
Abstract
X-ray and cryo-EM structures of tetrameric and pseudo-tetrameric P-loop channels are used to elaborate homology models of mammalian voltage-gated sodium channels with drugs and neurotoxins. Such models integrate experimental data, assist in planning new experiments, and may facilitate drug design. This chapter outlines sodium channel models with local anesthetics, anticonvulsants, and antiarrhythmics, which are used to manage pain and treat sodium channelopathies. Further summarized are sodium channel models with tetrodotoxin, mu-conotoxins, batrachotoxin, scorpion toxins, and insecticides. Possible involvement of sodium ions in the action of some ligands is discussed.
Similar content being viewed by others

Keywords
- Antiarrhythmics
- Anticonvulsants
- Batrachotoxin
- Conotoxins
- Homology modeling
- Insecticides
- Ligand docking
- Local anesthetics
- Pyrethroids
- Tetrodotoxin
1 Structure of Sodium Channels
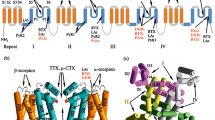
Voltage-gated sodium channels play key roles in physiology. They are targets for naturally occurring toxins (Stevens et al. 2011), various drugs used to treat pain and health disorders (Catterall 2014; Catterall and Swanson 2015), and insecticides (Silver et al. 2014). The α1 subunit of these channels folds from a polypeptide chain of four homologous repeats that circumvent the ion-permeating pore (Fig. 1). Each repeat contains six transmembrane helical segments (S1–S6), which are connected by extracellular and intracellular loops, and contributes to the pore domain an outer helix (S5), a pore-lining inner helix (S6), and an extracellular membrane re-entering P-loop (P) with descending (P1) and ascending (P2) helices. Residues at the C-ends of S6s contribute to the activation gate. Segments S1–S4 form voltage-sensing domains. Upon membrane depolarization S4s move in the extracellular direction. S4–S5 linker helices follow this motion initiating the activation gate opening. D, E, K, and A residues in homologous positions of the P1–P2 linkers contribute to the selectivity filter. The DEKA ring separates the outer pore, which is exposed to the extracellular space, and the inner pore, which opens to the cytoplasm upon the channel activation.

Ligand binding sites in sodium channels. (a) Transmembrane topology of eukaryotic channels. (b) Side view of the NavAb X-ray structure (Payandeh et al. 2011) with two subunits removed for clarity and helices colored as in (a). (c) Intracellular view of NavAb. Individual subunits have different colors. Reprinted from Zhorov and Tikhonov (2016) with permission from Elsevier
Universal labels help appreciate homologous positions of residues in P-loop channels (Zhorov and Tikhonov 2004). A label shows repeat number, segment type (p, P-loop; i, Inner helix), and relative residue number in the segment. For example, F4i15 designates phenylalanine in repeat four inner helix, position 15 (Fig. 2).

Aligned sequences of P-loops and S6 segments in NavMs and Nav1.4 channels. Residues, which are mentioned in the text, are underlined. Rows above the P-loop and S6 sequences indicate segment types and relative residue positions, which are used in universal labels of the residues
X-ray structures of closed prokaryotic sodium channels NavAb (Payandeh et al. 2011) and NavRh (Zhang et al. 2012b) confirmed common folding with potassium channels and revealed specific features such as P2 helices, wide selectivity filters, and large subunit interfaces (fenestrations). The latter may provide hydrophobic access pathway for drugs. Recently, X-ray structures of the open prokaryotic channels NavAb and NavMs (Lenaeus et al. 2017; Sula et al. 2017), and cryo-EM structure of the closed eukaryotic channel NavPaS (Shen et al. 2017) become available.
In the X-ray structure of the NavMs channel with a LA-like molecule PL1, a single bromine atom of PL1 is seen in a fenestration and modeling suggests a horizontal PL1 orientation and electrostatic mechanism of the channel block (Bagneris et al. 2014). In the X-ray structure of a bacterial calcium channel CavAb with a verapamil derivative (Tang et al. 2016) the pore-facing carbonyls at the C-ends of P1 helices are 5.6–7 Å from the center of the ligand ammonium group. The latter apparently displaces a calcium ion, which is seen between these carbonyls in the ligand-free CavAb (Tang et al. 2014). The X-ray structure of the homotetrameric NavAb/Nav1.7 chimera was used to design a small-molecule antagonist that specifically binds to the extracellular loops of VSD-IV in Nav1.7 (Ahuja et al. 2015).
2 Homology Modeling and Ligand Docking
In the absence of high-resolution structures of eukaryotic sodium channels with drugs and toxins computer-based homology modeling is used to predict such structures. The approach is based on the assumption that homologous proteins have similar folding. Computational methods use semi-empirical energy functions (force fields) and energy optimizations that eliminate unrealistic atomic clashes and favor atom–atom attractions. The search for optimal geometry involves random sampling of many 3D structures by using molecular dynamics (MD) or/and Monte Carlo energy minimizations (MCM). MD provides numerical solutions of the Newton equations of motion of atoms. All atoms move simultaneously, an important advantage to simulate large systems, e.g., ion channels in the lipid and water environment. However, a very small (femtosecond) time step requires huge computational resources to simulate millisecond trajectories (Jensen et al. 2012). During MCM, the molecular system “jumps” over energy barriers and this facilitates docking of ligands (Garden and Zhorov 2010). A promising approach is combining MCM to find energetically optimal structures and MD to simulate motions of the structures (Marzian et al. 2013).
Data from mutational, electrophysiological, and other experimental studies greatly facilitate ligand docking. However, results should be treated with caution due to several problems. Firstly, a residue substitution can affect ligand action either directly or indirectly (allosterically). Secondly, the sequence alignment of the query and template channels may be ambiguous. Thirdly, experimental data may be consistent with various low-energy structures. Furthermore, a computational model may inadequately represent the environment, precision of energy calculations is limited, and energy sampling protocols may overlook important structures. Reliable models are expected to integrate diverse experimental data and provide testable predictions. Some models, which seem consistent with various experimental data, are outlined below.
3 Inner Pore Blockers
The inner pore of sodium channels is blocked by various drugs including local anesthetics (LAs), anticonvulsants, and antiarrhythmics (Fig. 3). Typical LAs, e.g., lidocaine, are flexible molecules with an aromatic ring and protonatable amino group. Bulky semi-rigid molecules such as cocaine and quinidine also block the channel apparently at the site that overlaps with the LA receptor (Ragsdale et al. 1996; O’Leary and Chahine 2002). Mutational analyses suggest that these drugs interact with phenylalanine F4i15 (Ragsdale et al. 1994; Yarov-Yarovoy et al. 2002; Ahern et al. 2008) and residues in the pore-lining helices IS6, IIIS6, and IVS6 (Mike and Lukacs 2010), P1 helices (Yamagishi et al. 2009), and the selectivity-filter DEKA locus (Sunami et al. 1997). In contrast to protonatable LA-like molecules, typical anticonvulsants, e.g., phenytoin and carbamazepine, are electroneutral molecules with an aromatic moiety and non-ionizable polar groups (Liu et al. 2003). Intriguingly, despite highly diverse chemical structures, various drugs have a common binding region within the inner pore (Ragsdale et al. 1996; Kuo 1998) and similar mechanisms of action (Catterall 1987, 2012).

Inner pore-blocking drugs
Early Models of Ligand–Channel Complexes
Computational docking of cationic ligands in homology models allowed to rationalize various experimental data (Lipkind and Fozzard 2005; Scheib et al. 2006; Tikhonov and Zhorov 2007; Bruhova et al. 2008; Browne et al. 2009). The drugs are proposed to block the ion permeation by the electrostatic mechanism (Lipkind and Fozzard 2005; Tikhonov et al. 2006). Two principal binding modes were proposed. In the “vertical” mode, the ligand positive charge approaches the outer pore and the hydrophobic moiety extends along the inner pore (Tikhonov and Zhorov 2007). In the “horizontal” mode the charged moiety also approaches the outer pore, while the opposite end protrudes into the III/IV repeat interface, which is proposed to serve a hydrophobic access pathway for ligands to the closed channel (Tikhonov et al. 2006; Bruhova et al. 2008). Existence of the hydrophobic pathway has long been predicted (Hille 1977) and such pathways are now seen as wide fenestrations in the X-ray structure of NavAb (Payandeh et al. 2011) and other prokaryotic sodium channels. In contrast to cationic drugs, electroneutral anticonvulsants were proposed to block the ion permeation by sterically occluding the inner pore (Lipkind and Fozzard 2010).
Electrostatics of Drug-Channel Interactions
In the low-dielectric environment of membrane proteins, electrostatic interactions are expected to provide large contributions to the ligand binding energy. Therefore, the fact that cationic ligands target the cation-attractive permeation pathway is not surprising. But how electroneutral ligands could block the cation-attractive pore? Permeant ions are hypothesized to interact with electronegative groups of such ligands (Zhorov and Ananthanarayanan 1996; Tikhonov et al. 2006; Zhorov and Tikhonov 2013). These interactions are weak in the bulk solvent, but they may be strengthened in the low-dielectric environment of the confined permeation pathway where the ligand and a permeant ion can be favorably positioned against each other due to interactions with the channel residues. Experimental structures of ion channels with ion-bound ligands are lacking. In the X-ray structure of the NavMs channel, a completely hydrated sodium ion (NaIII) is seen between four backbone carbonyls p48 at the C-end turns of P1 helices (Naylor et al. 2016). This structure was used to build a homology model of the Nav1.4 channel and dock cationic and electroneutral ligands (Tikhonov and Zhorov 2017). Intensive docking using Monte Carlo-energy minimizations predicted ensembles of low-energy binding modes rather than unique ligand–channel complexes. Representative ligand–channel complexes are briefly described below.
Cationic Ligands
In the channel model with lidocaine and a sodium ion, the organic and inorganic cations repelled each other and were scattered over the pore domain. However, in the sodium-free model, the lidocaine ammonium group often occurred at site NaIII due to electrostatic attraction to backbone carbonyls p48 and side chains p49 (Fig. 4a, b). In alternative binding modes, the ammonium group formed cation-π contacts with F4i15 or approached a putative position NaIV at the focus of P1 helices, which is analogous to site 5 for potassium ions in the KcsA potassium channel (Zhou et al. 2001). The aromatic moiety usually formed face-to-face or edge-to-face contacts with F4i15 and approached Y4i22. In addition to the vertical orientations, horizontal orientations were found with the ammonium group below site NaIII and aromatic moiety extending in the III/IV fenestration as predicted before (Bruhova et al. 2008). In most of the binding modes, lidocaine would electrostatically block the ion permeation. Rather low lidocaine affinity (Bean et al. 1983) is consistent with the diversity of predicted binding modes. These results justified docking of other cationic ligands in the sodium-free channel model.

NavMs-based model of the Nav1.4 channel with ligands. Repeats I, II, III, and IV are cyan, green, yellow, and magenta, respectively. Backbone carbonyls in position p48 are thick sticks, a sodium ion is an orange sphere and side chains F4i15 and Y4i22 are thin sticks. (a) Extracellular view of lidocaine. (b–d) Side views of lidocaine, carbamazepine, and lamotrigine, respectively
QX-314 is a quaternary lidocaine analog that blocks sodium channels (Qu et al. 1995). In the low-energy binding modes, the triethylammonium group of QX-314 fits between four backbone carbonyls p48 or approached side chains in position p49 in agreement with the data that mutations Q1p49C and F3p49C decrease the ligand potency (Yamagishi et al. 2009). Mutational data suggest that bulky cocaine and quinidine bind in the inner pore (Ragsdale et al. 1996; O’Leary and Chahine 2002). In the channel models with these ligands, the protonated amino group approached site NaIII, but it was also found at level i15. The aromatic group bound between F4i15 and Y4i22. The hydroxyl group of quinidine interacted with polar groups in positions p48 and p49. Sipatrigine is a protonatable neuroprotective drug that blocks the inner pore of sodium channels (Liu et al. 2003). In the vertical binding mode, the long sipatrigine molecule bound parallel to the pore axis with the ammonium group at level NaIII and the opposite end reaching level i23 in the activation-gate region. In the horizontal binding mode, sipatrigine extended from the inner pore to the interface between helices IIIP, IIIS6, and IVS6, establishing multiple contacts with the channel residues. Numerous protonatable ligands, which are structurally similar to the above drugs, are expected to block the channel in similar ways.
Electroneutral Ligands
Experimental data suggest the inner pore as the binding region for various electroneutral ligands (Ragsdale et al. 1996; Kuo 1998; Liu et al. 2003; O’Reilly et al. 2012). In the sodium-free channel, carbamazepine binding modes were scattered over the pore domain as observed in MD simulations of the NavAb channel with electroneutral ligands (Boiteux et al. 2014). In the channel model with a sodium ion, the carbonyl oxygen of carbamazepine often bound the sodium ion at the pore axis and tricyclic moiety approached F4i15 (Fig. 4c). These results justified docking of other electroneutral ligands in the channel model with a sodium ion. The latter was initially placed at the NaIII site, but was not constrained to it and was free to move during MC-minimizations.
In the predicted complexes of the sodium channel with lamotrigine (Fig. 4d), the triazine-ring plane interacted with the sodium ion, the amino groups donated H-bonds to the backbone carbonyls p48, and the aromatic ring bound between F4i15 and Y4i22 and contacted V4i18 in agreement with the data that alanine substitutions of these residues affect lamotrigine action (Liu et al. 2003). In low-energy complexes of the channel with phenytoin, the sodium ion approached the ligand aromatic ring and bound to the carbonyl oxygen between two NH groups, which donated H-bonds to residues p48 and p49. For example, NH groups donated H-bonds to the T3p48 backbone and Q1p49 side chain. Ligands like phenytoin, carbamazepine, and lamotrigine have conserved mutual disposition of the O=C–NH or =N–C–NH moieties (Fig. 3). The models predict that the electronegative atoms would bind NaIII, while polar hydrogens would donate H-bonds to residues in positions p48 and p49. In such binding modes, a ligand would displace water molecules from the NaIII hydration shell and bridge NaIII to the channel oxygen atoms, thus strengthening the channel block.
Lacosamide does not block batrachotoxin-activated Nav1.5 channels (Wang and Wang 2014) indicating that its binding site overlaps with that of batrachotoxin, which is located in the inner pore (Tikhonov and Zhorov 2005c; Du et al. 2011). In some binding modes, (R)-lacosamide chelated a sodium ion at the NaIII site by two carbonyl oxygens, the NH groups approached oxygen atoms in positions p48 and p49, while the aromatic ring bound between F4i15 and Y4i22.
Bisphenol A is a pollutant whose action is sensitive to mutation F4i15A (O’Reilly et al. 2012). In the lowest-energy structure, two aromatic rings chelated NaIII by π-cations, whereas two hydroxyl groups donated H-bonds to diagonally opposed backbone carbonyls in position p48.
Common Binding Modes of Cationic and Electroneutral Ligands
Superposition of the channel-bound ligands (Fig. 5a) shows the ammonium nitrogen (blue) or a ligand-bound sodium ion (yellow) approaching the NaIII site and the most remote from this site atom (gray) below side chain of F4i15. Schematically these binding modes are shown in Fig. 5b where the ligand ammonium groups (top row) of ligand-bound sodium ion (bottom row) are at the level of cation-attractive site NaIII (red rectangle), whereas hydrophobic groups are at the levels of predominantly hydrophobic residues in the inner pore (gray rectangle). These binding modes suggest the electrostatic mechanism of the channel block. The ammonium group of a charged ligand would displace a permeant ion, whereas a neutral ligand would clamp an ion at its binding site. In any case, the ligand-associated charge would repel permeant cations and thus block the ion permeation. The binding modes are consistent with the pharmacophore model for LAs (Khodorov 1981), where the ammonium group and an aromatic ring are separated by four bonds (Fig. 5c). Many inner-pore blockers, e.g., ranolazine, do not fit this pharmacophore. However, compounds, which are structurally similar to the above-considered, are expected to have analogous binding modes and mechanisms of the channel block. It should be noted that the proposed mechanism is a hypothesis that needs testing by further experimental and computational studies.

(a) Cytoplasmic and side views at superposed channel-bound ligands. Blue and yellow spheres show, respectively, the ammonium nitrogens and ligand-bound sodium ions, which are clustered at the NaIII site. Gray spheres show carbon atoms, which are most remote from the NaIII site. These atoms are clustered below F4i15 (except for bisphenol A). (b) Scheme of ligand–channel interactions. The ligand-bound sodium ion or ammonium nitrogen are attracted to the NaIII site (red rectangle) and aromatic groups to the inner pore hydrophobic residues (gray rectangle). (c) A common pharmacophore of lidocaine and sodium-bound carbamazepine. Reproduced from Tikhonov and Zhorov (2017) (https://doi.org/10.1085/jgp.201611668) where coordinates of the models are available
Inner Pore Blockers in Therapy of Sodium Channelopathies
More than a thousand disease-causing mutations are identified in Nav1.X channels (Huang et al. 2017). The mutations are mapped in the pore domain, voltage-sensing domains, and extracellular and intracellular loops (Catterall 2014; Shen et al. 2017). For most of the channelopathy mutations, atomic mechanisms of the channel malfunctioning remain to be elucidated. Regardless, many sodium channelopathies are treated with small-molecule drugs (El-Sherif and Boutjdir 2015; Imbrici et al. 2016) whose channel-bound models are outlined above. For example, antiepileptic drugs like carbamazepine, phenytoin, and lamotrigine, which reduce neuronal hyperexcitability, are used not only for the therapy of various epileptic syndromes caused by mutations in the Nav1.1, Nav1.2, Nav1.3, or Nav1.6 channels, but also to treat migraine (Nav1.1 mutations), skeletal-muscle channelopathies (Nav1.4 mutations), and painful syndromes (mutations in Nav1.7, Nav1.8, or Nav1.9). Cardiac channelopathies are caused by mutations in various genes, including Nav1.5-encoding SCN5A. The first-choice treatment for the long-QT syndrome is β-adrenoceptor antagonists, but sodium channel blockers including mexiletine or ranolazine can be used as add-on therapy.
New gene- and mutation-specific drugs are desired. The 3D models of drug-channel complexes can assist in the drug development at the stages of lead discovery and lead optimization. Indeed, high-throughput in silico screening of millions purchasable molecules is used to select promising lead candidates for experimental high-throughput screening. The proposed sodium ion involvement in the action of electroneutral ligands suggests that different models should be used for in silico high-throughput ligand screenings. Models with sodium ions at the NaIII or NaIV sites should be used to find electroneutral lead hits, whereas models without ions at these sites should be used to find lead hits among cationic ligands. Repurposing of marketed drugs is another promising approach to treat sodium channelopathies. Such drugs do not need expensive safety tests, and their 3D complementarity to the sodium channel models can be tested in silico.
4 Neurotoxins
Tetrodotoxin binds in the outer pore of TTX-sensitive sodium channels. A sodium channel model with TTX and STX was proposed before any crystal structures of P-loop channels become available (Lipkind and Fozzard 1994). The model is based on mutational data that residues D1p50, E2p50, E1p54, E2p54, and D4p54 are important for the action of TTX and STX (Terlau et al. 1991). These data were further used to build TTX- and STX-bound sodium channel models based on X-ray structures of potassium channels (Lipkind and Fozzard 2000; Tikhonov and Zhorov 2005a). More recently, the same experimental data (Terlau et al. 1991) necessitated introduction of insertions–deletions in the NavAb/Nav1.4 sequence alignment (Fig. 2), which underlines a NavAb-based model of Nav1.4 with TTX (Tikhonov and Zhorov 2012). Superposition with the NavPaS structure (Shen et al. 2017) supports this model (Fig. 6a–c).

Intracellular (a), extracellular (b), and side (c) views at the TTX-bound model of Nav1.4 (gray bonds) and the NavPaS cryo-EM structure (green bonds) (Shen et al. 2017). TTX is space-filled and backbones are indicated as alpha carbon tracings. Coordinates of the TTX-bound model are available (Tikhonov and Zhorov 2012). (d) Cytoplasmic view at BTX in the inner pore. For clarity, only middle parts of S5s and S6s helices are shown. Semi-transparent van der Waals surfaces of the helices and BTX are gray and yellow, respectively. A sodium ion (orange sphere) binds between BTX oxygens and S1i15. BTX fits into the inner pore, but does not block it [Figure d was originally published in the Journal of Biological Chemistry (Du et al. 2011). © The American Society for Biochemistry and Molecular Biology]
μ-Conotoxins
The TTX receptor model (Tikhonov and Zhorov 2012) was further employed to dock peptide toxins GIIIA, PIIIA, and KIIIA (Korkosh et al. 2014). The mutant cycle analysis, which provides energies of pairwise interactions between individual residues in the channel and toxin (Chang et al. 1998; Choudhary et al. 2007), facilitated docking of GIIIA and resulted in the model where computed and experimental pairwise energies correlate. The four outer carboxylates (E1p53, E2p53, D3p54, and D4p53), which reportedly contribute to binding of permeant ions (Khan et al. 2002), are involved in the GIIIA binding. Computations further predicted how deep the charged residues of PIIIA penetrate from the extracellular space into the outer pore (Korkosh et al. 2014) and these estimates are in good agreement with the experimental data (McArthur et al. 2011b). The intriguing data that TTX and KIIIA can simultaneously bind to the sodium channel (Zhang et al. 2009) are rationalized in the model where TTX can bypass the channel-bound KIIIA and reach its receptor in the outer pore (Korkosh et al. 2014). The model further explained why some native and mutant conotoxins incompletely block the channel (Hui et al. 2002; McArthur et al. 2011a; Wilson et al. 2011). A toxin completely blocks the current if its basic residues salt-bridge with the four outer carboxylates of the channel. If a toxin lacks some of the basic residues, at least one of the outer carboxylates does not salt-bridge with the toxin and sodium ions can permeate, but slower than in the toxin-free channel.
Batrachotoxin (BTX) is a sodium channel agonist that has long been used in electrophysiological studies. The bulky steroidal molecule initially was thought to bind at the channel-lipid interface and allosterically activate the channel. However, BTX-sensing residues were found in all the four pore-lining inner helices, partially overlapping with LA-sensing residues (Wang and Wang 2003). Subsequent modeling study proposed that BTX and other sodium channel agonists (veratridine and aconitine) would bind in the inner pore, prevent the activation-gate closure, and allow sodium ions to move between the agonist hydrophilic face and the channel hydrophilic residues (Tikhonov and Zhorov 2005b). The model predicted BTX-binding residues within the pore and experiments confirmed these predictions (Wang et al. 2006, 2007a, 2007b). In particular, when the pore-facing N2i15 was replaced with lysine, BTX irreversibly blocked the channel (Wang et al. 2007a) implying involvement of N2i15 in the BTX action. Later, additional BTX-sensing residues were found and a model was elaborated to integrate available experimental data (Du et al. 2011). In this model, the horseshoe-shaped BTX is engaged in cation-π interactions with F3i16 and contacts BTX-sensing residues in all four repeats (Fig. 6d). Oxygen atoms at the horseshoe inner surface constitute a transient binding site for permeant ions, while the bulky BTX would resist the activation gate closure. To some extent, the model of BTX in the sodium channel resembles a surgical stent in a blood vessel.
Peptide Toxins Targeting VSDs
Peptide toxins produced by scorpions, spiders, and sea anemones bind to the extracellular loops in VSDs thus modifying the channel gating. α-Toxins produced by scorpions (Wang et al. 2011; Zhang et al. 2011, 2012a), spiders (Bosmans and Swartz 2010; Minassian et al. 2014), and sea anemones (Xiao et al. 2014) slow down inactivation, whereas β-toxins trap VSD-II in the activated state, causing a negative shift of activation. The extracellular loops of VSD-II and VSD-IV are involved in binding of α- and β-scorpion toxins, respectively (Thomsen and Catterall 1989; Rogers et al. 1996; Cestele et al. 2006) (Fig. 1). β-Toxin sensing residues are also found in loop IIIP2–S6 (Zhang et al. 2012a). Mutational studies helped elaborate models of scorpion toxin action (Zhang et al. 2012a). β-Toxin CssIV is proposed to bind deeply in a slot between loops IIS1–S2 and IIS3–S4, whereas α-toxins appear to fit a tapered slot between extracellular loops IVS1–S2 and IVS3–S4 (Wang et al. 2011). The different effects of α-toxins (inhibition of fast-inactivation) and β-toxins (enhancement of activation) may be due to functional asymmetry of the channel repeats. Thus, the VSD-I and VSD-II rapidly move in response to depolarization, whereas VSD-IV moves slowly (Chanda and Bezanilla 2002). A model with Huwentoxin-IV in the cleft between extracellular loops IIS1–S2 and IIS3–S4 (Minassian et al. 2014) is similar to that with the α-scorpion toxin.
Insecticides
Pyrethroid insecticides are synthetic analogs of natural pyrethrins from the pyrethrum daisy flowers. They are widely used to control deleterious arthropod pests and mosquito-borne diseases, including malaria and dengue (Dong et al. 2014). Intensive use of DDT and pyrethroids has led to the development of knockdown resistance (kdr) in many arthropod pests (Silver et al. 2014), motivating development of new insecticides. Structural models integrate mutational data, including kdr mutations, and predict two pyrethroid receptors, PyR1 and PyR2, in the lipid-exposed interfaces between repeats II/III and I/II, respectively (O’Reilly et al. 2006; Du et al. 2013, 2015). Ligand binding to both PyR1 and PyR2 seems necessary to activate the channel. DDT is further predicted to bind at two sites that overlap with PyR1 and PyR2 (Du et al. 2016). The model-driven mutagenesis allowed to discover new pyrethroid- and DDT-sensing residues (Du et al. 2015, 2016).
Indoxacarb, its active metabolite DCJW, and metaflumizone are electroneutral non-ionizable ligands that belong to a relatively new class of sodium channel blocker insecticides (SCBIs). In the open insect sodium channel model, SCBIs bind in the inner pore, interact with a sodium ion at the focus of P1 helices, and extend an aromatic moiety into the III/IV fenestration. Model-driven mutagenesis revealed new SCBI-sensing residues, including insect-specific ones (Zhang et al. 2016).
5 Conclusion
In the absence of experimental 3D structures of eukaryotic sodium channels with ligands, computational structural modeling can be used to visualize ligand–channel complexes, provide mechanistic rationale for mutational, electrophysiological, ligand-binding, and other experimental data, help design new experiments, and may assist in development of new drugs. However, precision of ligand-bound sodium channel models is limited and some models may be incorrect. A key criterion of a model validity is its consistency with a large body of experimental data and, especially, the ability to rationalize experimental data, which were not used at the stage of the model building.
Abbreviations
- DDT:
-
Dichlorodiphenyltrichloroethane
- EM:
-
Electron microscopy
- LAs:
-
Local anesthetics
- MC:
-
Monte Carlo
- MCM:
-
MC minimization
- MD:
-
Molecular dynamics
- PyR:
-
Pyrethroid receptor
- SCBIs:
-
Sodium channel blocker insecticides
- STX:
-
Saxitoxin
- TTX:
-
Tetrodotoxin
- VSD:
-
Voltage-sensing domain
References
Ahern CA, Eastwood AL, Dougherty DA, Horn R (2008) Electrostatic contributions of aromatic residues in the local anesthetic receptor of voltage-gated sodium channels. Circ Res 102(1):86–94
Ahuja S, Mukund S, Deng L, Khakh K, Chang E, Ho H et al (2015) Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science 350(6267):aac5464
Bagneris C, DeCaen PG, Naylor CE, Pryde DC, Nobeli I, Clapham DE et al (2014) Prokaryotic NavMs channel as a structural and functional model for eukaryotic sodium channel antagonism. Proc Natl Acad Sci U S A 111(23):8428–8433
Bean BP, Cohen CJ, Tsien RW (1983) Lidocaine block of cardiac sodium channels. J Gen Physiol 81(5):613–642
Boiteux C, Vorobyov I, French RJ, French C, Yarov-Yarovoy V, Allen TW (2014) Local anesthetic and antiepileptic drug access and binding to a bacterial voltage-gated sodium channel. Proc Natl Acad Sci U S A 111(36):13057–13062
Bosmans F, Swartz KJ (2010) Targeting voltage sensors in sodium channels with spider toxins. Trends Pharmacol Sci 31(4):175–182
Browne LE, Blaney FE, Yusaf SP, Clare JJ, Wray D (2009) Structural determinants of drugs acting on the Nav1.8 channel. J Biol Chem 284(16):10523–10536
Bruhova I, Tikhonov DB, Zhorov BS (2008) Access and binding of local anesthetics in the closed sodium channel. Mol Pharmacol 74(4):1033–1045
Catterall WA (1987) Common modes of drug action on Na+ channels: local anesthetics, antiarrhythmics and anticonvulsants. Trends Pharmacol Sci 8(2):57–65
Catterall WA (2012) Voltage-gated sodium channels at 60: structure, function and pathophysiology. J Physiol 590(11):2577–2589
Catterall WA (2014) Sodium channels, inherited epilepsy, and antiepileptic drugs. Annu Rev Pharmacol Toxicol 54:317–338
Catterall WA, Swanson TM (2015) Structural basis for pharmacology of voltage-gated sodium and calcium channels. Mol Pharmacol 88(1):141–150
Cestele S, Yarov-Yarovoy V, Qu Y, Sampieri F, Scheuer T, Catterall WA (2006) Structure and function of the voltage sensor of sodium channels probed by a beta-scorpion toxin. J Biol Chem 281(30):21332–21344
Chanda B, Bezanilla F (2002) Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol 120(5):629–645
Chang NS, French RJ, Lipkind GM, Fozzard HA, Dudley S Jr (1998) Predominant interactions between mu-conotoxin Arg-13 and the skeletal muscle Na+ channel localized by mutant cycle analysis. Biochemistry 37(13):4407–4419
Choudhary G, Aliste MP, Tieleman DP, French RJ, Dudley SC Jr (2007) Docking of mu-conotoxin GIIIA in the sodium channel outer vestibule. Channels (Austin) 1(5):344–352
Dong K, Du Y, Rinkevich F, Nomura Y, Xu P, Wang L et al (2014) Molecular biology of insect sodium channels and pyrethroid resistance. Insect Biochem Mol Biol 50:1–17
Du Y, Garden D, Wang L, Zhorov BS, Dong K (2011) Identification of new batrachotoxin-sensing residues in segment IIIS6 of sodium channel. J Biol Chem 286(15):13151–13160
Du Y, Nomura Y, Satar G, Hu Z, Nauen R, He SY et al (2013) Molecular evidence for dual pyrethroid-receptor sites on a mosquito sodium channel. Proc Natl Acad Sci U S A 110(29):11785–11790
Du Y, Nomura Y, Zhorov BS, Dong K (2015) Rotational symmetry of two pyrethroid receptor sites in the mosquito sodium channel. Mol Pharmacol 88(2):273–280
Du Y, Nomura Y, Zhorov BS, Dong K (2016) Evidence for dual binding sites for 1,1,1-Trichloro-2,2-bis(p-chlorophenyl)ethane (DDT) in insect sodium channels. J Biol Chem 291(9):4638–4648
El-Sherif N, Boutjdir M (2015) Role of pharmacotherapy in cardiac ion channelopathies. Pharmacol Ther 155:132–142
Garden DP, Zhorov BS (2010) Docking flexible ligands in proteins with a solvent exposure- and distance-dependent dielectric function. J Comput Aided Mol Des 24(2):91–105
Hille B (1977) Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol 69(4):497–515
Huang W, Liu M, Yan SF, Yan N (2017) Structure-based assessment of disease-related mutations in human voltage-gated sodium channels. Protein Cell 8(6):401–438
Hui K, Lipkind G, Fozzard HA, French RJ (2002) Electrostatic and steric contributions to block of the skeletal muscle sodium channel by mu-conotoxin. J Gen Physiol 119(1):45–54
Imbrici P, Liantonio A, Camerino GM, De Bellis M, Camerino C, Mele A et al (2016) Therapeutic approaches to genetic ion channelopathies and perspectives in drug discovery. Front Pharmacol 7:121
Jensen MO, Jogini V, Borhani DW, Leffler AE, Dror RO, Shaw DE (2012) Mechanism of voltage gating in potassium channels. Science 336(6078):229–233
Khan A, Romantseva L, Lam A, Lipkind G, Fozzard HA (2002) Role of outer ring carboxylates of the rat skeletal muscle sodium channel pore in proton block. J Physiol 543(Pt 1):71–84
Khodorov BI (1981) Sodium inactivation and drug-induced immobilization of the gating charge in nerve membrane. Prog Biophys Mol Biol 37(2):49–89
Korkosh VS, Zhorov BS, Tikhonov DB (2014) Folding similarity of the outer pore region in prokaryotic and eukaryotic sodium channels revealed by docking of conotoxins GIIIA, PIIIA, and KIIIA in a NavAb-based model of Nav1.4. J Gen Physiol 144(3):231–244
Kuo CC (1998) A common anticonvulsant binding site for phenytoin, carbamazepine, and lamotrigine in neuronal Na+ channels. Mol Pharmacol 54(4):712–721
Lenaeus MJ, Gamal El-Din TM, Ing C, Ramanadane K, Pomes R, Zheng N et al (2017) Structures of closed and open states of a voltage-gated sodium channel. Proc Natl Acad Sci U S A 114(15):E3051–E3060
Lipkind GM, Fozzard HA (1994) A structural model of the tetrodotoxin and saxitoxin binding site of the Na+ channel. Biophys J 66(1):1–13
Lipkind GM, Fozzard HA (2000) KcsA crystal structure as framework for a molecular model of the Na(+) channel pore. Biochemistry 39(28):8161–8170
Lipkind GM, Fozzard HA (2005) Molecular modeling of local anesthetic drug binding by voltage-gated sodium channels. Mol Pharmacol 68(6):1611–1622
Lipkind GM, Fozzard HA (2010) Molecular model of anticonvulsant drug binding to the voltage-gated sodium channel inner pore. Mol Pharmacol 78(4):631–638
Liu G, Yarov-Yarovoy V, Nobbs M, Clare JJ, Scheuer T, Catterall WA (2003) Differential interactions of lamotrigine and related drugs with transmembrane segment IVS6 of voltage-gated sodium channels. Neuropharmacology 44(3):413–422
Marzian S, Stansfeld PJ, Rapedius M, Rinne S, Nematian-Ardestani E, Abbruzzese JL et al (2013) Side pockets provide the basis for a new mechanism of Kv channel-specific inhibition. Nat Chem Biol 9(8):507–513
McArthur JR, Ostroumov V, Al-Sabi A, McMaster D, French RJ (2011a) Multiple, distributed interactions of mu-conotoxin PIIIA associated with broad targeting among voltage-gated sodium channels. Biochemistry 50(1):116–124
McArthur JR, Singh G, O’Mara ML, McMaster D, Ostroumov V, Tieleman DP et al (2011b) Orientation of mu-conotoxin PIIIA in a sodium channel vestibule, based on voltage dependence of its binding. Mol Pharmacol 80(2):219–227
Mike A, Lukacs P (2010) The enigmatic drug binding site for sodium channel inhibitors. Curr Mol Pharmacol 3(3):129–144
Minassian NA, Gibbs A, Shih AY, Liu Y, Neff RA, Sutton SW et al (2014) Analysis of the structural and molecular basis of voltage-sensitive sodium channel inhibition by the spider toxin huwentoxin-IV (mu-TRTX-Hh2a). J Biol Chem 288(31):22707–22720
Naylor CE, Bagneris C, DeCaen PG, Sula A, Scaglione A, Clapham DE et al (2016) Molecular basis of ion permeability in a voltage-gated sodium channel. EMBO J 35:820–830
O’Leary ME, Chahine M (2002) Cocaine binds to a common site on open and inactivated human heart (Na(v)1.5) sodium channels. J Physiol 541(Pt 3):701–716
O’Reilly AO, Khambay BP, Williamson MS, Field LM, Wallace BA, Davies TG (2006) Modelling insecticide-binding sites in the voltage-gated sodium channel. Biochem J 396(2):255–263
O’Reilly AO, Eberhardt E, Weidner C, Alzheimer C, Wallace BA, Lampert A (2012) Bisphenol A binds to the local anesthetic receptor site to block the human cardiac sodium channel. PLoS One 7(7):e41667
Payandeh J, Scheuer T, Zheng N, Catterall WA (2011) The crystal structure of a voltage-gated sodium channel. Nature 475(7356):353–358
Qu Y, Rogers J, Tanada T, Scheuer T, Catterall WA (1995) Molecular determinants of drug access to the receptor site for antiarrhythmic drugs in the cardiac Na+ channel. Proc Natl Acad Sci U S A 92(25):11839–11843
Ragsdale DS, McPhee JC, Scheuer T, Catterall WA (1994) Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 265(5179):1724–1728
Ragsdale DS, McPhee JC, Scheuer T, Catterall WA (1996) Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc Natl Acad Sci U S A 93(17):9270–9275
Rogers JC, Qu Y, Tanada TN, Scheuer T, Catterall WA (1996) Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J Biol Chem 271(27):15950–15962
Scheib H, McLay I, Guex N, Clare JJ, Blaney FE, Dale TJ et al (2006) Modeling the pore structure of voltage-gated sodium channels in closed, open, and fast-inactivated conformation reveals details of site 1 toxin and local anesthetic binding. J Mol Model 12(6):813–822
Shen H, Zhou Q, Pan X, Li Z, Wu J, Yan N (2017) Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 355(6328):eaal4326
Silver KS, Du Y, Nomura Y, Oliveira EE, Salgado VL, Zhorov BS et al (2014) Voltage-gated sodium channels as insecticide targets. In: Cohen E (ed) Advances in insect physiology, vol 46. Academic Press, Oxford
Stevens M, Peigneur S, Tytgat J (2011) Neurotoxins and their binding areas on voltage-gated sodium channels. Front Pharmacol 2:71
Sula A, Booker J, Ng LC, Naylor CE, DeCaen PG, Wallace BA (2017) The complete structure of an activated open sodium channel. Nat Commun 8:14205
Sunami A, Dudley SC Jr, Fozzard HA (1997) Sodium channel selectivity filter regulates antiarrhythmic drug binding. Proc Natl Acad Sci U S A 94(25):14126–14131
Tang L, Gamal El-Din TM, Payandeh J, Martinez GQ, Heard TM, Scheuer T et al (2014) Structural basis for Ca2+ selectivity of a voltage-gated calcium channel. Nature 505(7481):56–61
Tang L, Gamal El-Din TM, Swanson TM, Pryde DC, Scheuer T, Zheng N et al (2016) Structural basis for inhibition of a voltage-gated Ca2+ channel by Ca2+ antagonist drugs. Nature 537(7618):117–121
Terlau H, Heinemann SH, Stuhmer W, Pusch M, Conti F, Imoto K et al (1991) Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett 293(1–2):93–96
Thomsen WJ, Catterall WA (1989) Localization of the receptor site for alpha-scorpion toxins by antibody mapping: implications for sodium channel topology. Proc Natl Acad Sci U S A 86(24):10161–10165
Tikhonov DB, Zhorov BS (2005a) Modeling P-loops domain of sodium channel: homology with potassium channels and interaction with ligands. Biophys J 88(1):184–197
Tikhonov DB, Zhorov BS (2005b) Sodium channel activators: model of binding inside the pore and a possible mechanism of action. FEBS Lett 579(20):4207–4212
Tikhonov DB, Zhorov BS (2007) Sodium channels: ionic model of slow inactivation and state-dependent drug binding. Biophys J 93(5):1557–1570
Tikhonov DB, Zhorov BS (2012) Architecture and pore block of eukaryotic voltage-gated sodium channels in view of NavAb bacterial sodium channel structure. Mol Pharmacol 82(1):97–104
Tikhonov DB, Zhorov BS (2017) Mechanism of sodium channel block by local anesthetics, antiarrhythmics, and anticonvulsants. J Gen Physiol 149(4):465–481
Tikhonov DB, Bruhova I, Zhorov BS (2006) Atomic determinants of state-dependent block of sodium channels by charged local anesthetics and benzocaine. FEBS Lett 580(26):6027–6032
Wang SY, Wang GK (2003) Voltage-gated sodium channels as primary targets of diverse lipid-soluble neurotoxins. Cell Signal 15(2):151–159
Wang GK, Wang SY (2014) Block of human cardiac sodium channels by lacosamide: evidence for slow drug binding along the activation pathway. Mol Pharmacol 85(5):692–702
Wang SY, Mitchell J, Tikhonov DB, Zhorov BS, Wang GK (2006) How batrachotoxin modifies the sodium channel permeation pathway: computer modeling and site-directed mutagenesis. Mol Pharmacol 69(3):788–795
Wang SY, Tikhonov DB, Mitchell J, Zhorov BS, Wang GK (2007a) Irreversible block of cardiac mutant Na+ channels by batrachotoxin. Channels (Austin) 1(3):179–188
Wang SY, Tikhonov DB, Zhorov BS, Mitchell J, Wang GK (2007b) Serine-401 as a batrachotoxin- and local anesthetic-sensing residue in the human cardiac Na+ channel. Pflugers Arch 454(2):277–287
Wang J, Yarov-Yarovoy V, Kahn R, Gordon D, Gurevitz M, Scheuer T et al (2011) Mapping the receptor site for alpha-scorpion toxins on a Na+ channel voltage sensor. Proc Natl Acad Sci U S A 108(37):15426–15431
Wilson MJ, Yoshikami D, Azam L, Gajewiak J, Olivera BM, Bulaj G et al (2011) Mu-conotoxins that differentially block sodium channels NaV1.1 through 1.8 identify those responsible for action potentials in sciatic nerve. Proc Natl Acad Sci U S A 108(25):10302–10307
Xiao Y, Blumenthal K, Cummins TR (2014) Gating-pore currents demonstrate selective and specific modulation of individual sodium channel voltage-sensors by biological toxins. Mol Pharmacol 86(2):159–167
Yamagishi T, Xiong W, Kondratiev A, Velez P, Mendez-Fitzwilliam A, Balser JR et al (2009) Novel molecular determinants in the pore region of sodium channels regulate local anesthetic binding. Mol Pharmacol 76(4):861–871
Yarov-Yarovoy V, McPhee JC, Idsvoog D, Pate C, Scheuer T, Catterall WA (2002) Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel alpha subunit in voltage-dependent gating and drug block. J Biol Chem 277(38):35393–35401
Zhang MM, McArthur JR, Azam L, Bulaj G, Olivera BM, French RJ et al (2009) Synergistic and antagonistic interactions between tetrodotoxin and mu-conotoxin in blocking voltage-gated sodium channels. Channels (Austin) 3(1):32–38
Zhang JZ, Yarov-Yarovoy V, Scheuer T, Karbat I, Cohen L, Gordon D et al (2011) Structure-function map of the receptor site for beta-scorpion toxins in domain II of voltage-gated sodium channels. J Biol Chem 286(38):33641–33651
Zhang X, Ren W, DeCaen P, Yan C, Tao X, Tang L et al (2012a) Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 486(7401):130–134
Zhang JZ, Yarov-Yarovoy V, Scheuer T, Karbat I, Cohen L, Gordon D et al (2012b) Mapping the interaction site for a beta-scorpion toxin in the pore module of domain III of voltage-gated Na(+) channels. J Biol Chem 287(36):30719–30728
Zhang Y, Du Y, Jiang D, Behnke C, Nomura Y, Zhorov BS et al (2016) The receptor site and mechanism of action of sodium channel blocker insecticides. J Biol Chem 291(38):20113–20124
Zhorov BS, Ananthanarayanan VS (1996) Structural model of a synthetic Ca2+ channel with bound Ca2+ ions and dihydropyridine ligand. Biophys J 70(1):22–37
Zhorov BS, Tikhonov DB (2004) Potassium, sodium, calcium and glutamate-gated channels: pore architecture and ligand action. J Neurochem 88(4):782–799
Zhorov BS, Tikhonov DB (2013) Ligand action on sodium, potassium, and calcium channels: role of permeant ions. Trends Pharmacol Sci 34(3):154–161
Zhorov BS, Tikhonov DB (2016) Computational structural pharmacology and toxicology of voltage-gated sodium channels. Curr Top Membr 78:117–144
Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R (2001) Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 a resolution. Nature 414(6859):43–48
Acknowledgments
This work was supported by grant 17-15-01292 from the Russian Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Zhorov, B.S. (2017). Structural Models of Ligand-Bound Sodium Channels. In: Chahine, M. (eds) Voltage-gated Sodium Channels: Structure, Function and Channelopathies. Handbook of Experimental Pharmacology, vol 246. Springer, Cham. https://doi.org/10.1007/164_2017_44
Download citation
DOI: https://doi.org/10.1007/164_2017_44
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-90283-8
Online ISBN: 978-3-319-90284-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)