
Abstract
Cnidarians have historically served as excellent laboratory models for regenerative development given their capacity to regrow large portions of the adult organism. This capacity is notably absent or poorly developed in the powerful genetic laboratory models Drosophila, C. elegans, and mouse. Increasingly, development of genetic and genomic resources and the application of next-generation sequencing-based techniques in cnidarian systems has further expanded the potential of cnidarian regenerative models. Here, we present a workflow for the characterization of the regenerative response in the sea anemone Nematostella vectensis utilizing fluorescence-activated cell sorting and a plate-based single-cell RNA-sequencing pipeline. This approach can characterize the transcriptional response during regeneration in distinct populations of cells, thus providing a quantitative view of a whole organism process at cellular resolution.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
The ability to regenerate is widespread among marine invertebrates. Understanding the molecular and developmental basis of regeneration in a diversity of taxa has the potential to provide insight into shared principles of the regenerative response. Cnidarians (Hydra, jellyfish, sea anemones, corals) belong to one of the earliest-branching metazoan phyla and are well known for their extensive regenerative abilities [1,2,3]. The sea anemone Nematostella vectensis is an experimentally tractable Cnidarian species for studies into mechanisms of regeneration. Significant advances in genetic and genomic resources, ease of culture, and conservation of gene content with Bilaterian taxa contribute to the utility of this sea anemone as a developmental and regenerative model system [4,5,6].
An experimentally bisected Nematostella vectensis polyp can regenerate two whole animals in about a week [1]. How the sea anemone regenerates lost tissue, maintains axial coordinates, and generates the appropriate number and distribution of constituent cell types during regeneration or asexual reproduction is not well understood. Studies focusing on the cellular dynamics following oral pole regeneration in juvenile polyps have highlighted the involvement of cellular proliferation and apoptosis [1, 7]. Whole-animal transcriptomic time-courses have been employed to uncover genes modulated during the regeneration process [8,9,10]. However, the behaviors of individual cell populations during regeneration have not been molecularly or functionally characterized in Nematostella vectensis thus far [11]. In order to better characterize the process of regeneration, additional tools that allow for the characterization of behaviors of single cells are needed. As regeneration is dynamic, gaining an understanding of the molecular events happening in individual cells throughout this process is critical.
Over the past few years, newly developed single-cell RNA-Sequencing (scRNA-Seq) techniques have enabled investigators to sample the transcriptome of single cells from complex tissues and whole organisms, to assess their developmental trajectories and reconstruct gene regulatory networks governing cell identity and function [12]. Different single-cell sequencing technologies have been developed ranging from plate-based, droplet or microfluidic platforms, each with distinct advantages and limitations [13]. Recently, single-cell RNA-Seq of whole Nematostella vectensis polyps and planula larvae via a plate-based approach, MARS-seq, has enabled the generation of a molecular atlas of the cell types present in the sea anemone [14]. In combination with targeted genetic labeling and trajectory inference methods, scRNA-Seq data from regenerating animals could enable the reconstruction of key cellular trajectories, as well as the relative contributions of different cell populations to the newly regenerated tissues.
From cell suspension, most scRNA-seq protocols rely on a similar workflow, where single cells are physically isolated and lysed, in wells or droplets, captured mRNA molecules are reverse transcribed and amplified, and multiplexed dsDNA libraries are generated and sequenced [13]. Complete, high-viability dissociation of starting tissue is a critical pre-requisite of scRNA-seq. Single cells can then be isolated through Fluorescence-activated cell sorting (FACS ), which enables a single cell to be separated from a cell suspension with a high degree of purity. Following single-cell isolation, mature mRNAs are often captured through their polyadenylated tails using barcoded oligos bearing polyT stretches, then reverse transcribed into cDNAs before being amplified via PCR or in vitro transcription [15]. During capture or in subsequent steps, cell- and/or pool-specific barcodes are added, to enable attribution of the sequenced reads to a specific cell and sample of origin. To limit the impact of amplification artifacts, unique molecular identifiers (UMIs), short stretches of random DNA bases, are attached to the captured mRNA molecules [15, 16]. Duplicate reads from the same mRNA molecule can be merged by UMI. For data processing and analysis, several quality control and clustering analysis packages have been developed [17,18,19].
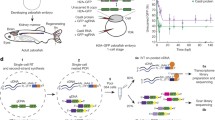
In this protocol , we focus on a single-cell RNA-sequencing experiment in a regenerating Nematostella vectensis polyp including the dissociation of samples of interest into live single-cell suspensions, the flow cytometer parameters and gating strategy to sort live single cells into separate wells of a 384-well plate, as well as an overview of quality control steps after library sequencing, read demultiplexing and mapping, QCs, and general notes and considerations specific to single-cell RNA-Seq Nematostella vectensis data (Fig. 1). Briefly, polyps along a regeneration time-course following oral pole amputation are dissociated into single-cell suspensions at timepoints of interest (Fig. 1a), then live single cells are sorted on a flow cytometer into single wells of a 384-well plate (Fig. 1b). Following cell lysis and mRNA capture with barcoded oligonucleotides in individual wells, multiplexed cDNA libraries are generated using the MARS-Seq single-cell RNA-Seq method [15, 20] (Fig. 1c). The scRNA-seq library preparation method has been adapted from an established workflow and an extensive protocol detailing the experimental steps, and an associated computational pipeline has recently been published [20]. After library sequencing, reads are filtered, demultiplexed, and mapped using the MARS-Seq computational pipeline onto the Nematostella genome to generate count tables, listing the number of recovered molecules from each gene in each sequenced cell [20, 21] (Fig. 1d). The count tables can then be filtered and clustered using various clustering methods for biological analyses [22] (Fig. 1d). Here, we generate an example clustering using the MetaCell clustering package, a published pipeline for the analysis of scRNA-seq data [17].

Overview of single-cell RNA Sequencing in the regenerating sea anemone Nematostella vectensis . (a) DIC images of the oral region of uncut control, 2 and 6 day post-amputation (2 dpa and 6 dpa) juvenile Nematostella vectensis polyps (~5 weeks) at room temperature. Amputation site is indicated with a red line. (b) Enzymatic dissociation of sample into a single-cell suspension and single-cell FACS sorting into 384-well plates. (c) Simplified overview of the labeling strategy for single-cell-derived mRNA which includes barcoding for each cell in each well using capture oligonucleotides with a polyT-tract, a random unique molecular identifier (UMI) and a cell barcode. (d) Multiplexed single-cell RNA-seq short-read libraries are sequenced, reads are filtered, mapped, and demultiplexed to attribute recovered molecules, through their unique UMIs tags, to their cell of origin. Cells are then clustered using unsupervised clustering based on gene expression for downstream analyses
2 Materials
All solutions should be prepared using ultrapure water and stored at room temperature (RT) unless otherwise stated.
-
1.
Nematostella medium: 12.8 g/L artificial seawater salt in distilled water.
-
2.
24 h starved 5- to 8-week-old juvenile animals.
-
3.
Six-well plates.
-
4.
Sterile scalpel blades.
-
5.
3-mL plastic Pasteur pipets.
-
6.
7% (w/v) magnesium chloride solution in ultrapure water.
-
7.
Low-binding 1.5-mL tubes (e.g., Biozym #710176).
-
8.
Calcium-Magnesium-Free Artificial Seawater (CMFSW) for Nematostella dissociation medium: 495 mM NaCl, 9.7 mM KCl, 27.6 mM NaHCO3, 50 mM Tris–HCl pH 8. Filter sterilize.
-
9.
Live cell marker: 1 μg/μL calcein AM resuspended in dimethyl sulfoxide (DMSO). Protect from light and store at −20 °C (see Note 1).
-
10.
Dead cell marker: 1 mg/mL propidium iodide ready-to-use solution. Protect from light and store at 4 °C (see Note 1).
-
11.
Collagenase solution: 5 mg/mL LiberaseTM resuspended in nuclease-free water. Store aliquots at −20 °C.
-
12.
Gelatin solution: 1 mg/mL gelatin. Heat to dissolve, filter.
-
13.
Gelatin-coated P200 tips: pipette up and down gelatin solution, let the tips dry overnight, store at RT.
-
14.
Gelatin-coated P1000 tips.
-
15.
Trimmed P200 gelatin-coated tips: remove 1–2 mm of dry coated tip length using a scalpel blade.
-
16.
Dissociation medium: 32.4 mL CMFSW, 17.6 mL ultrapure H2O, filter at 0.4 μm. Check salinity against sample salinity.
-
17.
Staining medium: 2 μL/mL live cell marker, 3 μL/mL dead cell marker in dissociation medium. Prepare fresh. Protect from light.
-
18.
Fluorescence-Activated Cell Sorter (e.g., BD FACS ARIAIII), with laser/filters combinations appropriate for the dyes used (see Note 2).
-
19.
Flow cytometer sizing beads: 1.34 μm, 3 μm, and 6 μm diameter.
-
20.
Frozen cell capture plates: 384-wells plates filled with 2 μL polyT barcoded capture oligos in lysis solution stored at −80 °C (see Note 3).
-
21.
Nematostella vectensis genome and gene annotation (see Note 4).
-
22.
MARS-Seq library preparation pipeline (see Note 5).
-
23.
DNA electrophoresis equipment (e.g., TapeStation, Agilent).
-
24.
DNA concentration fluorometer (e.g., Qubit, Thermo Fisher Scientific).
-
25.
MARS-Seq computational pipeline (see Note 6).
-
26.
scRNA-seq data clustering pipeline (see Note 7).
3 Methods
3.1 Cell Dissociation of Regenerated and Control Samples
-
1.
Select 5- to 8-week-old siblings of similar size (see Note 8).
-
2.
Transfer one polyp into the lid of a 5-cm plastic dish filled with 1.5 mL Nematostella medium.
-
3.
Repeat step 2 for the remaining siblings, in separate lids.
-
4.
Wait for ~20 min for the polyp to relax, until the tentacles are fully extended.
-
5.
Add drop-by-drop, on the side of the lid, 1.5 mL of 7% MgCl2.
-
6.
Wait for 30 s for full immobilization (see Note 9).
-
7.
Amputate the oral end by cutting perpendicularly to the oral-aboral axis below the pharynx in one straight cut. See red line in Fig. 1a for the amputation plane used here.
-
8.
Gently transfer the aboral end to a well of a six-well plate filled with ~3 mL Nematostella medium using a plastic Pasteur pipette with a wider opening (see Note 10).
-
9.
Keep the plate at 22 °C.
-
10.
Repeat steps 5–8 three more times to transfer the replicates to separate wells.
-
11.
Transfer the two control polyps to the remaining wells.
-
12.
Keep the plate in an incubator at 22 °C.
-
13.
Do not feed control or regenerating animals.
-
14.
Wait until the desired timepoint is reached (see Note 11).
-
15.
Transfer each sample in a separate 1.5-mL low-binding tube using a plastic pipette.
-
16.
Spin down each sample by brief centrifugation (<1 s) on a tabletop centrifuge.
-
17.
Remove the superficial liquid layer, while keeping each sample immersed.
-
18.
Add 1 mL of dissociation medium to wash.
-
19.
Repeat steps 16–18 for another wash step.
-
20.
Adjust total volume to 300 μL with dissociation medium (see Note 12).
-
21.
Add 3 μL of thawed liberaseTM solution.
-
22.
Gently pipette up and down slowly at RT (>1.5 s per pipetting cycle) using a trimmed P200 gelatin-coated tip (see Note 13). Use different tips for each sample to avoid cross-contamination.
-
23.
Transition to an uncut gelatin-coated P200 tip with a narrower opening when the diameter of the remaining particles is small enough to pass through the new opening.
-
24.
Add 30 μL of 0.5 M EDTA to stop the dissociation.
-
25.
Add 420 μL of dissociation medium.
-
26.
Gently pipette up and down slowly with gelatin-coated P1000 tips to homogenize.
-
27.
Fill the P1000 tip with the cell suspension.
-
28.
Place the tip directly atop the filtering cap of the 5-mL FACS tube, at an angle.
-
29.
Gently eject the cell through the filter, aiming at the side of the tube (see Note 14).
-
30.
Add 750 μL of staining medium.
-
31.
Slowly pipette up and down with a gelatin-coated P1000 tip to mix.
-
32.
Protect from light and place tubes in a tube holder atop ice.
-
33.
Bring tubes to the flow cytometer facility (see Note 15).
3.2 Live Single-Cell Sorting Using Flow Cytometry for Single-Cell RNA-Sequencing
-
1.
Initialize flow cytometer according to manufacturer’s instructions , including laser alignment testing and time delay setup for sorting (see Note 16).
-
2.
Adjust plate holder arm coordinates so that the sort stream deposits a single droplet within the center of each well of a cell capture plate (see Note 17).
-
3.
Display the forward scatter-area (FSC-A) signal and side scatter-area (SSC-A) signal in the biexponential scale for all samples and adjust the photomultipliers (PMT) voltages and thresholds to visualize the dynamic range of particles (Fig. 2a, b) (see Note 18).
-
4.
Select singlet particles from all the events by excluding multiplets based on their forward scatter-width vs. forward scatter-height signal (gated population is in blue in Fig. 2c).
-
5.
Set the photomultiplier voltages for the channels associated to the live and dead cell dyes fluorescent channels using positive (stained) and negative (fluorescence-minus one—FMO) populations (see Note 19).
-
6.
Select live singlet particles from the singlet population (in blue in Fig. 2c) by gating the populations according to the live cell marker and dead cell marker (gated population in purple in Fig. 2d).
-
7.
Select the live single cell-sized particles (~3–20 μm) to sort from the live singlet population (in purple in Fig. 2d) based on their SSC-A vs. FSC-A signal (gated population in cyan in Fig. 2e). The setup of the cell-sized gate is presented in Fig. 3a–g (see Note 20).
-
8.
Thaw the 384-well capture plates.
-
9.
Sort one single cell of interest (precision mode: single cell with yield mask: 0, purity mask: 32 and phase mark: 16) per well in each 384-well capture plate, leaving four control wells empty.
-
10.
Spin down the plate at 1000 rcf for 1 min and seal the plate.
-
11.
Freeze on dry ice until the rest of the plates have been sorted.
-
12.
Store at −80 °C until processing (see Note 21).

FACS gating strategy for live cell-sized singlets with live/dead double-staining. (a) Gating strategy to select live single cells from sample stained with calcein AM and propidium iodide (PI). (b) Overall events SSC-A vs. FSC-A profile. Note that we are using bi-exponential scaling on both FSC-A and SSC-A because of the size heterogeneities in our samples. (c) Singlet gate to exclude multiplets based on FSC-H vs. FSC-W profile. Singlet proportion is usually in the 90–95% range. (d) From the singlet population, the live cell gating strategy is based on calcein (live) signal inclusion and propidium iodide (permeabilized dead cells) signal exclusion. (e) From the live singlet population, setup of the cell-sized particle gate, to keep only live single cells for sorting. For setting up this cell-sized gate boundaries, see Fig. 3a, g. The proportion of cells that fall within the represented gate is indicated in red

FACS gating strategy setup for live cell-sized singlets with absolute size comparison. (a) Gating strategy to select live single cells. (b) All events gate from a whole animal cell suspension (SSC-A vs. FSC-A). Note that we are using bi-exponential scaling on both FSC-A (except in panel g) and SSC-A given the inherent size heterogeneities of Nematostella vectensis whole organism cell suspensions. (c) Overlay of three sizing beads FCS-A profiles (1.34 μm, 3.1 μm, and 6 μm beads) to set the FSC-A boundary of the cell-sized gate presented in panel g. (d) Singlet gate to exclude multiplets based on their FSC-H vs. FSC-W profile. (e) Live cell gating strategy based on calcein Violet signal to keep only cells with calcein (live) signal. (f) and (g) Cell-sized particle gate. Gate boundary is based on beads sizing gate, including all particles ~3 μm and above to maximize cells and minimize debris. Representation in a biexponential scale enables clearer demarcation of cell-sized live particles. The proportion of cells that fall within the represented gate is indicated in red.
3.3 Single-Cell RNA-Seq Library Generation, QC, and Initial Characterization
-
1.
Process plates through the MARS-Seq library preparation pipeline with N = 17 cycle of PCR amplification at the last step.
-
2.
Check library fragment size distribution using the DNA electrophoresis equipment, according to manufacturer’s instructions. MARS-Seq libraries should have a homogeneous fragment size distribution of around 410–440 bp (Fig. 4a for an example profile).
-
3.
Check library concentration using the DNA concentration fluorometer, according to manufacturer’s instructions. Nematostella libraries from a half-plate (190 cells) are routinely in the 1–5 ng/μL range, for a total yield of ~20–100 ng per library.
-
4.
Pool libraries equimolarly and sequence using an high output sequencer according to the manufacturer’s protocol (see Note 22).
-
5.
Run the MARS-Seq computational pipeline to filter, demultiplex and map sequencing reads onto the Nematostella genome according to the user manual (see Note 23).
-
6.
Check that sequencing depth across libraries is homogeneous (Fig. 4b). Library sequencing depth depends on the number of libraries pooled and on the sequencing kit used. Here, we sequenced libraries to a median depth of ~40,000 reads per well (see Note 24).
-
7.
Check the distribution of UMIs in each library (Fig. 4c). Median cell UMI number in Nematostella samples is usually around 400–800 UMIs per library, depending on sequencing depth, cell, and sample type. UMI recovery per cell correlates with the overall cell size, with bigger cells yielding higher UMI counts.
-
8.
Check noise level from the recovered UMIs in the control empty wells. UMI recovery from empty well are significantly lower than for wells with cells (red Xs on Fig. 4c).
-
9.
Filter out cells based on a UMI threshold. Threshold should be over the noise level. We usually use 100–300 UMIs as a lower boundary. Here, boundaries were set at 120–3000 UMIs (Fig. 5a).
-
10.
Cluster cells passing QC using the clustering pipeline, parameters will depend on cell number and sample cell type diversity. See 2D projection of a pilot experiment on ~1200 cells in Fig. 5b–d, with a subset of key known markers to propose a cell cluster annotation.

Library quality metrics of a sequenced single-cell RNA-seq experiment. (a) Example of a typical library tapestation profile of a single MARS-Seq half-plate library (red arrow). Expected fragment size is usually in the 410–440 bp range. Black arrowheads indicate the upper and lower lane markers on a D1000 high sensitivity tape. (b) Sequencing depth statistics across sequenced libraries, by half-plate (192 cells) library. Median read depth per library is indicated in dark red at the top. Median sequencing depth target is 40,000 reads/cell when sequencing 32 MARS-Seq libraries on a NextSeq 500 high output kit. Note the two libraries flagged in orange whose median sequencing depth is much lower, indicating potentially either a sample issue, or library undersequencing or library quality issues. As their matched other half-plates show the expected sequencing depth, it seems likely to be a technical issue arising during processing or library pooling. The other libraries pass this check. (c) Unique Molecular Identifiers (UMI) recovery statistics across half-plate libraries. Same libraries as shown in b. Note that flagged libraries (in orange in b and c) also display a lower UMI distribution compared to their other half-plate. The two empty wells kept as negative controls (red Xs) per half-plate display very low UMI recovery (noise) compared to wells that held one cell (blue dots)

UMI distribution in cells passing QC and clustering example from a N. vectensis regeneration experiment. (a) UMI distribution across all cells. Here, cells from which between 120 and 3000 (red lines) UMIs were recovered were kept for downstream analyses. (b) 2D-projection of 1300 cells clustered here using the MetaCell analysis package, colors are used to demarcate individual MetaCell clusters. (c) Gene expression (UMIs per 1000 UMIs in the cluster) for known marker genes from [14] are used to annotate the clusters. (d) 2D-projection of control and regenerated oral regions at 4/6 days post-amputation. Overall 2D clustering is split by sample origin (control or regenerated), and colored according to a preliminary MetaCell annotation based on known markers published in the N. vectensis cell atlas [14]. Most cells are split according to their prospective cell state, with newly regenerated oral side samples falling within all broad cell types
4 Notes
-
1.
For cells labeled with a fluorophore overlapping with the calcein AM or propidium iodide (e.g., transgenic fluorescent reporter), it is possible to use the calcein violet (live dye) instead of calcein AM, and SytoxRed (dead cell dye) instead of propidium iodide.
-
2.
For the calcein AM signal: fluorescein (FITC) channel, laser excitation: 488 nm, emission filters: LP 502 nm, and BP 530/30 nm. Propidium iodide (PI) signal: phycoerythrin (PE) channel, laser excitation: 561 nm, emission filters: BP 582/15 nm. If sorting mOrange-labeled cells, the following channel combinations can be used: calcein violet signal (live dye): laser excitation, 405 nm, emission filters: BP 450/40 nm. mOrange signal: phycoerythrin (PE) channel, laser excitation: 561 nm, emission filters: BP 582/15 nm. “Green” autofluorescence signal: fluorescein (FITC) channel, laser excitation: 488 nm, emission filters: LP 502 nm, and BP 530/30 nm. In that case, mOrange-positive cells should be sorted from live singlets from a mOrange signal/Green autofluorescence signal visualization, by selecting mOrange signal-positive cells that are not also GFP-like positive.
-
3.
A detailed step-by-step protocol to efficiently prepare the MARS-Seq capture plates, with lysis buffer and specific poly-T barcoded capture oligos has been published previously [15].
-
4.
Genome file [21] can be downloaded from the JGI website at https://genome.jgi.doe.gov/Nemve1/Nemve1.home.html. Annotation according to [17].
-
5.
We have been following the step-by-step MARS-Seq 2.0 protocol published by the Amit and Tanay’s labs [20] to generate sequencing-ready libraries.
-
6.
This protocol uses MARS-seq2.0 [20] as provided on the website of Amos Tanay’s group; http://compgenomics.weizmann.ac.il/tanay/?page_id=672.
-
7.
We have been using MetaCell, a clustering algorithm based on K-nn graph partitioning [17].
-
8.
A single polyp will yield ample cells to populate all the sixteen 384-well plates for a full MARS-seq run. However, as FACS-sorted plates can be associated with index information, multiple replicate polyps can be processed in the same sequencing run.
-
9.
If the animal is not fully relaxed, wait for it to relax before adding MgCl2. If the animal contracts upon MgCl2 addition, wash it out of the MgCl2 solution by moving it into bigger volumes of Nematostella medium and use another animal.
-
10.
It is best to cut off the tip of a 3-mL plastic Pasteur pipette to avoid damaging the polyp with the smaller opening while transferring it.
-
11.
4 days and 6 days post-amputation and control non-amputated animals were used here.
-
12.
For processing a larger amount of starting material, the dissociation volume can be scaled up as well as all the subsequent reagents, or/and samples can be split across several tubes.
-
13.
Liberase activity is optimal at 37 °C; however, to avoid heat stress, all steps are carried out at RT (22–24 °C), and dissociation usually takes between 10 and 30 min depending on sample density, type, and stage.
-
14.
If the cell suspension is very dense or large fragments of extracellular matrix remain, the filter may become clogged. In that case, changing the filter and rediluting the sample prior to refiltering is recommended.
-
15.
Cell concentration, morphology, presence of multiplets, and viability should be checked on a ~10 μL aliquot of the cell suspension at a microscope prior to FACS-sorting.
-
16.
It is recommended that the FACS be set up and calibrated prior to starting the dissociation. CS&T beads™ Fluorospheres (BD Biosciences) are used to perform optical QC (delay performance measurements) as well as establish laser delay. For additional settings for single-cell deposition, we have used Accudrop™ fluorescent beads. Sheath fluid—BD FACS Flow—was filtered twice through a 0.2-μm filter. The sheath flow formed stable droplets using a 70-μm nozzle.
-
17.
Single-cell deposition efficiency in each well of the 384-well plate should be controlled prior to experiments using a colorimetric method [23]. Briefly, this uses 3,3′,5,5′-tetramethylbenzidine (TMB) and horseradish peroxidase (HRP) to verify whether a droplet from the cell sorter has successfully reached the fluid in the bottom of the well. The droplet contains the enzyme HRP, which is deposited into a well containing the substrate TMB. When HRP and TMB come into contact, TMB is oxidized and rapidly turns to blue color. Parameters adjusted on the Aria III before single-cell deposition sorting include forward scatter area, side scatter area, and fluorescent area parameters. Forward scatter-area, forward scatter-width, forward scatter-height, side scatter-area, side scatter-width, and side scatter-height are used to exclude multiple cell containing droplets and ensure single cells were deposited. Higher acquisition rates will generally increase the likelihood that droplets will contain multiple cells; therefore, low flow rate is kept constant throughout sorting.
-
18.
Using a biexponential scale for the FSC-A signal is usually more informative because of the very broad size range (~3–25 μm, with a majority of 4–15 μm cells) seen in Nematostella cells. The default FSC-A machine threshold may need to be lowered as some Nematostella cells are much smaller.
-
19.
If sorting for a GFP-like fluorophore reporter, whose spectrum overlaps with the calcein AM live cell dye, the live dye can be substituted for calcein violet (e.g., 405 nm, emission filters: BP 450/40 nm), if there is no spectral overlap. Calcein AM and calcein violet signal are completely coherent (i.e., a double-stained sample will exhibit signal only in the diagonal in the calcein AM/calcein violet visualization plane).
-
20.
Multiple sizing beads (ideally with 1, 3, 6, and 10 μm sizing beads) are used as an absolute size reference for the FSC-A signal, which correlates with particle size. On a FSC-A vs. SSC-A display, most live single cells should be in the 3–20 μm range, with the majority in the 4–15 μm range. Alternatively, a solution of freshly spawned Nematostella vectensis mature sperm displays a homogeneous FSC-A vs. SSC-A profile and can be used as a ~3 μm threshold reference.
-
21.
Plates of captured sorted cells can be stored over at least several months at −80 °C as some samples have been processed and gave similar yields after more than 1.5 years in storage.
-
22.
We routinely used a NextSeq500 high output kit from Illumina but similar coverage can be obtained using several Illumina HiSeq lanes.
-
23.
The pipeline generates UMI count tables used for subsequent clustering as well as a quality control (QC) report whose panels are presented and commented in depth in the MARS-Seq protocol [20].
-
24.
If specific libraries are undersequenced, that could indicate a library pooling issue, or a library quality issue. Repeat DNA electrophoresis analysis of the library to check library profile and re-assess library concentration, if the profile is as expected and the concentration in the expected range for the number of amplification cycles used, use the new concentration to repool and sequence with other libraries. If read number is heterogeneous within a library, that can be due to technical issues during cell sorting or plate processing. Check the distribution of reads or UMIs across the plate, sometimes plate borders or plate sides display overall lower recovered UMI counts linked to well localization on the plate because of a plate processing issue.
References
Amiel AR, Johnston HT, Nedoncelle K et al (2015) Characterization of morphological and cellular events underlying oral regeneration in the sea anemone, Nematostella vectensis. Int J Mol Sci 16:28449–28471
Bradshaw B, Thompson K, Frank U (2015) Distinct mechanisms underlie oral vs aboral regeneration in the cnidarian Hydractinia echinata. eLife 4:e05506
Vogg MC, Galliot B, Tsiairis CD (2019) Model systems for regeneration: Hydra. Development 146(21):dev177212
Layden MJ, Rentzsch F, Röttinger E (2016) The rise of the starlet sea anemone Nematostella vectensis as a model system to investigate development and regeneration. Wiley Interdiscip Rev Dev Biol 5:408–428
Rentzsch F, Juliano C, Galliot B (2019) Modern genomic tools reveal the structural and cellular diversity of cnidarian nervous systems. Curr Opin Neurobiol 56:87–96
Rentzsch F, Technau U (2016) Genomics and development of Nematostella vectensis and other anthozoans. Curr Opin Genet Dev 39:63–70
Amiel AR, Foucher K, Ferreira S et al (2019) Synergic coordination of stem cells is required to induce a regenerative response in anthozoan cnidarians. bioRxiv 2019.12.31.891804
Schaffer AA, Bazarsky M, Levy K et al (2016) A transcriptional time-course analysis of oral vs. aboral whole-body regeneration in the Sea anemone Nematostella vectensis. BMC Genomics 17:718
Warner JF, Guerlais V, Amiel AR et al (2018) NvERTx: a gene expression database to compare embryogenesis and regeneration in the sea anemone Nematostella vectensis. Development 145(10):dev162867
Warner JF, Amiel AR, Johnston H et al (2019) Regeneration is a partial redeployment of the embryonic gene network. bioRxiv:658930
Watanabe H, Hoang VT, Mättner R et al (2009) Immortality and the base of multicellular life: lessons from cnidarian stem cells. Semin Cell Dev Biol 20:1114–1125
Kolodziejczyk AA, Kim JK, Svensson V et al (2015) The technology and biology of single-cell RNA sequencing. Mol Cell 58:610–620
Ziegenhain C, Vieth B, Parekh S et al (2017) Comparative analysis of single-cell RNA sequencing methods. Mol Cell 65:631–643.e4
Sebé-Pedrós A, Saudemont B, Chomsky E et al (2018) Cnidarian cell type diversity and regulation revealed by whole-organism single-cell RNA-seq. Cell 173:1520–1534.e20
Jaitin DA, Kenigsberg E, Keren-Shaul H et al (2014) Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343:776–779
Kivioja T, Vähärautio A, Karlsson K et al (2012) Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 9:72–74
Baran Y, Bercovich A, Sebe-Pedros A et al (2019) MetaCell: analysis of single-cell RNA-seq data using K-nn graph partitions. Genome Biol 20(1):206
Stuart T, Butler A, Hoffman P et al (2019) Comprehensive integration of single-cell data. Cell 177:1888–1902.e21
Kiselev VY, Kirschner K, Schaub MT et al (2017) SC3: consensus clustering of single-cell RNA-seq data. Nat Methods 14:483–486
Keren-Shaul H, Kenigsberg E, Jaitin DA et al (2019) MARS-seq2.0: an experimental and analytical pipeline for indexed sorting combined with single-cell RNA sequencing. Nat Protoc 14:1841–1862
Putnam NH, Srivastava M, Hellsten U et al (2007) Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317:86–94
Satija R, Farrell JA, Gennert D et al (2015) Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33:495–502
Rodrigues OR, Monard S (2016) A rapid method to verify single-cell deposition setup for cell sorters. Cytometry A 89:594–600
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Plessier, F., Schmutz, S., Novault, S., Marlow, H. (2022). Single-Cell Transcriptomic Analysis in the Regenerating Cnidarian Nematostella vectensis . In: Blanchoud, S., Galliot, B. (eds) Whole-Body Regeneration. Methods in Molecular Biology, vol 2450. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2172-1_30
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2172-1_30
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2171-4
Online ISBN: 978-1-0716-2172-1
eBook Packages: Springer Protocols