
INTRODUCTION
Inflammation and pain are some of the main health problems worldwide [1], which makes nonsteroidal anti-inflammatory drugs (NSAIDs) the most dispensed pharmacological group worldwide [1].
Although the use of NSAID drugs is quite popular and available over the counter, chronic use of these types of drugs can cause a wide variety of side effects, gastric bleeding being one of the leading problems [2,3]. The gastric problems caused by these drugs are derived from their mechanism of action, by inhibiting the cyclooxygenase (COX) enzyme [3], responsible for the synthesis of structural prostaglandins at the gastrointestinal level from arachidonic acid, which produces the formation of the protective mucosa of the endothelium, avoiding gastritis and gastric ulcerations [4].
Ketorolac is an NSAID that has been used more frequently in the treatment of acute and chronic pain; moreover, is useful in pain after dental or postoperative procedures [5], and its use is even recognized in the treatment of pain in infants [6], but ever monitoring chronic use, often not recommended due to gastrointestinal, cardiovascular, and renal problems [7].
To date, the studies in this well-known pharmacological group continue to search for new active nuclei based on the already known subgroups; evidence of this is the current discovery of a new active nucleus based on the mixture of the active nucleus of piroxicam and celecoxib in search of new anti-inflammatory active substances but low probabilities of adverse effects [8], as well as the search for new modified aspirin drugs [9]; however, the increased activity of NSAID drugs is due to the presence of two aromatic rings linked by a keto in search of the noncoplanarity of the rings so that the increased activity would occur in derivatives of drugs such as ketorolac [10].
These actions give rise to the continuous search for new ketorolac derivatives, which, although it is a drug discovered many years ago [11], its current and growing use [6], as well as its structural properties, make it a solid basis for the search for new chemical variants; therefore, from search techniques for new chemical entities under changes by bioisosteres, new ketorolac derivatives with greater anti-inflammatory activity and fewer adverse effects are proposed through in silico assays.
METHODS
Ketorolac derivatives were designed under bioisosteric replacement, in such a way that it allows maintaining biological pharmacological activities but decreases the toxicological activities implicit in ketorolac.
Prediction of physicochemical properties
The physicochemical properties of the ketorolac derivatives were predicted under the parameters of the SwissADME server [12].
Activity predictions
The prediction of the anti-inflammatory activity, as well as the possible complementary activities and those responsible for adverse actions, were predicted under the parameters of the Way2Drug server [13], which allowed the probabilistic quantification of anti-inflammatory, analgesic, antipyretic, and fibrinolytic effects.
2D quantitative structure-activity relationship (QSAR) analysis
The algorithm for the 2D QSAR analysis has been conducted by analyzing the work of Muchowski et al. [11], that information was obtained from the ChemBL database [14]. The data was parameterized and stored in a .CSV file, which was used for the design of the algorithm, which was written in R v.4.3.0 using the R studio [15]. The 2-D descriptors were those related to the Lipinski rules, Veber rules, and QSAR characteristics, such as lipophilicity, and electronic and steric effects, which were determined using PADEL-descriptor [16].
Molecular modeling
The crystalline structure of COX-2 was obtained from the protein data bank database under the code 5IKR, which went through a cleaning process of ions, ligands, and water molecules, which were removed under the use of PyMOL Molecular Graphics System 2.5 software programs [17]. For molecular docking, the structure of the selected protein was parameterized using the AutoDock Tools program [18], polar hydrogens were added, nonpolar hydrogens were removed, and Kollman charges were added.
The docking protocol was performed using AutoDock Vina [19] based on VINA’s genetic algorithm and default procedures for directed and rigid docking. The amino acid residues Serine 530, Arginine 120, Tyrosine 355, and Valine 349 and 523 were defined as constituents of the potential binding site [20], proceeding to create a grid box in the identified catalytic site whose coordinates were: Center_x = 43.375, Center_y = 6.133, Center_z = 61.379, Size_x = 12.0, Size_y = 15.0, and Size_z = 15.0; each of the ketorolac derivatives with the increased anti-inflammatory activity according to the 2D QSAR analysis proceeded to couple to this site of the enzyme, determining the most probable and energetically favorable union conformations. The completeness was 100 runs for each ligandprotein. The resulting structures and junctional docking poses were graphically inspected for interactions using the PyMOL Molecular Graphics System 2.5 programs and BIOVIA Discovery Studio [17,21].
The conformations that presented the lowest affinity energy were selected; a more negative value means a higher affinity, the highest number of hydrogen bonding interactions, and total interactions.
RESULTS
Under the knowledge of bioisosteres, modifications were made in each of the parts of the molecule (carboxylic group, amino bicyclo, keto, and benzene ring), looking for similar groups that maintain or decrease the acidity of the molecule. With this, 42 bioisosteres were obtained, view Figure 1, in addition to ketorolac (molecule 43).
At the same time, the prediction of the physicochemical characteristics of the molecules was conducted using the SwissADME server, taking as important data the molecular weight (MW), lipophilicity (CLogP), and topological polar surface area (TPSA), which are three-dimensionally plotted in Figure 2.
The prediction of the activity of the 42 ketorolac derivatives was made under the parameters of the Way2Drug server (Table 1); in addition, the ketorolac (molecule 43) was analyzed as a blank standard.
After the analysis, it is observed that the absorptivity characteristics of Lipinski and Veber analyzed in Figure 1, only 12 molecules comply with this characteristic; likewise, it is shown in Figure 2 that 16 molecules have fibrinolytic activity, and, of the 12 molecules have molecular properties for oral activities according to Lipinski and Veber rules, 4 molecules would not have anti-inflammatory activity, with only molecules 1, 13, 21, 32, 34, 37, 38, and 40, the molecules to analyze.
Similarly, 59 molecules derived from 5-aroyl-1,2-dihydro-3H-pyrrolo [1,2-a]pyrrole-1-carboxylic acid were analyzed, from which their molecular descriptors were obtained using Padel Descriptor. With the statistical analysis in R v.4.3.0, the 2D QSAR equation of the descriptors that maintain linearity with the activity was obtained, which is reflected in Equation (1). Equation (1) maintains a standard error of 0.2149, an adjusted R2 of 0.9022, and p-value was 1,301 × 10−06.
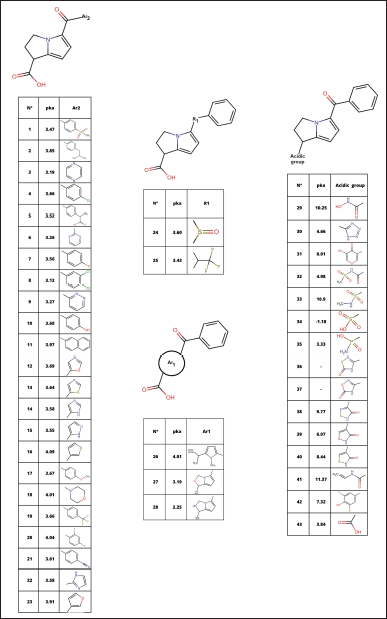 | Figure 1. Ketorolac bioisosteres. [Click here to view] |
Activity = −7.42E+03 + 2.41E-01 ATS3e + −1.31E-01 ATS7e + −4.64E-01 ATS4p + −8.93E-03 ATS3i + −3.16E-03 ATS4i + −4.26E-04 ATS6i + 8.78E-03 ATS7i + −4.62E-03 ATSC5v + 6.87E-01 ATSC5p + −5.90E+00 AATSC4v + −3.65E+02 AATSC5p + 3.71E+02 MATS4v + 1.62E+01MATS4p + 7.79E+01 MATS5p + −9.21E+00 MATS7p + 6.96E+00 MATS4i + 5.38E+00 MATS5i + 9.84E+01 GATS4v + 6.73E+00 GATS6e + 2.88E-02 VR1_Dzs + −6.02E-01 VR2_Dzs + 9.63E+01 SpMin2_Bhm + 1.36E+02 SpMin2_Bhv + −1.62E+02 SpMin2_Bhe + −5.05E+02 SpMax1_Bhi + 1.01E+02 SpMin2_Bhi + 2.29 C2SP3 + 2.17E+05 SCH-5 + −4.33E+03 SCH-6 + −2.36E+02 SCH-7 + −2.56E+05 VCH-5 + 1.44E+04 VCH-6 + 1.34E+02 VCH-7 + −2.35E+01 VP-5 + 1.50E+03 AVP-6 + −1.51 nHCsats + 1.71E+02 maxHBd + −2.15E+01 maxHBint2 + 2.84E+01 ETA_EtaP + −1.85E+04 n5Ring + 1.24E+04 nT8Ring + −4.21E+02 SRW7 + 2.59E+03 SRW9 (1)
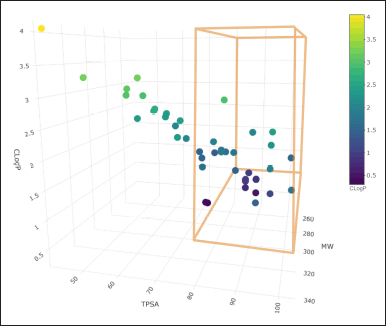 | Figure 2. Physicochemical properties of ketorolac derivatives. [Click here to view] |
With the equation, the anti-inflammatory activity of the 42 molecules derived from ketorolac was predicted. Molecule 43 (ketorolac) was analyzed under this equation as a target to determine the potency of the equation (Table 2), finding that of the eight molecules that meet the characteristics of Lipinski and Veber, and demonstrate anti-inflammatory activity without fibrinolytic activity, molecules 32, 34, and 37 are more active than ketorolac under this analysis.
The three molecules selected under the 2D QSAR analysis have been analyzed using molecular docking to determine their affinity energy and the possible interactions they conduct, determining computationally if COX-2 inhibition is carried out, showing the 2D impressions of the molecular modeling in Figure 3 and the analysis data in Table 3.
DISCUSSION
The search for new chemical substances with anti-inflammatory activity is based on the discovery of new chemical entities through natural active substances [22] designs based on chemical entities already in use, and other methods [23]. The design of the 42 ketorolac derivatives used in this article (Fig. 1) was based precisely on this second point, designing them under bioisostere replacement tools through the chemical structure of ketorolac, dividing its structure into four fundamental parts for change: acidic group, bicyclic (Ar1), keto (R1), and aromatic ring (Ar2).
Bioisosteric replacement is a strategy that avoids drug design failures due to metabolism and pharmacokinetic problems, preserving the potency/efficacy of the leading compound. Structural changes by bioisosteric replacement seek to alter the shape, size, chemical, and physical characteristics, keeping its pharmacodynamic properties like the lead compound and improving its pharmacokinetic and toxicological properties [23,24]. Bioisostere replacements have been used as a tool in the discovery and design of new drugs in other pharmacological groups, such as anti-HIV drugs [24]; even their use in heterocyclic rings is well-known, both for their activity-enhancing properties and their pharmacokinetic limitations [25], a crucial point when designing new drug candidates. Likewise, changes due to bioisosteres have recently been used in the design of new aspirin derivatives [9]; however, the absence of a second ring limits the potency of these compounds, which is only compensated by a CF3 in one of the molecules, which results precisely in the most active of the derivatives. Although, in this research, the bioisosteric replacement was handmade, tools such as MolOpt facilitate changes in the design of new drugs using bioisostere transformations [26].
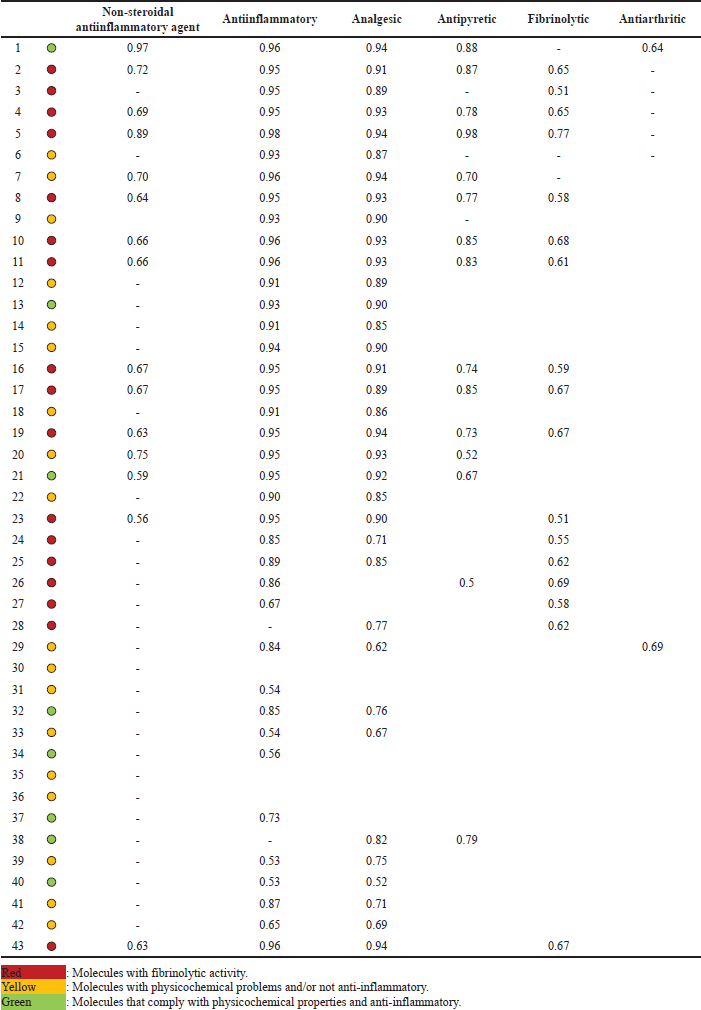 | Table 1. Predictive analysis of the activity of ketorolac derivatives. [Click here to view] |
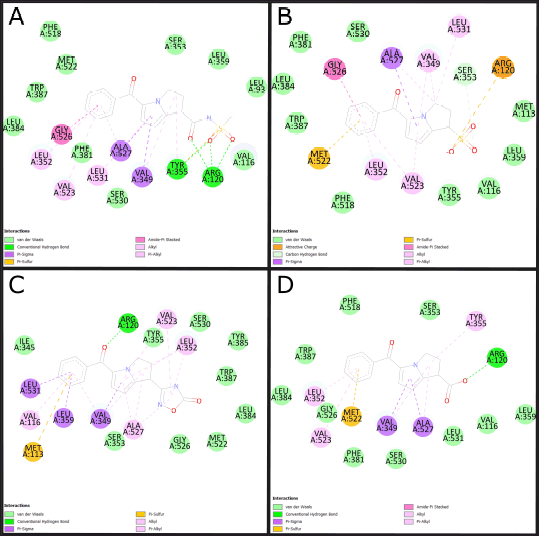 | Figure 3. 2D molecular docking of ketorolac derivatives with increased anti-inflammatory activity. (A) Molecular docking of molecule 32. (B) molecular docking of molecule 34. (C) Molecular docking of molecule 37. (D) Molecular docking of molecule 43. Source: BIOVIA Discovery Studio Program. [Click here to view] |
The relationship of the physicochemical characteristics, such as MW, TPSA, and lipophilicity (CLogP), allow us to predict the oral absorption of drug candidates. MW and CLogP are used in Lipinski’s rule of five, where he mentions that MW must not be greater than 450 Da and CLogP must not be less than 1 nor more than 5 [27]; likewise, Veber’s rules state that the TPSA must not be greater than 140 Å2 nor less than 80 Å2, since a substance with more than 140 Å2 is very hydrophilic substances and less than 80 Å2 are very lipophilic, which would limit its gastrointestinal absorption [28].
CLogP has a direct effect on the bioavailability of a molecule, and this, in turn, on potency and efficacy. This parameter is essential in the design or modification of compounds with biological activity. Those molecules with optimal intestinal absorption have a CLogP value greater than 1 and less than 4. It is worth mentioning that the ketorolac derivatives designed are within this parameter [29].
The physicochemical parameters used at the time of computer-assisted molecule design are the same as those proposed by Lipinski, where MW and CLogP may be the most important but also the most limiting because these are the ones that direct entry into the cell after oral administration. Although ketorolac is one of the drugs with specific utility for moderate pain, this has also been characterized by the manifestations of heartburn, among other gastropathy. The MW and CLogP are the physicochemical characteristics influencing absorption and affinity towards the receptor. The substituent functional groups on the rings influence the hydrogen bonding interactions (HBD and HBA), which affect the polarity, solubility, and interaction strength (ΔG°) with its receptor protein. The ketorolac derivatives show a considerable number of HBA and HBD, highlighting molecule 12 and molecule 13 for presenting a higher number of HBA [30,31].
The TPSA relates the cell permeability through the membranes and parameters such as N° HBD and CLogP. TPSA values <140 Å2 ensure good intestinal permeability, although its ability to penetrate the blood–brain barrier with affinity and show activity in the central nervous system should show TPSA values <80 Å2; however, this parameter also indicates potential toxic risks. The values of the number of rotatable bonds (N° RB) or conformational flexibility are related to the molecular coupling to the receptor; even if the value observed in ketorolac is three, several molecules exceed this value [32–35].
Of the designed molecules, only 12 ketorolac derivatives meet these restrictions (Fig. 2), so the rest of the parameters were only developed for these molecules that meet probable oral absorption and low probabilities of toxicity.
Way2Drug [13] was the server used to predict the activities of the ketorolac derivatives. These results shown in Table 1 allow us to observe that not all the molecules present predictive anti-inflammatory and analgesic activity. The lack of possible activity in molecule 36 can be explained from the point of view that the molecules must present an acidic center for better interaction with the rest of Arginine 120, which demands a direct inhibition of the COX-2 enzyme responsible for the proliferation of inflammatory mediators [36].
The fibrinolytic activity is of foremost importance since it limits the probability of myocardial infarctions, even NSAIDs such as aspirin have been used for many years for this type of effect [37,38]; however, this effect is based on the inhibition of the COX-1 isoform, so an increased fibrinolytic activity is also based on probable increased gastric effects [38]. This implies that molecules with a low fibrinolytic effect would be the ones that present the least adverse gastric reaction, with the molecules highlighted in red being those with probable increased gastric toxicity. Of these, molecules 5, 16, 35, and 36 that maintain good physicochemical characteristics were discarded due to this probability or for not presenting predictive anti-inflammatory and analgesic activity, keeping only eight molecules marked green in Table 1.
The in silico anti-inflammatory activity was confirmed using 2D QSAR analysis techniques [15], the most accurate technique for drug discovery by relating pharmacological activities and structural changes. For its development, a multiple regression equation of molecular descriptors must be integrated with the quantitative activity of similar substances already studied, for which the substances studied by Muchowski et al. [11] were used. For the development of the descriptors, Padel Descriptor was used [16], resulting in Equation (1), which was used in the molecules derived from ketorolac for the quantification of their activity, which is shown in Table 2, resulting in an activity value of −11.91 for ketorolac (molecule 43); therefore, substances with an activity value above it would be better, being three highly active molecules (molecule 32, 34, and 37), contrastable with a lower probability of adverse reactions and a better physicochemical profile. The 2D QSAR analysis was carried out for the 42 derivatives since the fact of presenting an increased activity of some of the bioisosteres can give innovative ideas of changes for the attempt to improve the physicochemical profiles; because, although they can indeed produce adverse gastric reactions or low oral absorption, they do have an anti-inflammatory activity.
Molecular modeling (Table 3 and Fig. 3) allows the identification of the probable positioning of the linked molecules in the active site [39], which is important because not every substance that binds to the active site will exert an action; this is due to the presence of fundamental amino acids identified in active site, whose participation in the interaction allows predicting the activation or inhibition of the base receptor, such as Arginine 120, Serine 530, and Tyrosine 355, in the case of the COX-2 enzyme [1]. Based on this, we can predict that all except molecule 37 comply with a clear position in the active site, well the acid functional group attached to the bicyclic must interact with Arginine 120, and the ketonic bridge that joins the two rings must interact with Serine 530 [20], molecule 37 does not comply with this parameter, most likely due to a lack of acidity in the carboxylic acid bioisostere. The ligand efficiency (LE) parameters are the capacity of the ligand to produce a biological response when it binds to the target receptor and the quantitative magnitude of this response [40], a parameter widely used in drug design and which must be >−0.4, even though an LE of −0.39 is accepted for cases of leading compounds, so molecules 34 and 37 would comply with it, despite having a low LE, molecule 32 is likely to maintain very high activity, this due to the high-affinity binding in Arg120 and high formation of hydrogen bonds. Molecule 34 meets to a high degree all the parameters established in the study; however, its pka below the values of ketorolac (Fig. 1) limits the probability of no formation of gastrointestinal effects in in vivo tests.
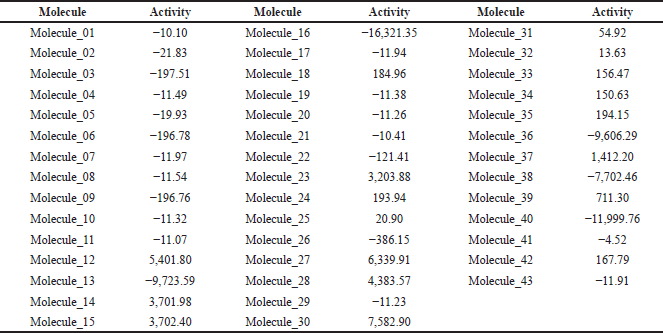 | Table 2. Activity prediction of ketorolac derivatives under 2D QSAR analysis. [Click here to view] |
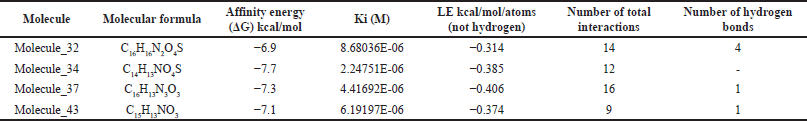 | Table 3. Affinity energy and number of interactions of ketorolac derivatives with greater anti-inflammatory activity. [Click here to view] |
The discovery of new anti-inflammatory active substances with a decreased gastro-injurious effect is still ongoing; under these predictive parameters, molecule 32 meets all the requirements set out in this research; even so, these results must be contrasted with in vivo experiments in preclinical phases, before their studies in humans.
CONCLUSION
Molecule 32 complies with the parameters established in this study; therefore, of the 42 ketorolac derivatives proposed, this molecule would be the most active with anti-inflammatory and analgesic effects and with decreased gastro-injurious effects; even so, these findings must be contrasted with in vivo experiments in preclinical trials. The employ in silico techniques has allowed the reduction of large numbers of experimental animals in the next study phase, rationalizing their use.
LIST OF ABBREVIATIONS
CLogP: lipophilicity, COX-2: cyclooxygenase 2, HBA: H-bond acceptors, HBD: H-bond donors, LE: ligand efficiency, MW: molecular weight, NSAIDs: nonsteroidal anti-inflammatory drugs, QSAR: quantitative structure-activity relationship, RB: rotatable bonds, TPSA: topological polar surface area.
ACKNOWLEDGMENTS
The authors thank the Bioinformatics and Chemoinformatics Research Group (BIOQUIM) by the Universidad Nacional de Trujillo (Resolución Vicerrectoral de Investigación N°060-2023-VIN-UNT).
AUTHOR CONTRIBUTIONS
CDGS: Wrote the first draft. CNRS: Performed physicochemical analyses and predictions with Way2Durg. ACM and LAC: Performed Molecular Docking analyses. VEVLT and CRSC: Performed 2D QSAR predictive design. All authors reviewed, edited, read, and approved the final manuscript.
FUNDING
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Atkinson TJ, Fudin J. Nonsteroidal antiinflammatory drugs for acute and chronic pain. Phys Med Rehabil Clin N Am. 2020;31(2):219–31. CrossRef
2. Lee MW, Katz PO. Nonsteroidal antiinflammatory drugs, anticoagulation, and upper gastrointestinal bleeding. Clin Geriatr Med. 2021;37(1):31–42. CrossRef
3. Connors JM. Non-steroidal antiinflammatory drug gastroenteropathy. xPharm: the comprehensive pharmacology reference. Amsterdam: Elsevier; 2007. pp 1–10. CrossRef
4. Tai FWD, McAlindon ME. Non-steroidal anti-inflammatory drugs and the gastrointestinal tract. Clin Med (Lond). 2021;21(2):131–4. CrossRef
5. Motov S, Yasavolian M, Likourezos A, Pushkar I, Hossain R, Drapkin J, et al. Comparison of intravenous ketorolac at three single-dose regimens for treating acute pain in the emergency department: a randomized controlled trial. Ann Emerg Med. 2017;70(2):177–84. CrossRef
6. Ziesenitz VC, Welzel T, van Dyk M, Saur P, Gorenflo M, van den Anker JN. Efficacy and safety of NSAIDs in infants: a comprehensive review of the literature of the past 20 years. Paediatr Drugs. 2022;24(6):603–55. CrossRef
7. McNicol ED, Ferguson MC, Schumann R. Single-dose intravenous ketorolac for acute postoperative pain in adults. Cochrane Database Syst Rev. 2021;5(5):CD013263. CrossRef
8. Vo NB, Ngo QA. Synthesis, antiinflammatory, and cytotoxic activity of novel pyrazolo[4,3-c][2,1]benzothiazine 4,4-dioxide derivatives. J Heterocycl Chem. 2022;59(10):1813–23. CrossRef
9. Uzzaman M, Mahmud T. Structural modification of aspirin to design a new potential cyclooxygenase (COX-2) inhibitors. In Silico Pharmacol. 2020;8(1):1. CrossRef
10. Hadjipavlou-Litina D. Quantitative structure-activity relationship (QSAR) studies on non steroidal anti-inflammatory drugs (NSAIDs). Curr Med Chem. 2000;7(4):375–88. CrossRef
11. Muchowski JM, Unger SH, Ackrell J, Cheung P, Cooper GF, Cook J, et al. Synthesis and antiinflammatory and analgesic activity of 5-aroyl-1,2-dihydro-3H-pyrrolo[1,2-a]pyrrole-1-carboxylic acids and related compounds. J Med Chem. 1985;28(8):1037–49. CrossRef
12. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. CrossRef
13. Zakharov AV, Lagunin AA, Filimonov DA, Poroikov VV. Quantitative prediction of antitarget interaction profiles for chemical compounds. Chem Res Toxicol. 2012;25(11):2378. CrossRef
14. Gaulton A, Hersey A, Nowotka M, Patrícia Bento A, Chambers J, Mendez D, et al. The ChEMBL database in 2017. Nucleic Acids Res. 2016;45:945–54. CrossRef
15. Gajewicz-Skretna A, Kar S, Piotrowska M, Leszczynski J. The kernel-weighted local polynomial regression (KwLPR) approach: an efficient, novel tool for development of QSAR/QSAAR toxicity extrapolation models. J Cheminform. 2021;13(1):1–20. CrossRef
16. Yap CW. Software news and update PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem. 2011;32:1466–74. CrossRef
17. Schrödinger L, DeLano W. PyMOL [Internet]. 2020. Available from: http://www.pymol.org/pymol
18. Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19(14):1639–62. CrossRef
19. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–61. CrossRef
20. Beale J, Block J. Wilson and Gisvold’s textbook of organic medicinal and pharmaceutcal chemistry. 20th ed. Philadelphia, PA: Wolters Kluwer Health; 2011.
21. BIOVIA. Dassault systèmes BIOVIA Discovery Studio v21.1.0.20298. San Diego, CA: BIOVIA; 2020.
22. Fernández-Flores N, Rojas-Cardenas N, Vasquez-Quispe A, Chavez-Flores JE, Justil-Guerrero HJ, Parreno-Tipian J, et al. Protection of erythrocytes against lipoperoxidation and antiinflammatory effects of ethanolic extract of Encelia canescens Lam leaves in mice. Pharmacogn J. 2020;12(4):798–804. CrossRef
23. Gareth T. Medicinal chemistry—an introduction; fundamentals of medicinal chemistry. 2nd ed. England, UK: John Wiley & Sons Ltd; 2007.
24. Dick A, Cocklin S. Bioisosteric replacement as a tool in anti-HIV drug design. Pharmaceuticals (Basel). 2020;13(3):36. CrossRef
25. Subbaiah MAM, Meanwell NA. Bioisosteres of the phenyl ring: recent strategic applications in lead optimization and drug design. J Med Chem. 2021;64(19):14046–128. CrossRef
26. Shan J, Ji C. MolOpt: a web server for drug design using bioisosteric transformation. Curr Comput Aided Drug Des. 2020;16(4):460–6. CrossRef
27. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. CrossRef
28. Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–23. CrossRef
29. Tsopelas F, Giaginis C, Tsantili-Kakoulidou A. Lipophilicity and biomimetic properties to support drug discovery. Expert Opin Drug Discov. 2017;12(9):885–96. CrossRef
30. Hopkins AL, Keserü GM, Leeson PD, Rees DC, Reynolds CH. The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov. 2014;13(2):105–21. CrossRef
31. Doak BC, Over B, Giordanetto F, Kihlberg J. Oral druggable space beyond the rule of 5: insights from drugs and clinical candidates. Chem Biol. 2014;21(9):1115–42. CrossRef
32. Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRX. 2005;2(4):541–53. CrossRef
33. Barret R. Importance and evaluation of the polar surface area (PSA and TPSA). In: Barret R, editor. Therapeutical chemistry. Oxford, UK: Elsevier; 2018, pp 89–95.
34. Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem. 2000;43(20):3714–7. CrossRef
35. Rossi Sebastiano M, Doak BC, Backlund M, Poongavanam V, Over B, Ermondi G, et al. Impact of dynamically exposed polarity on permeability and solubility of chameleonic drugs beyond the rule of 5. J Med Chem. 2018;61(9):4189–202. CrossRef
36. Halim PA, El-Nassan HB, El-Dash YS. Design and synthesis of novel ibuprofen derivatives as selective COX-2 inhibitors and potential anti-inflammatory agents: evaluation of PGE2, TNF-α, IL-6 and histopathological study. Med Chem. 2022;18(4):427–43. CrossRef
37. Krittanawong C, Hahn J, Kayani W, Jneid H. Fibrinolytic therapy in patients with acute ST-elevation myocardial infarction. Interv Cardiol Clin. 2021;10(3):381–90. CrossRef
38. Stiller CO, Hjemdahl P. Lessons from 20 years with COX-2 inhibitors: importance of dose-response considerations and fair play in comparative trials. J Intern Med. 2022;292(4):557–74. CrossRef
39. Sanchez G. Automated prediction of ligand-binding sites in proteins. Proteins. 2008;70(4):1506–17. CrossRef
40. Meneses L, Cuesta S. determinación computacional de la afinidad y eficiencia de enlace de antinflamatorios no esteroideos inhibidores de la ciclooxigenasa-2. Rev Ecuat Med Cienc Biol. 2015;36(1–2):17–25. CrossRef