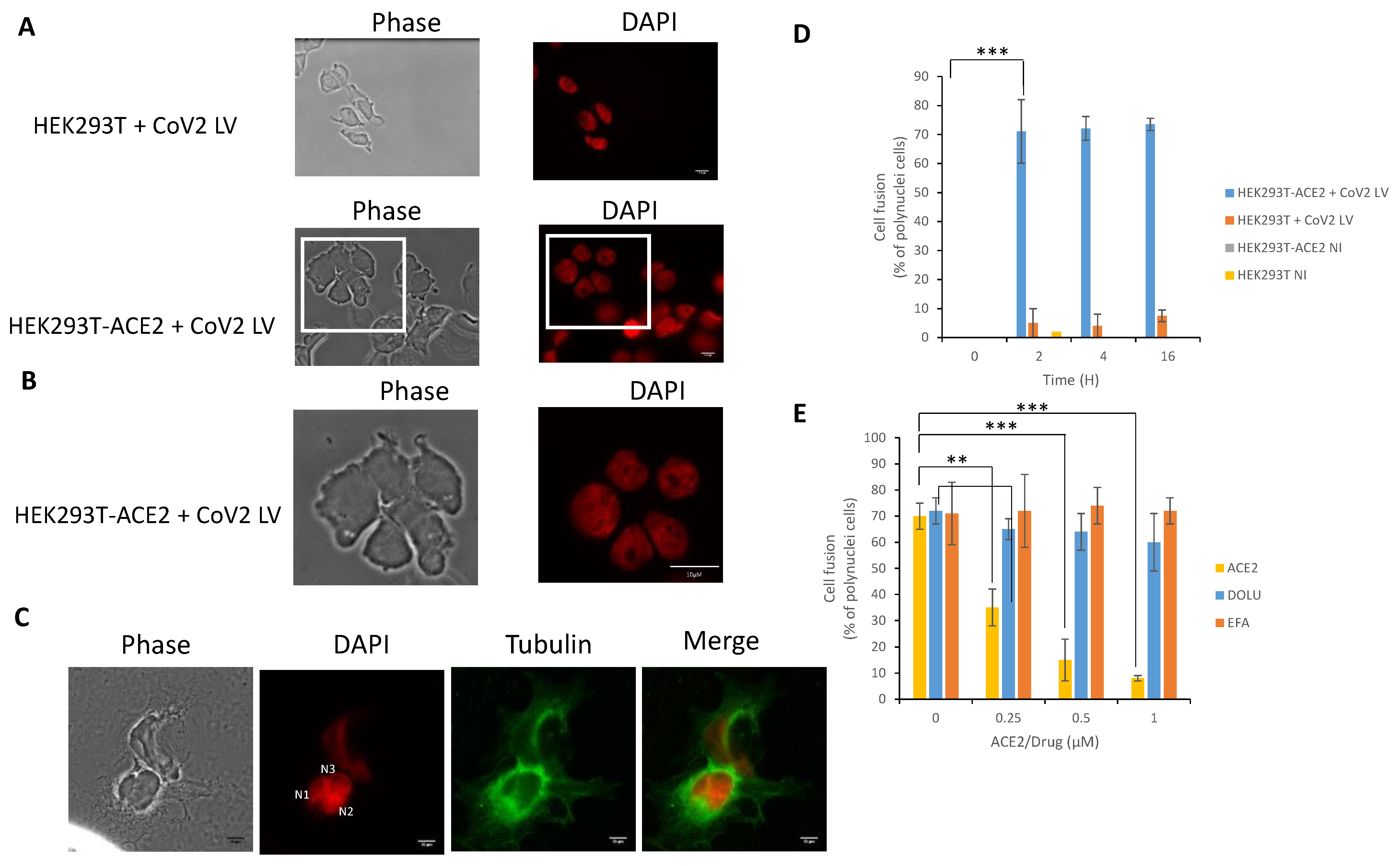
3.4. In Cellulo Monitoring of the S/ACE2-Mediated Cell-Cell Fusion
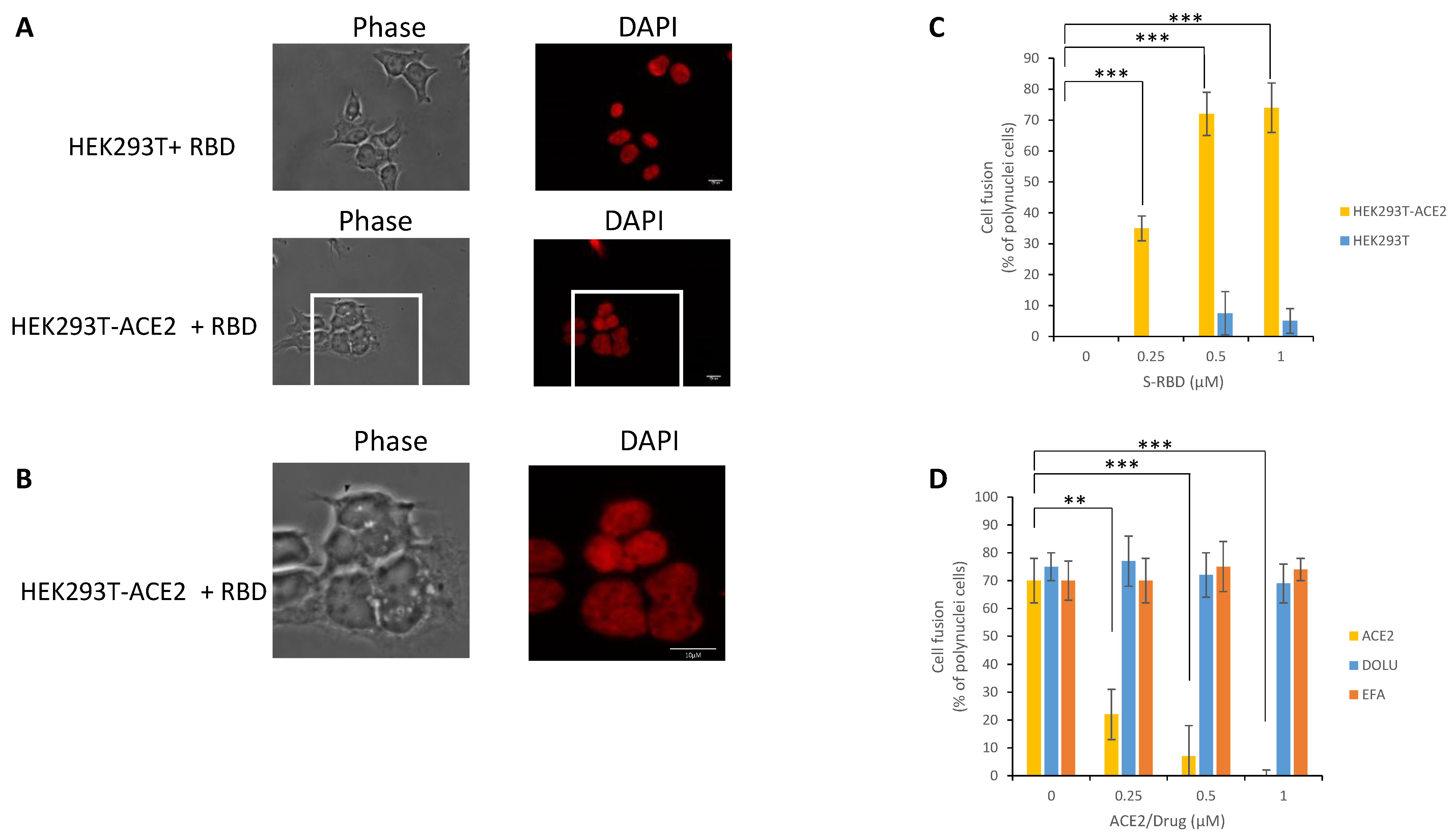
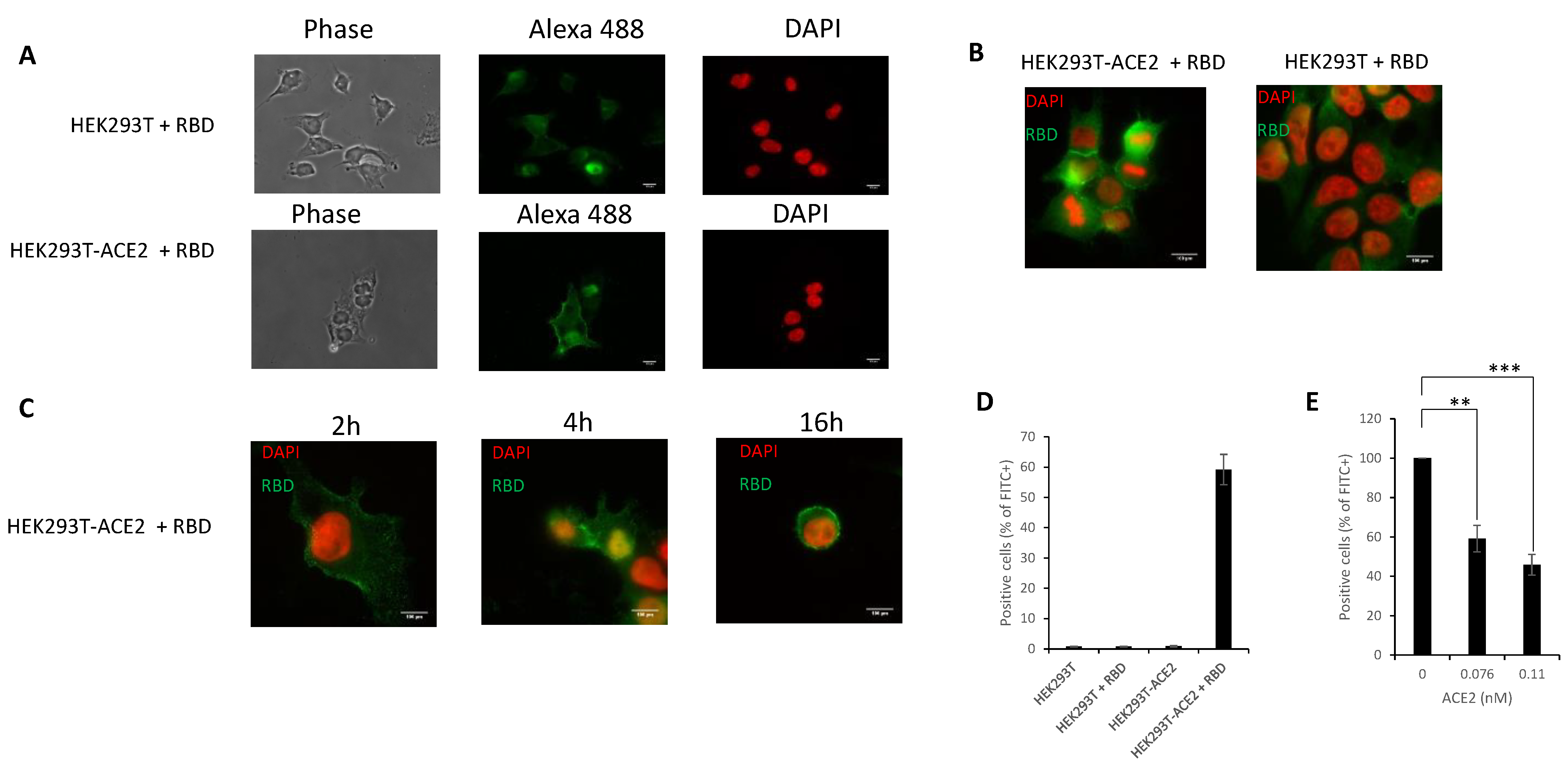
3.5. In Cellulo Monitoring and Quantification of the S/ACE2 Interaction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boopathi, S.; Poma, A.B.; Kolandaivel, P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lazartigues, E. Expression of ACE2 in Human Neurons Supports the Neuro-Invasive Potential of COVID-19 Virus. Cell Mol. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.; Navis, G.; Van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; He, L.; Zhang, Q.; Huang, Z.; Che, X.; Hou, J.; Wang, H.; Shen, H.; Qiu, L.; Li, Z.; et al. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: Implications for pathogenesis and virus transmission pathways. J. Pathol. 2004, 203, 622–630. [Google Scholar] [CrossRef]
- Gu, J.; Gong, E.; Zhang, B.; Zheng, J.; Gao, Z.; Zhong, Y.; Zou, W.; Zhan, J.; Wang, S.; Xie, Z.; et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 2005, 202, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Prado, A.V.; Skriabine, S.; Lu, P.; Weizman, O.-E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. BioRxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef]
- Barrantes, F.J. Central Nervous System Targets and Routes for SARS-CoV-2: Current Views and New Hypotheses. ACS Chem. Neurosci. 2020, 11, 2793–2803. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Crawford, K.H.D.; Eguia, R.; Dingens, A.S.; Loes, A.N.; Malone, K.D.; Wolf, C.R.; Chu, H.Y.; Tortorici, M.A.; Veesler, D.; Murphy, M.; et al. Protocol and Reagents for Pseudotyping Lentiviral Particles with SARS-CoV-2 Spike Protein for Neutralization Assays. Viruses 2020, 12, 513. [Google Scholar] [CrossRef]
- Moreira, R.A.; Chwastyk, M.; Baker, J.L.; Guzman, H.V.; Poma, A.B. Quantitative determination of mechanical stability in the novel coronavirus spike protein. Nanoscale 2020, 12, 16409–16413. [Google Scholar] [CrossRef]
- Silva de Souza, A.; Rivera, J.D.; Almeida, V.M.; Ge, P.; de Souza, R.F.; Farah, C.S.; Ulrich, H.; Marana, S.R.; Salinas, R.K.; Guzzo, C.R. Molecular Dynamics Reveals Complex Compensatory Effects of Ionic Strength on the Severe Acute Respiratory Syndrome Coronavirus 2 Spike/Human Angiotensin-Converting Enzyme 2 Interaction. J. Phys. Chem. Lett. 2020, 11, 10446–10453. [Google Scholar] [CrossRef] [PubMed]
- Sakkiah, S.; Guo, W.; Pan, B.; Ji, Z.; Yavas, G.; Azevedo, M.; Hawes, J.; Patterson, T.A.; Hong, H. Elucidating Interactions Between SARS-CoV-2 Trimeric Spike Protein and ACE2 Using Homology Modeling and Molecular Dynamics Simulations. Front. Chem. 2020, 8. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef]
- Buchrieser, J.; Dufloo, J.; Hubert, M.; Monel, B.; Planas, D.; Rajah, M.M.; Planchais, C.; Porrot, F.; Guivel-Benhassine, F.; Van der Werf, S.; et al. Syncytia formation by SARS-CoV-2-infected cells. EMBO J. 2020, 39, e106267. [Google Scholar] [CrossRef]
- Moreira, R.A.; Guzman, H.V.; Boopathi, S.; Baker, J.L.; Poma, A.B. Characterization of Structural and Energetic Differences between Conformations of the SARS-CoV-2 Spike Protein. Materials 2020, 13, 5362. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef]
- Artese, A.; Svicher, V.; Costa, G.; Salpini, R.; Di Maio, V.C.; Alkhatib, M.; Ambrosio, F.A.; Santoro, M.M.; Assaraf, Y.G.; Alcaro, S.; et al. Current status of antivirals and druggable targets of SARS CoV-2 and other human pathogenic coronaviruses. Drug Resist. Updates 2020, 53, 100721. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | Total SASA (Å2) | Polar SASA (Å2) | Non-Polar SASA (Å2) | Volume (Å3) | ACE2 Main Residues | S1 RDB Main Residues | |
|---|---|---|---|---|---|---|---|
| X-ray | 1 | 141.0 | 75.8 | 65.2 | 351.1 | Asn33, His34, Glu37, Lys353 | Arg403, Lys417, Tyr453, Tyr495, Tyr505, |
| 2 | 193.8 | 98.4 | 95.4 | 738.9 | Gln42, Leu45, Asn49, Asn61, Asn64, Lys68 | Gly446, Tyr449 | |
| 3 | 241.4 | 115.8 | 125.6 | 827.5 | Thr324, Phe356, Arg393 | Gly502, Val503, Tyr505 | |
| Cluster 1 | 1 | 223.5 | 134.1 | 89.4 | 828.7 | Asn33, His34, Glu37, Asp38, Lys353, Pro389, Phe390 | Arg403, Glu406, Lys417, Ile418, Tyr495, Tyr505 |
| 2 | 155.7 | 79.6 | 76.1 | 543.4 | Phe28, Lys31, Phe32, Glu35, Asp38, Leu39, Phe72, Gln76, Leu79 | Glu484, Phe486, Asn487, Tyr489 | |
| 3 | 152.0 | 43.3 | 108.7 | 525.8 | Thr324, Gln325, Asn330, Asp355, Phe356 | Val503, Tyr505 | |
| Cluster 2 | 1 | 304.7 | 173.1 | 131.6 | 919.7 | His34, Glu37, Asp38, Lys353, Pro389, Arg393 | Arg403, Gln409, Lys417, Ile418, Tyr495 |
| 2 | 174.4 | 112.2 | 62.2 | 549.8 | Lys31, Gln76, Leu79 | Glu484, Tyr489, Phe490 | |
| Cluster 3 | 1 | 207.3 | 98.6 | 108.7 | 603.1 | Lys353, Phe356, Arg393 | Asp405, Tyr505 |
| 2 | 257.4 | 142.6 | 114.7 | 750.6 | Thr324, Gln325, Glu329, Asp355 | Thr500, Gly502, Val503 | |
| Cluster 4 | 1 | 213.1 | 128.6 | 84.5 | 642.8 | Asn33, His34, Pro389, Arg393 | Arg403, Asp405, Lys417 |
| Cluster 5 | 1 | 232.1 | 122.2 | 109.9 | 784.6 | Asn33, His34, Pro389, Phe390 | Asp405, Lys417, Ile418, Tyr495, Tyr505 |
| Cluster 6 | 1 | 155.8 | 96.6 | 59.2 | 535.4 | Asp30, Asn33, His34, Glu37, Asp38, Lys353, Phe390 | Arg403, Lys417, Tyr495, Tyr505 |
| 2 | 187.7 | 75.4 | 112.3 | 566.0 | Asp350, Phe356, Tyr385, Arg393 | Gly502, Tyr505 | |
| Cluster 7 | 1 | 175.3 | 107.7 | 67.6 | 565.9 | Glu37, Asp38, Leu39, Lys353 | Leu452, Arg403, Gln493, Tyr495, Tyr505 |
| 1 | 285.5 | 128.5 | 157.0 | 833.7 | Asn33, Thr324, Gln325, Phe356, Pro389, Arg393 | Arg403, Asp405, Val503 | |
| Cluster 8 | 1 | 252.0 | 144.5 | 107.5 | 799.2 | Thr20, Glu23, Gln24, Lys26, Thr27, Asp30 | Lys417, Tyr421, Phe456, Tyr473, Ala475 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lapaillerie, D.; Charlier, C.; Fernandes, H.S.; Sousa, S.F.; Lesbats, P.; Weigel, P.; Favereaux, A.; Guyonnet-Duperat, V.; Parissi, V. In Silico, In Vitro and In Cellulo Models for Monitoring SARS-CoV-2 Spike/Human ACE2 Complex, Viral Entry and Cell Fusion. Viruses 2021, 13, 365. https://doi.org/10.3390/v13030365
Lapaillerie D, Charlier C, Fernandes HS, Sousa SF, Lesbats P, Weigel P, Favereaux A, Guyonnet-Duperat V, Parissi V. In Silico, In Vitro and In Cellulo Models for Monitoring SARS-CoV-2 Spike/Human ACE2 Complex, Viral Entry and Cell Fusion. Viruses. 2021; 13(3):365. https://doi.org/10.3390/v13030365
Chicago/Turabian StyleLapaillerie, Delphine, Cathy Charlier, Henrique S. Fernandes, Sergio F. Sousa, Paul Lesbats, Pierre Weigel, Alexandre Favereaux, Véronique Guyonnet-Duperat, and Vincent Parissi. 2021. "In Silico, In Vitro and In Cellulo Models for Monitoring SARS-CoV-2 Spike/Human ACE2 Complex, Viral Entry and Cell Fusion" Viruses 13, no. 3: 365. https://doi.org/10.3390/v13030365