
Abstract
Innate immune responses to pathogens are evolutionarily ancient and are found in the most primitive organisms. These are highly conserved and are not pathogen-specific, but are in response to classes of molecular structures. Infections can be perceived both extracellularly and intracellularly by Pathogen Associated Molecular Patterns (PAMPs) and their host cell ligands, Pathogen Recognition Receptors (PRRs), among them, Toll-Like Receptors (TLRs). The innate immune response to infection includes the release of soluble preformed mediators, or synthesis of cytoplasmic enzymes, cytokines, chemokines, interferons (IFNs), lipid mediators, proteins of the complement cascade, neurotransmitters, nucleotides, and components of transcription factors (High Mobility Group B1, receptors for sex hormones/steroids). Directed cellular migration of parenchymal astrocytes and microglia, as well as recruitment across the blood brain barrier (BBB) of circulating neutrophils, natural killer, monocytes, macrophages, dendritic cells, and ultimately T lymphocytes to the site of infection are also hallmarks of innate responses to infections. These responding cells contribute their own secreted effector molecules and effector activities (such as phagocytosis). Distinct viruses are capable of infecting every cell type (endothelial cells, ependymal cells, perivascular macrophages and pericytes, astrocytes, microglia, oligodendrocytes, Schwann cells, and neurons) in the central nervous system (CNS). These CNS infections challenge the host with a different set of problems than do peripheral viral infections. Among the complications are (a) neurons that rarely express Class I or Class II Major Histocompatibility Complex (MHC) molecules and are thus not suitable targets for either CD4+ or CD8+ MHC-restricted T cells, (b) an enclosed volume that is constrained from swelling during inflammation, as well as poorly developed lymphatic drainage, and (c) the immunologic privilege of the CNS which leads to extremely limited immune surveillance for pathogens. Therefore, the role of innate immunity, both from CNS-resident cells and their products, and from circulating inflammatory cells and molecules which traverse the BBB are essential to “buy time,” inhibiting viral replication and dissemination, until the host can marshal an adaptive immune response. The immune responses are crucial for host survival from the infection. Consequently, successful pathogens, especially those that persist, have developed a wide variety of evasive approaches to limit the inhibition of replication. Many of these pathways are highlighted in individual chapters that precede this one. These evasive measures range from neutralizing host secreted molecules (cytokines and chemokines) with soluble receptors, encoding anti-inflammatory proteins in their genome, preventing signal transduction, blocking inhibition of protein synthesis, degradation of essential antiviral molecules, preventing apoptosis, and blockade of the nuclear pore complex. In this chapter, I will attempt to cover the breadth of the innate immune response to viral infection, but will also devote more space to generally under-considered aspects than to the well-known components. There are some caveats to consider, as most experiments have been performed in the murine model and not in man; further, conclusions from experiments using in vitro cultured cells (whether primary or established lines) may not reflect physiological conditions in an undisturbed CNS. Lastly, we now appreciate the complexities imposed on hosts by polymorphisms in genes of critical pathways, leading to increased susceptibility or resistance of that individual but not others.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Innate immunity
- Neuroimmunology
- Viral infections
- Interferon
- Lipid mediators
- Resolution
- Blood–brain barrier
Host Recognition of Pathogen Associated Molecular Patterns (PAMPs) and Damage Associated Molecular Patterns (DAMPs)
Viruses trigger host responses by engaging several different families of receptors, both surface and within cells; these receptors recognize generic patterns, and not specific sequences (such as a peptide of viral surface protein or genome). One of the best-known families of these pattern recognition receptors is Toll Like Receptors (TLRs), present on the cell surface or endosomal membrane (Lester and Li 2014). Intracellular RIG-I-Like Receptors (RLRs) having common domains called caspase recruitment domain (CARD) , helicase, and NACHT are RIG-I (retinoic acid inducible gene-I), MDA5 (melanoma differentiation associated gene 5), Caterpillar, NOD, NALP, NAIP, and CIIT (Fitzgerald et al. 2014b).
TLR bind a wide range of PAMPs , ranging from peptidoglycans of gram-positive and lipopolysaccharides of gram-negative bacteria (TLR 2 and TLR 4, respectively) to viral genomes dsRNA, ssRNA, and dsDNA (TLR3, 7, and 9, respectively). Signaling through TLR leads to production of IFN-β and proinflammatory cytokines (Trotta et al. 2014; Gay et al. 2014; Kawai and Akira 2011). Few cells in the CNS constitutively express high levels of TLR (Suh et al. 2009).
In neurotropic viral infections, TLR are critical for IFN and cytokine production for Enterovirus-71, Human Immunodeficiency Virus-Encephalitis (HIV-E), Herpes simplex virus-1 (HSV-1), Flaviviruses, Japanese encephalitis virus (JEV), Junin virus, LaCrosse virus, Lymphocytic choriomeningitis virus (LCMV), Rabies, Semliki Forest virus (SFV), Sindbis, Theiler’s murine encephalomyelitis virus (TMEV), and West Nile virus (WNV) (Han et al. 2014; Fadnis et al. 2013; Nazmi et al. 2014; Cuevas and Ross 2014; Denizot et al. 2012; El-Hage et al. 2011; Furr and Marriott 2012; McKimmie et al. 2005; Neal 2014; Olson and Miller 2004; Sabouri et al. 2014; Szretter et al. 2009; Taylor et al. 2014; Thomas et al. 2014; Wollish et al. 2013; Zhou et al. 2008; Zolini et al. 2014).
TLR signaling may regulate the expression of micro-RNAs including MiR-146 and MiR-155, leading to down-regulation of some inflammatory genes (Aalaei-andabili and Rezaei 2013). MiR-155 may regulate JEV-induced inflammation by controlling Src Homology 2-containing inositol phosphatase-1 (SHIP-1) (Thounaojam et al. 2014b) and MiR-29b targets TNF-α-induced protein 3 (Thounaojam et al. 2014a). However, enhanced production of MiR-155 may lead to BBB dysregulation (Lopez-Ramirez et al. 2014).
RIG- I binds 5′ uncapped single stranded RNA, an essential intermediate in RNA virus replication (Goubau et al. 2014; Hornung et al. 2006). MDA5 , in contrast, recognizes dsRNA (Wu et al. 2013), and is required for picornavirus responses (Kato et al. 2006). Like TLRs, RIG-I activation leads to activation of a protein variously known as Cardif/IPS-1/MAVS/VISA upstream of IRF3 and NF-kB activation, which transduce the signals with nuclear translocation, leading to the production IFN-β and all the downstream IFN-stimulated genes (ISGs) (Schneider et al. 2014). RIG-I is negatively regulated by a deubiquitinase, USP21 (Fan et al. 2014).
While these pathways have been well documented in many cell types, they may not always “work” in the CNS. For instance, while Vesicular stomatitis virus (VSV) replication is extremely sensitive to the antiviral effect of pretreatment of neurons with IFN-β, VSV infection of dendritic cells rapidly induces IFN, it fails to elicit IFN-β production in neurons (Trottier et al. 2005). The mechanism(s) by which IFNs alter cellular physiology to resist viral infection is distinct in neurons when compared to cell types that have been more frequently studied (D’Agostino et al. 2009a, b; Chesler et al. 2003). VSV also evades cell-autonomous responses through one of many actions of the viral M protein, essentially preventing mRNA export from the nucleus (Faria et al. 2005). In mice, VSV infection elicits IFN-β production by plasmacytoid dendritic cells in peripheral lymphoid compartments (Akira and Hemmi 2003), no detectable IFN-β is made in the CNS during the first week of VSV encephalitis (Trottier et al. 2007). In contrast, both Theiler’s encephalomyelitis virus (TMEV) and LaCrosse virus infections led neurons to produce Type I IFN (Delhaye et al. 2006). Both RIG-I and MDA5 are essential to detect and control WNV infection (Carty et al. 2014; Errett et al. 2013).
The Receptor for Advanced Glycation End-products (RAGE) , an activating receptor, is expressed on many cells including vascular smooth muscle cells, endothelial cells, monocytes, and microglia (Ramasamy et al. 2005). It was originally recognized as a contributor to the inflammation seen in diabetes, and binds, as its name suggests, proteins that have been posttranslationally modified with glucose. Engagement of RAGE by its ligands leads to signal transduction through NF-kB and synthesis of proinflammatory mediators, leading to neuroinflammation and oxidative stress (Tobon-Velasco et al. 2014). Other ligands for the receptor are S100 family proteins, HMGB1 and insoluble complexes of Aβ peptide, which are released during tissue damage in arthritis, atherosclerosis, aging, neurodegeneration, pulmonary diseases, sepsis, and ischemia (Chuah et al. 2013; Kang et al. 2014). This has led to the classification of RAGE as a Damage Associated Molecular Pattern (DAMP) receptor (Foell et al. 2007). Thus direct or indirect compromise of neurons and parenchymal cells during viral encephalitis leads to the release of HMGB1 (Wang et al. 2006) or S100 that can activate microglia, perivascular macrophages, and pericytes, as well as the microvascular endothelial cells (Jaulmes et al. 2006; Rong et al. 2005). Release of S100 or HMGB1 during VSV encephalitis did not contribute to the production of IFN-β by splenic plasmacytoid dendritic cells, since infusion of soluble RAGE did not suppress the response (Reiss and Schmidt, unpublished data). Thus, the BBB may be disrupted; cells within the CNS will secrete cytokines, chemokines, and other inflammatory mediators. This could be among the first of the sequential waves of innate immunity in response to the viral infection .
Interferon-Induced Antiviral Responses
The initial report of a factor made by cells which inhibited viral replication was made ~50 years ago (Isaacs and Lindenmann 1957). There are three Types of IFN . Type I is more diverse, produced by virtually all cells and includes IFN-α, IFN-β, and IFN-τ. Type II has only one member, IFN-γ; Type III comprises IL-28 and IL-29, a family of IFN-λ proteins (Reid and Charleston 2014; Guayasamin et al. 2014; Hermant and Michiels 2014). IFNs inhibit viral replication by pathways described below, may also lead to neurodegeneration and demyelination through the activation of microglial production of neurotoxins (Owens et al. 2014; Block et al. 2007; Mana et al. 2006).
IFN may also be a beneficial cytokine in LCMV and Lassa fever infections where viral pathogenesis may induce vascular leak (Baccala et al. 2014). In Langat virus and TBE infections, IFN is protective against fatal neurotropic disease (Weber et al. 2014a). In measles infections, neurons express IFN needed for early control (Cavanaugh et al. 2015), but in intranasal VSV infection, astrocytes are the source (Detje et al. 2015). Therefore, the beneficial and pathologic effects of IFNs may depend on the quantity and duration of expression.
Once IFNs have been induced and secreted, these cytokine bind ubiquitously expressed receptors and induce a signal transduction kinase cascade starting with Jaks and STATs, leading to nuclear translocation of phosphorylated STAT complexes that result in gene induction in virtually all cells (Nallar and Kalvakolanu 2014; Ivashkiv and Donlin 2014; Owens et al. 2014). As with most other signal transduction cascades, there are regulatory phosphatases that dampen the IFN-mediated induction, these include Suppressors of cytokine signaling (SOCS) and Protein inhibitor of activated STAT1 (PIAS) proteins. Resveratrol may upregulate SOCS-1, and thus dampen inflammation (Dragone et al. 2014). While the Jak-STAT pathway is predominant, secondary signal transduction pathways are also important for IFN’s activity (Ivashkiv and Donlin 2014). Although most of the consequences of IFN binding and signaling are transcriptional, not all of the inductive effects of IFNs require new mRNA production; I will discuss that below.
IFN responses are essential host components of intrinsic and cell autonomous immunity to viral infection . Many viruses block IFN signaling or downstream mediators such as Tick borne flaviviruses and IRF-1 signaling (Robertson et al. 2014). The exact pathways by which IFNs antagonize viral replication required are not yet fully elucidated. New techniques such as silencing are providing insights into the downstream mediators (Diamond and Farzan 2013; Fensterl et al. 2012; Schoggins et al. 2014), as are explorations of specific genes used by some viruses to evade antiviral pathways (Taylor and Mossman 2013) such as HSV γ34.5 (Rosato and Leib 2014) or Rabies P-protein (Wiltzer et al. 2014). JEV modulates SOCS in infected macrophages, inhibiting the production and release of proinflammatory cytokines (Kundu et al. 2013).
Many tumors have been shown to have disabled IFN responses; this has led to the development of several viruses for oncolysis , that is, infection to target tumors but spare normal tissue.
Expression of IFNs may be regulated by cellular micro-RNAs , targeting the IFN mRNA for destruction. MiR-548 suppresses IFN-λ1 expression (Li et al. 2013b) and MiR-466I targets IFN-α mRNA (Li et al. 2012), leading to increased viral replication.
Some IFN-stimulated genes (ISG) are critical for co-stimulation, antigen processing, and presentation, some for antiviral effects, others contribute to regulation of angiogenesis, cellular apoptosis, or stasis (Xiao et al. 2006), and other physiological processes. Hundreds of IFN-regulated genes have been identified using microarray analysis and functional assays (Schneider et al. 2014; Cho et al. 2013; Schoggins et al. 2014). Traditionally these were studied in isolation, and many antiviral pathways have been well characterized including Mx, PKR, RNAseL, OAS, and IDO. I will focus on a few of the more important antiviral pathways controlled by IFNs.
Inactivation of GTP
The first antiviral IFN-stimulated pathway studied in detail initially for myxovirus (Influenza) infections, Mx , was discovered in 1978 by Lindenmann and colleagues; they observed that some mice were spontaneously resistant to influenza virus replication and later showed that Mx had GTPase activity (Lindenmann et al. 1978; Isaacs and Lindenmann 1957; Kochs et al. 1998). Other GTPases including Very Large Inducible GTPase-1 and TGTP/Mg21/IRG-47 are induced by IFNs (Klamp et al. 2003). Guanylate binding proteins are also ISGs. These include GBP-1, a Dynamin superfamily member with GTPase activity (MacMicking 2004).
MxA was induced in HIV and Simian immunodeficiency virus (SIV) infection of the CNS (Singh et al. 2014; Zaritsky et al. 2012), and in Reovirus infections Mx was critical to limit viral replication (Dionne et al. 2011). WNV evades the antiviral activity of MxA (Hoenen et al. 2014). An isoform of MxA may lead to enhanced HSV-1 replication (Ku et al. 2011). There are polymorphisms in the promoter region of human MxA; these may lead to altered gene expression, and thus sensitivity to infections or to resistance to exogenous IFN (Tran Thi Duc et al. 2013).
Inhibition of Protein Synthesis
Probably the best known ISG is triggered when dsRNA, produced during viral infection, activates the kinase PKR, phosphorylating and inactivating the translation elongation factor eIF2α, inhibiting the production of new proteins in infected cells. This pathway is important in many neurotropic viral infections including MHV-A59, Sindbis, WNV, VSV, TMEV, and HSV-1 (Kapil et al. 2014; Geiss et al. 2003; Baltzis et al. 2004; Cheng et al. 2005; Gorchakov et al. 2004; Palma et al. 2003; Ryman et al. 2005; Ventoso et al. 2006).
Cellular stress associated with the Unfolded Protein Response, when viral glycoprotein synthesis dysregulates endoplasmic reticulum function, is a DAMP response (Smith 2014; Noack et al. 2014). UPR pathway inactivates eIF2α using two enzymes PERK and GCN2 (Berlanga et al. 2006). Flaviviruses such as JEV and Coronaviruses trigger this alarmin response (Noack et al. 2014).
Viperin /cig5/vig is an ISG and also induced during infection by cytomegalovirus (CMV), JC virus, or VSV, and suppresses synthesis of some viruses (Helbig and Beard 2014). Viperin restricts WNV pathogenesis (Szretter et al. 2011). Inhibition of viral protein synthesis is an effective host response to cripple viral infection.
Recognition, Degradation, and Sequestration of Viral RNA and Viral DNA
Recognition of viral RNA in infected cells involves many different pathways. The substrate of 2′,5′-Oligoadenylate Synthase (OAS)-Dependent RNAseL is viral dsRNA (Hornung et al. 2014). This pathway is important in the resistance to HSV-1, flaviviruses, LCMV, and VSV infections of the CNS (Bhattacharyya 2014). RNAseL may contribute to the apoptosis of infected cells (Castelli et al. 1998).
Adenosine deaminase which acts on dsRNA (ADAR1) is an IFN-γ-inducible antiviral enzyme which may be coupled with the PKR pathway (Taylor et al. 2005). ADAR1 restricts measles infection in the CNS (Ward et al. 2011).
IFN-induction of stress granules (also called P bodies ) may sequester ADAR1 (John and Samuel 2014). Some viruses Mengovirus, TMEV, WNV, JEV, Measles, and Junin prevent the formation of P bodies; while other viral infections including poliovirus, SFV, MHV-A59, and VSV induce the formation of stress granules (Pattnaik and Dinh 2013; Onomoto et al. 2014). TBEV replication is inhibited by sequestration of vRNA in stress granules (Albornoz et al. 2014).
Another antiviral protein that recognizes and sequesters viral mRNAs is Zinc finger Antiviral protein (Glasker et al. 2014). Zinc has been shown to contribute not only to this antiviral protein, but to other proteases including matrix metalloproteinases (MMPs) and metallothionein necessary for diapedesis (Rink and Haase 2007), for inflammatory cells crossing peripheral capillaries or for breaching the BBB, or for migration of CNS parenchymal cells in response to chemoattractants during viral infection. Zinc contributes to inhibition of polyprotein processing for many Picornaviruses (Krenn et al. 2005).
Stimulator of IFN genes (STING) [also known as mediator of IRF3 activation (MITA) ] is activated by IFN-γ inducible protein 16 (IFI16) binding to viral dsDNA and HN200; STING then activates TANK binding kinase-1 (TBK-1) phosphorylation of the transcription factor IRF3, resulting in expression of ISGs (Thompson et al. 2014). MITA/STING is an ER and mitochondrial membrane-bound cytoplasmic sensor for pathogen-induced cyclic dinucleotides (cyclic GMP-AMP); its C-terminal domain recruits TBK1 and IRF3 (Dubensky et al. 2013; Ran et al. 2014). The host enzyme cyclic GMP-AMP synthase, cGAS (also called MB21D1 ), is the DNA and also dsRNA sensor (Hornung et al. 2014; Schoggins et al. 2014). NLRC3 is a negative regulator of STING activation (Zhang et al. 2014). Diffusion of cGAMP to neighboring cells may lead to paracrine cell-autonomous innate immunity (Ablasser et al. 2013).
IFI16 senses and contributes to control of HIV-1 infection (Jakobsen et al. 2013). STING can also bind to reverse transcriptase intermediates of Human T cell leukemia virus (HTLV-1), and with IRF3 and SAMHD1, activate apoptosis (Sze et al. 2013). In many viral infections, there is an arms race between the host’s ability to shut down viral replication and the virus inactivating host antiviral pathways; that is observed with HSV-1 ICP0 and US3-PK and STING (Kalamvoki and Roizman 2014). Hepatitis C virus (HCV) NS4B blocks STING activation of TBK-1 (Ding et al. 2013). The Dengue virus (DENV) NS2B/3 protease cleaves STING (Aguirre et al. 2012; Green et al. 2014).
IFN-induced protein with tetratricopeptide repeats (IFIT) family proteins contribute to antiviral responses IFIT1 (ISG56), IFIT2 (ISG54), IFIT3 (ISG60), IFIT5 (ISG58) and are regulated by viral infection (Hyde et al. 2014; Zhang et al. 2013b). IFIT1 binds the 5′ capped 2′-O unmethylated RNA of JEV inhibiting its replication (Kimura et al. 2013), but VEE mutants evade IFIT1 (Hyde et al. 2014). IFIT2 protects mice from VSV neuropathogenesis (Fensterl et al. 2012) and VSV infection of the peripheral nervous system (Fensterl et al. 2014). Ifit2 is essential for host control of neurotropic Mouse Hepatitis Virus A59 (MHV) (Butchi et al. 2014), EMCV, MHV, and WNV (Fensterl and Sen 2015), and HBV (Pei et al. 2014). IFITM is an IFN-induced membrane associated protein. IFIT3 potentiates antiviral signaling by connecting TBK1 and MAVS (Liu et al. 2011). They have broad-spectrum antiviral activity (Diamond and Farzan 2013). However, some neurotropic RNA viruses including WNV can evade host restriction by IFIT family members (Daffis et al. 2010).
Altered Amino Acid Metabolism
The ISG Indoleamine 2,3-Dioxygenase (IDO) , a catabolic enzyme for tryptophan, generates kynurenines. IDO may have regulatory effects on T cell activity (Sakurai et al. 2002) and may be neuroprotective or neurotoxic: IDO contributes to alterations in serotonin metabolism, enhances astrocyte viability, but contributes to the formation of toxic quinolinic acid (Campbell et al. 2014).
IDO has antiviral activity against vaccinia virus, HTLV-1, measles, and HSV-1 (Adams et al. 2004; Maloney et al. 2000; Oberdorfer et al. 2003). However, it may be antagonistic to containment of HIV-1 (Miller and Bhardwaj 2013). IFN-induced alterations in amino acid metabolism can suppress infection, but clearly this is a two-edged sword.
Miscellaneous Antiviral ISGs
Although the phosphatase(s) and kinase(s) altered by IFN-β treatment in neurons were not identified, in IFN-β-treated neurons, the posttranslational modification of two of the five VSV proteins was profoundly altered; the M protein was hyper-phosphorylated and the P protein, a subunit of the RNA-dependent RNA polymerase, was hypo-phosphorylated. M protein lost affinity for the RNP complex, impairing assembly, and the RNA-dependent RNA polymerase activity was altered. Together, these modifications led to inhibition of productive VSV replication in neurons (D’Agostino et al. 2009a, b; D’Agostino and Reiss 2010).
There are many other ISGs that have antiviral activity, although they are less well studied. One of these is ISG12; it contributes to resistance to Sindbis encephalitis (Labrada et al. 2002). ISG12 is a nuclear envelope protein that binds nuclear receptors like peroxisome proliferation activating receptors (PPARs) (Uhrin et al. 2013). Cytokines and chemokines will be discussed below.
Ubiquitinases, Deubiquitinases, ISG15, Sumoylation
Posttranslational modifications of proteins are essential for their activities, their cellular localization, and also their half-life. One regulator of protein lifespan is the proteasome; proteins are targeted for degradation by addition of strings of ubiquitin (Ub), polyubiquitin tails, by a series of proteins that recognize the target (E1) bridge that complex (E2) to the ubiquitin ligase (E3). There are also host cellular enzymes capable of stripping Ub from proteins, deubiquitinases (Herrmann et al. 2007).
There are both host-cell beneficial applications of Ub-modification and pro-viral life cycle modifications. Malfunction of this pathway leads to accumulation of proteins that should have been degraded, and can result in neurodegenerative diseases (Atkin and Paulson 2014).
An example of viral-enabling modification is induction of autophagy, as viruses generate membranes for their cytoplasmic replication organelles (Suhy et al. 2000; Nchoutmboube et al. 2013). Autophagy can also be associated with presentation of viral glycoproteins to class I MHC molecules, facilitating recognition of the infected cell by CD8 cytolytic T cells (Tey and Khanna 2012).
Mono-ubiquitination is often associated with either targeted proteins on the cell surface directed to the cellular endosomal sorting complexes required for transport (ESCRT) pathway or in viral assembly, with viral proteins usurping the ESCRT pathway to deliver viral components to the cell surface for assembly and budding; neurotropic viruses employing the vacuolar protein sorting pathway for assembly include VSV, Rabies, LCMV, Japanese encephalitis virus (JEV), HIV, HSV-1, and Epstein-Barr virus (EBV) (Votteler and Sundquist Wesley 2013; Chen and Lamb 2008).
TRIM79α, an ISG, facilitates the ubiquitin-dependent degradation of TBE’s viral polymerase but does not recognize the closely related WNV protein (Taylor et al. 2011). As with so many other essential host antiviral pathways, viruses have devised novel evasion tools. HSV-1 and RNA viruses including VSV induce Siglec-G, that recruits SHP2 and the E3 Ub ligase c-Cbl to RIG-I, targeting RIG-I for proteolysis (Chen et al. 2013). This pathway, capable of preventing IFN induction by RIG-I, can in turn be antagonized by IFN, inactivating Siglec-G (Chen et al. 2013).
HSV-1 ICP0 induces depletion of CD83 in dendritic cells, diminishing their effectiveness to present viral proteins to T cells (Heilingloh et al. 2014). ICP0 also targets the ND10 DNA repair complex proteins hDaxx, Sp100 and PML to degradation via the Ub-ligases RNF8 and RNF168 (Lilley et al. 2011). Varicella zoster virus (VZV) ORF61 antagonizes IFN production by targeting IRF3 for degradation (Zhu et al. 2011).
HIV-1 has two proteins that target cellular proteins for proteosomal removal: Vpu prevents CD317/tetherin from retaining nascent viral particles on the cell surface (Schmidt et al. 2011), and Vif targets APOBEC3, the cytidine deaminase (Zhang et al. 2012).
There are two other host cell proteins that are similar to Ub and can cross-regulate: ISG15 and SUMO. ISG15 has been shown to play a central role in host-cell antiviral responses in VSV, LCMV, and HSV-1 infections (Campbell and Lenschow 2013; Lenschow 2010).
SUMO-modified proteins are often found in PML nuclear bodies, where proteins may be sequestered (Lallemand-Breitenbach and de The 2010). IFN regulation of MiRs including the Lin28/Let-7 pathway may enhance SUMO expression and inhibition of HIV-1 and HSV-1 infections (Sahin et al. 2014a). IFN treatment and oxidative stress may lead to changes in Sumoylation of target proteins in the PML bodies (Sahin et al. 2014a, b). SUMO conjugation can also be evaded by encephalomyelocarditis virus, HSV-1, VZV, and EBV (Mattoscio et al. 2013).
Tetherin/BST-2/CD317 is an unusual cell surface glycoprotein with both gpi and transmembrane domains holding the protein around lipid rafts (Billcliff et al. 2013). It is expressed by many cell types including neurons, and is induced by both IFN-α/β and IFN-γ (Sarojini et al. 2011). Tetherin is an ISG contributing to antiviral activity for VSV in neurons (Sarojini et al. 2011).
This protein is able to dimerize and tether, hold virus particles on the surface, preventing budding and release of viruses that exit the cell via lipid rafts (Gustin and Douglas 2013). This antiviral pathway is so important that many viruses have developed evasive pathways (Neil 2013; Sauter 2014). HIV Vpu antagonizes tetherin via Ub-modification and degradation, and protecting cells from antibody-dependent cell mediated cytotoxicity (ADCC) (Arias et al. 2014). Glycoproteins of Filoviruses, HSV-1, Sendai, and SIV also block this pathway (Nikovics et al. 2012; Bampi et al. 2013; Zenner et al. 2013; Gnirss et al. 2014). SIV nef and HSV-2 Env mediate endocytosis of tetherin and intracellular sequestration (Serra-Moreno and Evans 2012). HHV-8 K5 ubiquitinates tetherin (Mansouri et al. 2009). CD317/tetherin is an important host cell glycoprotein found at lipid rafts, whose expression is enhanced by IFNs, and can retain budding viruses.
Reactive Nitrogen and Oxygen Species
The production of superoxide (O2*), nitric oxide (NO), and peroxynitrite (ONOO−) contribute to elimination of many intracellular pathogens. There are three isoforms of the enzyme responsible for generating NO, nitric oxide synthase (NOS) (Bruckdorfer 2005). In the CNS, NOS-1 is constitutively found in neurons, NOS-2 induced microglia and inflammatory macrophages, and NOS-3 constitutively in astrocytes, ependymal and endothelial cells (Reiss and Komatsu 1998). NO is not only involved in long-term potentiation in the CNS, it also contributes to regulation of blood flow (Murad 2006). Astrocytes and endothelial cells release NO, resulting in dilation of capillaries and increased local perfusion (Moore 2000).
NO has been associated with some inflammatory neurological disorder (Siciliano et al. 2011; Banach et al. 2011; Bernstein et al. 2011). The mechanism of NO-mediated inhibition is covalent modification of viral proteins at cysteine, serine, and tyrosine, resulting in inappropriate folding, assembly, and/or enzyme activity. NO-mediated inhibition and/or pathology in the CNS contributes to the host response for Reovirus , TMEV, HIV-1, SIV, Adenovirus, Junin, Bornavirus, Venezuelan equine encephalitis (VEE), MAIDS, CMV, Murray Valley encephalitis, MHV, Sindbis, VSV, rabies, JEV, and dengue (Andrews et al. 1999; Brodie et al. 1997; Cheeran et al. 2000; Dietzschold and Morimoto 1997; Gendelman et al. 1994; Gomez et al. 2003; Goody et al. 2005; Hooper et al. 2001; Koeberle et al. 2004; Komatsu et al. 1999a; Liao et al. 2012; de Souza et al. 2013; Koustova et al. 2000; Lane et al. 1999; Lin et al. 1998; Mestre et al. 2005; Minagar et al. 2002; Molina-Holgado et al. 1999; Murphy 2000; Navarra et al. 2004; Schoneboom et al. 1999; Thongtan et al. 2010).
NOS-2 is not constitutively expressed but is rapidly induced when macrophages or microglia are exposed to inflammatory cytokines. Microglia produce reactive oxygen species, as well, contributing to neurotoxicity block (Block et al. 2007). However, unlike most of the IFN-regulated effector molecules described above, NOS-1 mRNA is not induced in neurons by IFNγ and other inflammatory cytokines , although treatment of neurons leads to accumulation of the enzyme and greater activity due to degradation of a protein inhibitor (Chesler et al. 2004a, b; Chesler and Reiss 2002; Komatsu et al. 1996; Yang et al. 2007, 2008). This is one instance where IFN-mediated of antiviral activity is at a posttranscriptional level.
Neuropeptides, Peptide Hormones, and Neurotransmitters
Neurons release a variety of peptides and hormones in order to “talk” to other neurons. Many of these proteins have activities outside the synaptic signaling, and can regulate immune responses. Among these molecules are Substance P, Neuropeptide Y (NPY), vasoactive intestinal peptide (VIP/PACAP), neurokinin1 (NK1), and α-melanocyte stimulating hormone (α-MSH) (Dantzer 2004; Brogden et al. 2005; Metz-Boutigue et al. 2003; Prod’homme et al. 2006). NK1 and NPY are inflammatory and may have Defensin-like activity [Defensins are discussed below]. Others like VIP/PACAP are negative modulators, which act principally on dendritic cell induction of regulatory T cells (Delgado et al. 2006; Gonzalez-Rey et al. 2007). Thus, the impact of many neurotrophins, peptides, and neurotransmitters is by modulation of adaptive immune response, as has been seen with CMV (Li et al. 2013a) and HIV-1 infection (Souza et al. 2014).
Adenosine signaling, through surface A1 and A2A adenosine receptors, has been shown to be neuroprotective (Perigolo-Vicente et al. 2014; Latini et al. 1996). The receptors also regulate pain (Sawynok and Liu 2003). In HIV infection, these receptors have been shown to play an anti-inflammatory role (Gilbert et al. 2007). However, A1 receptors may also contribute to neutrophil infiltration, although this is antagonized by A2 receptors (Cronstein et al. 1992). Expression of A2B receptors is induced by HIF-1-α, a cytokine-inducible transcription factor (Kong et al. 2006). ATP, released by cells, can attract neutrophils to tissues via engagement of A3 and P2Y2 receptors. There is reciprocal modulation of cannabinoid receptor expression by adenosine (Carrier et al. 2006), thus signaling by one neurotransmitter can alter the response of neurons to other neurotransmitters. Cannabidiol was shown to be protective in inflammation during TMEV infection via regulation of A2A receptors (Mecha et al. 2013).
Cannabinoids are both endogenously synthesized (endocannabinoids) lipid neurotransmitters and are also found in some plants (e.g., marijuana) or synthetic pharmaceuticals. Two serpentine 7-transmembrane receptors have been well described: CB1 expressed by neurons and CB2 expressed by cells of the reticuloendothelial system including microglia (Ullrich et al. 2007). The functions of these receptors are distinct, although the same signaling pathways are used; the serpentine 7-transmembrane receptor is G-protein coupled. These receptors (a) negatively regulate Ca2+ channels inhibiting Ca2+ release, (b) activate Raf-1, MEK, and ERK, as well as (c) adenyl cyclase which ultimately activates protein kinase A. The CB1 receptor is associated with hypothermia, immobility, euphoria, and hyperphagia, while the CB2 receptor is a negative regulator of monocyte and microglial activation, hence immuno-dampening. Thus selective receptor agonists can target either immune responses or neurons. However, this distinction is potentially murky when you consider the regulation of cell-autonomous innate immune responses to viral infections in neurons.
Cannabinoids have been shown to be neuroprotective in Huntington’s disease, Parkinson’s disease, and multiple sclerosis (Pryce and Baker 2012; Santos 2012; Sagredo et al. 2012; Di Iorio et al. 2013). Cannabinoids have a beneficial impact on TMEV infections and may regulate CD200-CD200R interactions (Loria et al. 2008; Mecha et al. 2013; Mestre et al. 2005, 2006, 2009). Cannabinoids may contribute to neurogenesis by antagonizing NO production (Kim et al. 2006b). In ischemia (Belayev et al. 1995) or persistent Bornavirus infections (Solbrig et al. 2013; Hooper et al. 2001) where NOS-2 is overactive, NO is associated with pathology, cannabinoids are beneficial. In contrast, in infections where host inflammation and NO are essential to control CNS disease, cannabinoids promoted pathology (Reiss 2010; Herrera et al. 2008). I speculate that cannabinoids may protect the BBB integrity in those settings.
An indirect anti-inflammatory effect of cannabinoids had been found with activation of the nuclear transcription factor peroxisome proliferation activating receptor (PPAR) family, described below. CB2 receptor activation may lead to release of endogenous opioids, which inhibit inflammatory pain (Ibrahim et al. 2006). Somewhat unexpectedly, the antinociceptive and anti-pyretic effects of acetaminophen (Tylenol™) may be due to binding CB1 receptors.
Δ9-Tetrahydrocannabinol treatment decreases host resistance to HSV-2 infection (Cabral et al. 1987), probably by inhibiting host inflammatory immune responses against the virally infected cells. In several models where inflammation contributes to pathology, such as TMEV, the synthetic cannabinoid WIN 55,212-2 ameliorates clinical disease (Croxford and Miller 2003); WIN 55 may also induce PGE2 production (Mestre et al. 2006). However, cannabinoids may contribute to syncytia formation in HIV-E (Noe et al. 1998), leading to pathology . In VSV infection of neuronal cells, activating the CB1 receptor leads to ~15-fold enhanced viral replication via inhibition of Ca2+-flux and thus impairing the activity of constitutive NOS-1 (Herrera et al. 2008). Therefore, there is no hard and fast rule about the impact of cannabinoid activity on viral infection [reviewed in (Reiss 2010)]. Caution is urged when considering use of these drugs; the effect(s) may be on reticuloendothelial cells or on neurons.
Lipids in Innate Immunity in the CNS
The first part of this section will be devoted to eicosanoids, lipid mediators derived from arachidonic acid, liberated from cell membranes by Phospholipase A2. These include prostaglandins (PG), leukotrienes (LT), lipoxins, epoxides, resolvins, marensins, and other bi-products. The second part of the section will include exogenous sources of these metabolites and lipid modification of cellular proteins. Cannabinoids were just discussed. PPAR agonists and Sex hormones will be discussed below; HPAI axis and neuroendocrine regulation are included here.
The sphingolipid Sphingosine-1-phosphate (S1P) regulates lymphocyte traffic from lymph nodes to circulation; it may be bound to the lipid complex HDL in blood (Wilkerson and Argraves 2014). Lymphocytes may express two different receptors S1PR1 and S1PR2; S1PR5 is found on endothelial cells (van Doorn et al. 2012). This pathway has been therapeutically targeted with the drug Fingolimod, an S1P agonist also called FTY720, to sequester proinflammatory T cells away from sites, such as myelin sheaths in multiple sclerosis (Martin and Sospedra 2014; Halmer et al. 2014). Females express a higher level of S1PR2 than males (Cruz-Orengo et al. 2014), and this may contribute to sex-bias in some autoimmune diseases and responses to infections. I propose that in proinflammatory viral infections of the CNS, where infiltration of cells from circulation contributes to pathology (example: LCMV , although data were not promising (Carr et al. 2013)), use of the S1P agonist may be indicated.
Prostaglandins (PG) : Cyclooxygenase (COX) 1 and 2 are the enzymes responsible for the pathway from arachidonic acid leading to the formation of distinct PGs and thromboxane (Ueno et al. 2005). PGJ2 will be discussed below as an agonist for the nuclear transcription factor PPAR. The family of receptors for PGs is among the 7-transmembrane serpentine surface molecules. The end products have many biologic effects ranging from platelet aggregation (TXA2) to inflammation and fever (PGE2) (Ushikubi et al. 2000), but also a profound consequence is immunoregulation, modulating dendritic cell maturation, differentiation, cytokine secretion, and antigen presentation (Harizi and Gualde 2006). The importance of these molecules in physiological processes and pathology has led to drug discovery efforts (Claria 2003). Nonsteroidal anti-inflammatory drugs that block the production of PGs have been shown to be anti-inflammatory in the CNS and somewhat protective for neurodegeneration and cognitive decline in neuroinflammatory diseases (Auriel et al. 2014) and schitzophrenia (Muller et al. 2013).
In the CNS, PGs compromise host responses to VSV, TMEV, JEE, Bornavirus, HSV-1, HIV, enterovirus 71, and EMCV infections (Mestre et al. 2006; Chen and Reiss 2002a; Chen et al. 2000, 2002; Reynolds and Enquist 2006; Steer and Corbett 2003; Lima et al. 2006; Hooks et al. 2006; Rohrenbeck et al. 1999; Tung et al. 2010; Bertin et al. 2012b). The mechanism of interference involves suppression of NO production by NOS isoforms (Chen et al. 2002). Therefore NSAIDs and COXIBs are beneficial not only to prevent fever, aches, and pains, but also to promote recovery from viral infection (Chen and Reiss 2002a; Steer and Corbett 2003).
Leukotrienes (LT) : 5-Lipoxygenase (5-LO) is the enzyme responsible for LT formation. In general, there is a dynamic ying-yang relationship between the balance of COX and 5-LO activity, since they both use the same initial substrate, arachidonic acid. There are two groups of LT that contribute to pathophysiology based on the receptors used and whether the LT contain cysteine. The CysLT (LTA4, LTC4, LTD4) are associated with fluid production, fibrosis, and airway inflammation in asthma and other pulmonary diseases, while LTB4 is a potent chemoattractant of neutrophils (Ogawa and Calhoun 2006). High levels of LT are seen secondary to mast cell infiltration or Th2-biased host responses patients infected with RSV, HIV, and CMV viruses (Fullmer et al. 2005; Flamand et al. 2004; Gosselin et al. 2005). More importantly, in the CNS, rather than contribute to pathology and BBB disruption, LT play a beneficial role in recruiting neutrophils and promoting recovery from VSV encephalitis (Chen et al. 2001). LT inhibit early stage HIV infection of microglia (Bertin et al. 2012a), but contribute to recruitment of CD4+ cells by astrocytes (Bertin et al. 2014). Those individuals who take nonsteroidal anti-inflammatory drugs will produce more LT, while those with asthma being treated with LOX inhibitors will have more PG; these common medications can have profound consequences on acute or latent viral infections.
Omega-3 fatty acids consumed in diets rich in cold-water fish (or by capsules) are also anti-inflammatory. They attenuate cytokine production and COX activity, downregulate adhesion molecules, and promote recovery from spinal cord injury (De Caterina et al. 2004; Morris et al. 2006; Serhan 2005b; King et al. 2006; Su et al. 2014). Dietary ω-3 fatty acids may prevent or delay Amyotrophic Lateral Sclerosis (Fitzgerald et al. 2014a). In infections, the data are mixed with benefit in HIV, RSV, HBV, influenza, and HSV keratitis (Razzini and Baronzio 1993; Wu et al. 2012; Bryan et al. 2005; Tam Vincent et al. 2013; Rajasagi et al. 2013), but more rapid death in lymphoma associated with the Murine leukemia virus RadLV (Potworowski et al. 1992). Thus, when host inflammation is essential for controlling viral infection, the immune dampening of ω-3 fatty acids contributes to disease.
Lipoxins (LX) are anti-inflammatory products of arachidonic acid; 15-epi-LXA4 is produced in the presence of aspirin. They are produced at temporally and spatially distinct sites from the inflammatory LT; LX signal through SOCS2 (Machado et al. 2006; Serhan 2005a). LXA4 and 15-epi-LXA4 were associated with attenuation of neural stem cell proliferation and differentiation, in contrast to the activity of LTB4, which induced proliferation (Wada et al. 2006). The literature is sparse concerning the contribution of LXs in the resolution of inflammation associated with viral infections (Shirey et al. 2014; Russell and Schwarze 2014); however, it is possible that LX are produced in the CNS during viral infections.
11,12-Epoxyeicosatrienoic acid (EET) and hydroxyleicosatetraenoic acid (HETE) are the products of Cytochrome P450 Epoxygenase, and are also anti-inflammatory, probably through activation of the PPAR nuclear transcription factor family (discussed below) (Node et al. 1999). However, HETEs can also be produced in oxidative damage; they were elevated in plasma of Dengue virus infected people (Seet et al. 2009). 15-HETE was anti-apoptotic in EBV-transformed B cells natoni (Belfiore et al. 2007). 15- and 20-HETE regulate cerebral blood flow, enhancing perfusion (Gebremedhin et al. 2000).
Resolvins : Additional anti-inflammatory lipid molecules are Resolvins and Protectins, which are produced late in inflammation and promote resolution , including in HSV infection (Bannenberg et al. 2005; Russell and Schwarze 2014). The mechanism by which resolvins promote resolution of inflammation is via induction of miRNA that downregulate proinflammatory mRNA (Recchiuti and Serhan 2012).
Protein Isoprenylation
Statins (HMG CoA inhibitors) were developed and licensed to block cholesterol biosynthesis; however, data indicate that statins diminish inflammation. Bisphosphonates block bone resorption by acting on osteoblasts. Farnesyl transferase (motif CAAX, found in some proteins with an unpaired Cys) inhibitors were hoped to inhibit cellular Ras family activity and thus be powerful cancer therapeutics. These three classes of drugs block distinct enzymes in the same lipid biosynthetic pathway and therefore contribute to regulation of cell autonomous and systemic innate immunity.
These inhibitors of protein-lipid modification can be anti-inflammatory, in part, because protein isoprenylation contributes to production of monokines like IL-1 (Mandey et al. 2006). TLR4 signaling is impaired by statins (Methe et al. 2005), as is LPS-induced AKT phosphorylation (Patel and Corbett 2004). Cytokine activation of microglia is negatively regulated by RhoA, which prevents NF-k B activation. RhoA negatively regulates COX-2 expression, leading to increased PGs levels when isoprenylation is inhibited (Degraeve et al. 2001). These drugs suppress both chemokine production and chemokine receptor expression (Veillard et al. 2006).
However, bisphosphonate treatment results in sustained activation of Rac, Cdc42, and Rho (Dunford et al. 2006), possibly because isoprenylation of phosphatases (PTPases) including the PRL family (phosphatase found in regenerating liver) regulates the activity of Rac (Fiordalisi et al. 2006). Rho/Rho-kinase activity modifies actin cytoskeletal proteins and results in dynamic cellular shape changes as well as the activation of NOS-3, resulting in the production of NO and thus, endothelial cell relaxation; inhibition of protein isoprenylation inhibits this cytoskeletal plasticity and changes in blood-flow dynamics (Rikitake and Liao 2005). These drugs may diminish inflammation by inhibiting diapadesis of inflammatory cells (Walters et al. 2002). Statin treatment is beneficial therapy in multiple sclerosis, not by inducing Th2 or Treg cells, but by inhibiting proliferation of inflammatory T cell (Weber et al. 2014b).
Protein isoprenylation is essential for formation of functional clusters of proteins tethered to cellular membranes (Liao and Laufs 2005). Among the functional complexes which require lipid modification for effective enzymatic activity are the small GTPase activating proteins (GAPs) including RhoGAP (Ligeti and Settleman 2006). Monocyte anti-bacterial activity associated with NADPH oxidase, activated by Rac guanine nuclear exchange factor is negatively regulated by statins (Mizrahi et al. 2005). Ras must be farnesylated to interact with phosphoinoside-3-kinase (Rubio et al. 1999).
With respect to viral infections, there have been several reports that protein isoprenylation is essential to replication of HBV, HCV, HDV, RSV, influenza, and HIV (Acheampong et al. 2007; Einav and Glenn 2003; Mehrbod et al. 2014; Gower and Graham 2001; Huang et al. 2006; Kapadia and Chisari 2005). VSV replication in neurons is inhibited ~15-fold by one of the drugs (D’Agostino, unpublished). In HIV-E, statin treatment was unsuccessful in inhibiting the release of virus to CSF (Probasco et al. 2008), and there was a slight increase in the risk of developing herpes zoster (Antoniou et al. 2014).
Bisphosphonates treatment was beneficial in RSV infections, but contributed to human metapneumovirus pathogenesis (Kolli et al. 2014). They were beneficial in HIV infection by blocking the retroviral integrase (Agapkina et al. 2014). Neuropathology associated with TMEV infection was controlled by bisphosphonates and by SHP-1, a protein tyrosine phosphatase (Christophi and Massa 2009).
Membrane fusion , which is important to initiate many virus infections or to release enveloped virus progeny, is inhibited by isoprenylated SNAREs (Grote et al. 2000). Isoprenylated proteins are not incorporated into lipid rafts (Melkonian et al. 1999). An IFN-inducible antiviral protein, human Guanylate-binding protein-1 (hGBP-1), a GTPase, is isoprenylated, and Golgi-associated, thus its activity is impaired in the presence of pathway inhibitors (Modiano et al. 2005). Dengue virus assembly was inhibited by statins (Martinez-Gutierrez et al. 2011). Thus, statins, bisphosphonates, or isoprenyl transferase inhibitors may inhibit viral replication, but may also suppress the host IFN-dependent antiviral pathway(s) and inflammation. Overall, inhibition of protein isoprenylation is beneficial to hosts in a wide range of viral infections .
Vitamin D
Plasma levels of Vitamin D and its biological activity are regulated by many factors including diet, sun exposure, and polymorphisms that regulate its receptor and plasma binding protein. In addition to regulating cytokine expression, Vitamin D positively regulates human antimicrobial peptides including Defensins (Wang 2014). Low levels of Vitamin D have been associated with inflammatory diseases including inflammatory bowel disease, rheumatoid arthritis, systemic lupus erythematosus, atherosclerosis, and asthma (Wobke et al. 2014). Low Vitamin D levels are also associated with increased susceptibility to infections including bacterial, parasitic and HCV, HIV, and influenza (Havers et al. 2014; Kitson et al. 2014; Bryson et al. 2014; Lang et al. 2013).
In the CNS, low Vitamin D levels are a risk factor for multiple sclerosis (Disanto et al. 2012), and normal levels were observed to be neuroprotective and beneficial for maintaining cognition (Anastasiou et al. 2014). Experimental sunlight or ultraviolet light exposure inhibited spinal cord inflammation and reduced demyelination (Wang et al. 2015). In ALS, Vitamin D supplementation may be therapeutic (Gianforcaro and Hamadeh 2014). Serum levels of Epstein-Barr virus were negatively correlated with levels of Vitamin D (Lucas et al. 2011); EBV infection is associated with susceptibility to MS in some individuals (Cocuzza et al. 2014). The mechanism of neuroprotection may be impairment of CD4 extravasation across the BBB (Grishkan et al. 2013). Vitamin D levels have not been studied in other neurotropic viral infections, but we may speculate that normal concentrations may be protective.
Protein Players in Innate Immunity
Many different classes of proteins are critically involved in innate immunity in the CNS. This section will briefly describe the roles of Defensins, Lactoferrin, Complement cascade components, Cytokines and Chemokines. IFNs were discussed earlier.
Defensins
Defensins are small, conserved antimicrobial peptides, produced by many cell types including epithelial and leukocytes, which are found in both very primitive species which lack adaptive immunity and mammals. While inflammatory infiltrating cells may also contribute, in the CNS parenchyma, defensins are synthesized by astrocytes, the choroid plexus , and the hypothalamus (Evans and Harmon 1995; Angeli et al. 1994; Williams et al. 2012, 2014b). Peripherally synthesized defensin molecules can cross the BBB (Schluesener and Meyermann 1995). They have been shown to contribute to elimination of both bacteria and to virus infections by many mechanisms (Wiens et al. 2014; Wilson et al. 2013). In viral infections of the CNS , these include HIV, VZV, HSV, Dengue, adenovirus, and JC (Klotman and Chang 2006; Crack et al. 2012; Rothan et al. 2012; Gwyer Findlay et al. 2013; Wang 2013; Zins et al. 2014). The release of defensins may be regulated by LTB4 (Flamand et al. 2004).
Lactoferrin is a small secreted, iron-complexed protein that has both anti-bacterial and antiviral activity. Lactoferrin is found in milk and plasma and secreted by neutrophils (Baynes and Bezwoda 1994). Recent studies suggest that it may contribute to inhibition of innate immune responses during CNS infections with picornaviruses, alphaviruses, papovaviruses, EBV, and HSV (Seganti et al. 2001; Waarts et al. 2005; McCann et al. 2003; Drobni et al. 2004; Valimaa et al. 2009; Zheng et al. 2012).
Complement cascade components and their receptors are expressed both constitutively and can be induced during immune responses in the CNS. Of course, in the classical cascade, specific antibody must first be synthesized and engage its epitopes, inducing conformational changes in IgG which result in exposure of the cryptic C1q binding site and the initiation of the cascade. The alternative and lectin pathways can also induce activation of complement. IgG can cross the BBB, and can, under circumstances of persisting immune responses, be synthesized in the CNS in tertiary lymph nodes (Phares et al. 2013). But that reflects adaptive and not innate immune responses.
The small anaphylatoxins, C3a, C4a, and C5a, which are proteolytic products of the zymogens, are potently active as activators of vascular permeability and chemoattractants for neutrophils. These molecules are produced in the CNS by astrocytes and microglia (Bruder et al. 2004). C5a is a potent recruiter of polymorphonuclear leukocytes (PMNs) . In VSV encephalitis C5a was not required for host responses (Chen and Reiss 2002b), the redundance of chemokines and LTB4 were sufficient to promote recovery.
Complement receptors include both serpentine 7-transmembrane molecules (which bind C3a, C4a, and C5a) G-protein coupled transmembrane glycoproteins. Endothelial cells and neurons express some complement receptors, including CD46, a measles virus, and Human herpes virus-6 (HHV-6) receptor (Santoro et al. 2003; Schneider-Schaulies et al. 2001; Shusta et al. 2002). Rabies virus infection of the CNS induces the expression of complement genes (Zhao et al. 2011). Complement activation has also been shown to be critical for the development of the adaptive Ab response for WNV (Mehlhop et al. 2005; Dietzschold et al. 1995). In the presence of antibody, exacerbated C3-dependent pathology was observed in MHV-A59, TMEV, and Coxsackie B3 infections (Burrer et al. 2007).
The complement lectin pathway contributes to protection from West Nile virus infection (Fuchs et al. 2011). Complement contributes to the neurovirulence of Sindbis, HIV, SIV, and Bornavirus infections (Bruder et al. 2004; Speth et al. 2004; Griffin et al. 1997; Dietzschold et al. 1995; Johnston et al. 2001; Phares et al. 2013). In TMEV infection, complement activation contributes to seizures (Libbey et al. 2010). HSV, vaccinia, and the murine gamma herpes virus MHV-68, have developed evasive proteins that prevent complement activity and contribute to their disease pathogenesis (Kapadia et al. 2002).
A vaccinia virus protein has been isolated and has been proposed as a therapeutic when host complement activation is pathogenic in the CNS (Pillay et al. 2005). Recombinant HSV-1 deficient in the complement-interacting γ134.5 gene product has been proposed as an effective vector for viral oncolysis (Broberg and Hukkanen 2005). Similarly, herbal proteins are also able to block complement and have been suggested as potential neuroprotective therapeutics (Kulkarni et al. 2005). In experimental infection with enterovirus 71 (EV71), a fusion protein of complement receptor 2 (CR2) and the inhibitor Crry prevented complement activation, alleviating local inflammation , and preventing severe disease associated with the picornavirus (Qiu et al. 2012).
Chemokines : IFNs induce expression of chemokines including IFN-inducible 10KD protein (IP-10; CXCL-10; which also has anti-angiogenic activity), Mig/Crg-2 (CXCL-9), and I-TAC (CXCL11). These proteins may also have defensin-like activity, nonspecifically arming anti-microbial responses (Cole et al. 2001). These chemokines recruit neutrophils, natural killer (NK) cells, monocytes, and T cells to the brain (Williams et al. 2014b). They have been shown to be important in the host’s response to LCMV, MHV, VSV, and TMEV infections (Rubio et al. 2006; Asensio et al. 1999; Ireland and Reiss 2006; Liu et al. 2000; Palma and Kim 2001). Fractalkine (C3XCL1) has been associated with recruitment of microglia, brain macrophages, and peripheral cells in HIV dementia (Cotter et al. 2002). Chemokine receptors CCR2 and CXCR3 are essential for recruitment of peripheral cells during SFV and WNV encephalitis (Michlmayr et al. 2014).
Many molecules have chemoattractant activity, but are not in the peptide families of chemokines. These include the cytokine IL-12, produced by antigen presenting cells, that can recruit NK cells (Michel et al. 2012), the bacterial tri-peptide f-MetLeuPhe, and the leukotriene LTB4 for neutrophil recruitment (Lefebvre et al. 2011).
Cytokines are comprised of dozens of different protein mediators, which are generally secreted by one cell and act on the secreting cell (autocrine), locally (paracrine), or systemically (endocrine) to either activate and differentiate another cell type, or regulate the activity of another cell. Generally, the receptors are heterodimers of surface glycoproteins, signaling through tyrosine kinase cascades. Although many investigators have tested the effects of individual molecules in experimental systems, in real life, cytokines are secreted as a part of a coordinated program with many different molecules (including receptor antagonists) released at the same time; it is the composite of these mediators’ signal transduction pathways that lead to the outcome. The differentiation state of the downstream cell, the quantity, and duration of exposure determine the response. Cytokines can downregulate their receptors by internalization, leading to desensitization, or induce enhanced expression of their own (and other) receptor and second messengers, which can positively regulate subsequent responses.
In the brain these molecules can be produced by both parenchymal cells and infiltrating inflammatory cells . Two excellent reviews of cytokines and the CNS are a book edited by Ransohoff and Benviste and an article by Campbell (Ransohoff and Benveniste 2005; Campbell 2005). It is the balance between proinflammatory cytokines such as TNF-α, IL-17, or IL-23 and the anti-inflammatory molecules such as IL-4, TGF-β, or IL-10 that ultimately determine the outcome.
Systemically produced cytokines can lead to CNS consequences ranging from the fever response to IL-1 (inducing the production of PGE2) or TNF-α secretion, resulting in transient disruption of the BBB. Excessive peripheral release of cytokines has been associated with “sickness behavior” (Dantzer 2005; Watkins and Maier 2000).
In infections, proinflammatory cytokines can lead to beneficial outcomes, such as IL-12, TNF-α, or IFN-γ induction of NO which leads to elimination of VSV infections (Komatsu et al. 1996, 1997, 1999a; Ireland and Reiss 2004; Ireland et al. 1999, 2005); however, IL-12/IL-23 and IL-18 are not necessarily critical even if synthesized or administered (Hodges et al. 2001; Ireland et al. 2005). These are important in Sindbis, TMEV, and MHV infections (So et al. 2006; Binder and Griffin 2003; Lane et al. 1996; Olson and Miller 2009; Rempel et al. 2004). Excessive expression of proinflammatory cytokines, in other circumstances, may contribute to pathology (neurovirulence or neurodegeneration) in Bornavirus, JEV, HTLV-1, HIV/SIV, MHV, TMEV, enterovirus 71, LCMV, canine distemper, rabies, and VEE. The proinflammatory cytokine IL-27 induced IL-10 production in MHV-A59 infection, leading to increased demyelination and reduced control of viral replication (de Aquino et al. 2014). Ironically, the anti-inflammatory cytokine IL-4 may be associated with resistance to human WNV infection in a GWAS study (Qian et al. 2014). Cytokines are essential mediators in viral infections of the CNS. Some cytokines are neuropoietic, promoting recovery (Bauer et al. 2007). The timing, quantity, and balance of these bioactive molecules determine the outcome: recovery or pathology.
Transcription Factors Regulating Inflammation
Many transcription factor families regulate responses, and the roles of IFN-inducible STATs have been discussed above. In this section, three classes of transcriptional factors will be described: Hypoxia-inducible factor, Peroxisome proliferation activating receptor, and High mobility group-1 protein. Hypoxia-inducible factor-1α (HIF-1α) is a transcription factor whose expression is triggered by transient hypoxia (or ischemia/stroke); it induces the expression of a number of genes including defensins, the adenosine A2B receptor, Vascular Endothelial Growth Factor (VEGF), COX-2, NOS-2, and NOS-3 (Kong et al. 2006; Hellwig-Burgel et al. 2005; Peyssonaux and Johnson 2004). VEGF regulates not only BBB permeability, but also angiogenesis. HIF-1α expression can be induced by IL-1, TNF-α, and possibly TLR agonists (Hellwig-Burgel et al. 2005; Argaw et al. 2006). It is expressed during inflammatory and demyelinating diseases (Aboul-Enein et al. 2003). In studies with viral vectors for oncolysis in the CNS, HIF-1α expression was enhanced (Shen et al. 2006; Post et al. 2004). Thus, HIF-1α expression , whether elicited by transient vascular compromise or cytokine expression, enhances the innate antiviral (and anti-bacterial) gene expression and may lead to increased BBB perfusion of the local area.
Peroxisome Proliferating Activating Receptors , nuclear hormone transcription factors, have three isoforms α, β, and γ, each with distinct activity and expression. PPAR-γ, the canonical nuclear hormone receptor involved in muscle glucose uptake and lipid homeostasis and cell differentiation, is translated as two splice variants, γ1 and γ2. PPAR-γ2, 30 amino acids longer than γ1, is expressed in high levels of adipose tissue. PPAR-γ ligands are polyunsaturated fatty acids, eicosanoids, FA oxidation products (13-HODE and 15-HETE), J-series prostaglandins (15-deoxy-D12,14-prostaglandin J2), some nonsteroidal anti-inflammatory drugs (NSAIDS), and insulin sensitizing thiazolidinediones (TZDs). PPAR-γ functions as an obligate heterodimer with Retinoic X receptor (RXR) to activate transcription by binding to 5′ promoters of target genes (Grygiel-Gorniak 2014; Fidaleo et al. 2014).
PPAR-γ agonists modulate inflammatory responses in the CNS, resulting in the reduction of iNOS in cerebellar granule cells. PPAR-γ signaling has anti-inflammatory function in EAE, PPAR-γ agonists alleviate symptoms with antagonists performing the opposite, indicating regulation of auto-reactive Th1 and Th17 cells (Kanakasabai et al. 2010). PPAR-γ also can regulate pathologic immune responses within the CNS in MS (Shukla et al. 2010). Alzheimer’s disease is a severe neurodegenerative disease characterized by the accumulation of amyloid plaques accompanied with activated microglia. TZD treatment in in vitro experiments with microglia and monocytes attenuated the secretion of proinflammatory cytokines. Medium from TZD-treated microglia was neuroprotective (Drew et al. 2006).
Infection by Adenovirus 36 induces PPAR expression, and increases insulin sensitivity (Pasarica et al. 2006). In vitro treatment of cells with PPAR agonists inhibited replication of RSV, HHV8, HCV, HIV, and VSV (Rakic et al. 2006; Bryan et al. 2005; Arnold and Konig 2006; Herrera et al. 2008), although the mechanism(s) by which this inhibition occurred have not been elucidated. However, HBV X-associated protein 2 complexes with PPAR and inactivates it (Sumanasekera et al. 2003), an evasive pathway. Thus treatment with PPAR agonists, such as TZDs, may be beneficial for treatment of viral encephalitis both as potential antiviral compounds, and as anti-inflammatory drugs in infections where pathology is associated with inflammation , such as Bornaviral disease and HIV infection (Kim et al. 2012).
High Mobility Group protein B1 (HMGB1) is unique among transcription factors as it is found not only in the nucleus, but also in the cytoplasm associated with α-Synuclein filaments (Lindersson et al. 2004) and is actively released as an alarmin. Its expression may be upregulated by IFN (Seeler et al. 2001). HMGB1 may engage AMIGO receptors of neurons where it regulates neurite outgrowth, TLR 2 and TLR 4, or the Receptor for Advanced Glycation Endproducts (RAGE) which results in a proinflammatory response by microglia, macrophages, and dendritic cell maturation (O’Connor et al. 2003).
HMGB1 release has been shown to be neurotoxic in ischemia, Alzheimer’s disease (Kim et al. 2006a) and in Bornavirus disease, HTLV-1, HIV-1, and WNV infections (Troseid et al. 2013; Kimura and Mori 2014; Zhao et al. 2006; Chu and Ng 2003). HMGB1 is a Janus molecule with both regulatory transcriptional activity and signaling of tissue damage; it may be important in eliciting innate immunity during viral encephalitis.
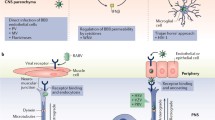
BBB
Within the CNS, there are anatomically distinct regions that have some constitutive vascular permeability (Circumventricular organ, choroid plexus, for example), but most areas are highly restricted in access to circulating cells and proteins. Astrocytes regulate the perfusion of the parenchyma by controlling vasodilation of the cerebrovascular capillaries through the activity of NOS-3 (Sporbert et al. 1999; Komatsu et al. 1999b). The BBB is associated with the immune privilege of the brain and separates the CNS from peripheral circulation and immune surveillance that is characteristic of the periphery. Entry of cells requires adhesion to the brain microvascular endothelium, release of MMPs to degrade tight junctions and the extracellular matrix, as well as migration along a gradient of chemoattracting molecules (Bechmann et al. 2007; Arima et al. 2013). The chemoattracting molecules for circulating cells range from LTB4 to complement products, chemokines, cytokines, FLT3L, and even ATP (all discussed in the relevant sections, above).
The BBB breakdown associated with infection results from excessive normal physiological process regulating blood flow within the CNS (Proescholdt et al. 1999; Abbott et al. 2006). Activation leads to NOS-3-expressing astrocytes to release NO, which induces guanadyl cyclase to produce cGMP, leading to endothelial and smooth muscle cell relaxation. Other mediators such as the small complement cascade mediators C3a, C4a and C5a, VEGF and PGE2 can also lead to the relaxation and increased permeability of the BBB .
A hallmark of many viral infections including VSV, Rabies, Flaviviruses, HIV, TMEV, and WNV (Daniels et al. 2014; Wang et al. 2013; Neal 2014; Johnson et al. 2014; Williams et al. 2014a; Chen et al. 2002) is the breakdown of the BBB. However the global breakdown of the BBB seen in fatal LCMV (Kang and McGavern 2010) and in fatal VSV infections is extreme, and unusual. In most cases, the overall integrity of the BBB is maintained, but in discrete regions, there is increased perfusion leading to entry of normally excluded proteins from circulation.
Apoptosis and Autophagy
Cells under stress from viral infection, TNF-family cytokines, CTL recognition, as well as many other stimulate can undergo programmed cell death (apoptosis) (Danial and Korsmeyer 2004). I will not review the cellular pathways which lead to genome fragmentation and membrane inversion, but will focus, instead, on associations between viral infection and apoptosis. Cells which commit suicide in this manner may spare the host from continued viral replication, a benefit, especially since most cells can be replaced by stem cells in that organ; however, if the infected cell undergoing apoptosis were a neuron , significant consequences might ensue (Perkins 2005).
Neurotropic viruses which elicit apoptosis include alphaviruses (Griffin 2005), Flaviviruses (Clarke et al. 2014), Picornaviruses (Ruller et al. 2012), VSV (Gaddy and Lyles 2007), Rabies (Fu and Jackson 2005), coronaviruses (Desforges et al. 2014), LCMV (Sun et al. 2014), reovirus (Dionne et al. 2013), JC (Merabova et al. 2012), HIV-1 (Geffin and McCarthy 2013), and HTLV-1 (Marriott and Semmes 2005). In fact, apoptosis is such an important cellular defensive response to viral infections that poxviruses have developed an evasive pathway, using serine protease inhibitors (serpins) (Taylor and Barry 2006). But, scientists are clever and have selected apoptosis as a tool for viral oncolysis.
At other times, viral infections or cellular stress from starvation can lead to recycling of large volumes of cytoplasmic contents by generation of vesicles which fuse with lysosomes (autophagy, self-eating) (Deretic 2005). This pathway can become dysregulated, resulting in inflammation and neurodegenerative diseases (Deretic et al. 2013; Noch and Khalili 2013; He and Klionsky 2006).
Some picornaviruses use this cellular response to develop additional membranes on which to replicate (Jackson et al. 2005). In the CNS infections caused by Coxsackie B3 (Tabor-Godwin et al. 2012), Sindbis (Sumpter and Levine 2011), HIV (Levine and Sodora 2006), LCMV (El-Azami-El-Idrissi et al. 2005), and HSV (Korom et al. 2013) autophagy-associated pathology has been reported. Thus, in general, autophagy is an innate host cellular response to suppress viral infection, however, some viruses, to their benefit, can manipulate it.
Parenchymal and Inflammatory Cells in Innate Immunity in the CNS
Infiltration of Peripheral Circulating Cells
Normally there are very few lymphocytes, neutrophils, and NK cells in brain parenchyma. Infiltration of inflammatory cells ranging from PMNs to NK cells to macrophages and finally lymphocytes takes place in response to a series of signals from both chemoattractant molecules and orchestrated binding to microvascular endothelial cell surface molecules (Williams et al. 2001; Luster et al. 2005). For cells to cross the endothelial vessel wall, they must diapedese and then digest the basement membrane with MMPs. This review will not discuss the infiltration of antigen-specific T cells or B cells, as it is limited to innate immune responses.
Neutrophils are the first cell to infiltrate sites of viral infection. Chemoattractants for neutrophils include Adenosine, ATP, f-MetLeuPhe, C5a, LTB4, and chemokines (Gabriel et al. 2013). As described above, they produce defensins, cytokines, mediators from aracodonic acid, and other bioactive compounds. In VSV encephalitis, LTB4 and chemokines, but not C5a, are essential (Ireland and Reiss 2006; Chen and Reiss 2002b; Chen et al. 2001). PMN infiltration is also characteristic of Murray Valley encephalitis, MHV, HSV-1, TMEV, Western equine encephalitis, and adenovirus infections (Libbey et al. 2011; Weiss et al. 2007; Matthews et al. 2000; Reed et al. 2005; Wakimoto et al. 2003; Welsh et al. 2004; Weinberg et al. 2007; Zhou et al. 2003; Campbell et al. 2001; Bell et al. 1996).
NK cells are generally the second cell type to diapedese in response to viral infections of the CNS in response to both chemokines and IL-12. NK cells nonspecifically recognize patterns of receptor expression on cells and are sensitive to low levels of MHC molecules, and, when activated, release IFN-γ, perforin and granzymes, like CD8+ CTL. NK cells have been associated with the host response to JEV, WNV, Sindbis, MHV, Bornavirus, EBV, HSV-1, VSV, SIV, CMV, TMEV, and enterovirus 71 (Hatalski et al. 1998; Christian et al. 1996; Wensman et al. 2011; Fernandes et al. 2011; Wang et al. 2013; Mott et al. 2011; Brehin et al. 2008; Larena et al. 2013; Ogura et al. 2013). NK cells contribute not only lytic activity against virally infected cells, but also are a significant source of IFN-γ secretion, both of which may regulate viral replication .
Antigen Processing and Presentation
Pioneering work by the late Helen Cserr and her colleagues including Paul Knopf explored lymphatic drainage of soluble antigens and their ability to evoke an acquired immune response (Knopf et al. 1995). This drainage is polarized from rostral to caudal, and is modest and does not include the hallmarks of peripheral tissue dendritic cells bearing antigens to the draining lymph nodes.
Antigen processing and presentation is at the interface between the innate and adaptive immune responses to pathogens. There is little constitutive expression of Class II MHC molecules in the undisturbed CNS; however, both astrocytes and microglia readily express these molecules in response to inflammatory cytokines, especially IFN-γ (Gresser et al. 2000). Infection indirectly induces the expression of MHC II and enhances the expression of MHC I by parenchymal cells (Berman et al. 1998; Abraham and Manjunath 2006; Aguirre and Miller 2002; Alldinger et al. 1996; Caplazi and Ehrensperger 1998).
Perivascular macrophages have been shown to be an important player in antigen presentation for the brain in infections and autoimmune disease (Williams and Hickey 2002). Macrophages and microglia may produce proinflammatory (M1) or anti-inflammatory (M2) cytokines and bioactive mediators; M2 microglia antagonize neuroinflammation (Cherry et al. 2014). Mi cells contain multimolecular complexes called inflammasomes, intracellular sensors for pathogens and danger signals; these inflammasomes generate substantial quantities of proinflammatory IL-1 and IL-18 (Walsh et al. 2014). M1 microglia are characteristic of VSV, neurotropic influenza virus, and TMEV infections (Jurgens et al. 2012; Son et al. 2009; Steel et al. 2014).
Mast cells are often overlooked except in studies of Type 1 hypesensitivities. Mast cells can participate with glia in neuroinflammation (Skaper et al. 2014). IL-33, produced by glia, would promote mast cells; in TMEV infections, IL-33 was produced (Hudson et al. 2008). In HIV-E with immune reconstitution syndrome after antiretroviral therapy, mast cells contribute to CNS pathology (Rushing et al. 2008).
Dendritic cells are the principal antigen presenting cells in the periphery and are a complex group of cells whose phenotypes rival T cell subsets (Guilliams et al. 2014; Cohn and Delamarre 2014). They are extremely difficult to detect in undisturbed brain tissue. During immune responses in the CNS, it is possible to find cells expressing dendritic cell markers (Matyszak and Perry 1997; Ambrosini et al. 2005). In addition to any chemokines, Flt3L has been shown to recruit dendritic cells to the CNS (Curtin et al. 2006). Parenchymal dendritic cells have numerous phenotypes (D’Agostino et al. 2012a), and VSV infection induces CD103+ CD11b+ cells (D’Agostino et al. 2012b). Dendritic cell responses are age dependent and may contribute to the susceptibility of immature hosts to some forms of viral encephalitis (Taylor et al. 2014). However, the microenvironment during which dendritic cells are exposed to virus can determine whether pro-inflammatory or Treg responses are found (Durrant et al. 2013; Martinez et al. 2014).
HPAI Axis and Neural-Endocrine Regulation
The hypothalamic-pituitary-adrenal-immune (HPAI) axis controls not only fight-or-flight in response to stressors, but also critical control of immune responses to infections. There are short-term and chronic stress manifestations of this (Eskandari and Sternberg 2002; Shanks et al. 1998), with long-term compromise of immune responses to viral infections (Silverman et al. 2005). This may be manifest as alterations in the humoral response to viral infection (Ijaz et al. 1990). Acute stress may also alter the integrity of the BBB (Esposito et al. 2001), thus potentially permitting entry to otherwise excluded viruses.
Sympathetic Nervous System
Chemical sympathectomy , achieved by infusion of 6-hydroxydopamine, has profound effects on the peripheral immune response, as there is sympathetic innervation of the spleen and lymph nodes (Callahan et al. 1998). Hosts are more susceptible to bacterial, VZV reactivation, and HSV-1 infections (Cao et al. 2002; Massad et al. 2004; Leo et al. 1998; Templeton et al. 2008), but when hosts are already immune suppressed, whether by malnutrition or by lentivirus infections, they are not further compromised (Kelley et al. 2002; Gonzalez-Ariki and Husband 1999). We tested whether chemical sympathectomy altered the ability of peripheral plasmacytoid dendritic cells to produce IFN-β in response to VSV infection of the CNS, and found no contribution of innervation of secondary lymphoid organs in this response (Trottier et al. 2007). However, production of proinflammatory cytokines and increased pathology were observed in influenza virus infection to be associated with the sympathetic response (Grebe et al. 2010).
Cholinergic pathways have been shown to be anti-inflammatory in bacterial model systems and ischemia-reperfusion injury (Tracey 2007), inhibiting cytokine production and tissue injury in models such as colitis (Sun et al. 2013). In a transgenic model, HIV-1 was associated with learning deficits; activation of the cholinergic pathway with nicotine did not ameliorate the loss vigorito (Vigorito et al. 2013). But, there are no published reports in the literature on the impact of this regulatory neurotransmitter pathway on neurotropic viral infections.
Leptin was originally identified as the gene product deficient in obese mice and was found to regulate energy balance, but like so many other effector molecules , has many other activities. Proinflammatory cytokines, induced during infections, can upregulate production of leptin (Yu et al. 2014), and lead to anorexia (Langhans 2000). Central leptin and insulin resistance has been associated with Adenovirus (SMAM-1 and Ad36) infection, leading to obesity (Wierucka-Rybak and Bojanowska 2014). Recent evidence suggests that this adipocyte-produced protein is also immunoregulatory and is, in fact, a proinflammatory cytokine (Lord 2006). Leptin is pathogenic in EAE (Matarese et al. 2002) by virtue of its action on dendritic cells, resulting in the induction of Th1 responses (Mattioli et al. 2005) and its inhibition of thymic apoptosis (Mansour et al. 2006). In experimentally induced obese mice, more severe influenza pathology was associated with leptin (Zhang et al. 2013a). Well-nourished infants, with elevated leptin levels, were more susceptible to Dengue hemorrhagic fever, than were thinner children (Libraty et al. 2014). High leptin levels were observed in HCV-infected people with chronic fatigue symptoms (El-Gindy et al. 2012). In contrast, low levels of leptin are observed in HIV infections, and exaggerated in those patients with lipodystrophy veloso (Veloso et al. 2012). Therefore, leptin may be a potential target for therapeutic intervention in persistent inflammatory infections of the CNS such as HIV-E and Bornaviral disease.
Sex hormones regulate more pathways than just those in secondary sexual organs.
Estrogen is neuroprotective in infection, Alzheimer’s disease, traumatic injury, and ischemia (Barreto et al. 2014; Cue et al. 2015). Estrogen has profound immuno-modulating effects ranging from induction of NOS-3, and thus increased vascular perfusion (Hayashi et al. 1997). Selective estrogen receptor modulators enhance neurogenesis and spine density (Khan et al. 2015). Movement disorders including Parkinson’s disease are more frequent in males (Lubomski et al. 2014). Estrogen positively regulates expression of IFN-γ (Fox et al. 1991); this is clearly linked with the increased frequency of females who have Th1-associated autoimmune diseases such as EAE/MS (Whitacre et al. 1999).
Additionally, sex hormones regulate PPARs, and can influence the severity of EAE (Dunn et al. 2007). Females may be more resistant to some viral infections due to enhanced Th1 responses, including VSV, TMEV, and HSV-1 (Markle and Fish 2014; Forger et al. 1991; Fuller et al. 2005; Peter and Sevall 2001).
Androgens may be immunosuppressive by inhibiting Th1 differentiation (Kissick et al. 2014). This may contribute to diminished efficacy of vaccination and the production of anti-influenza antibody by males (Furman et al. 2014). Hepatitis B and HCV disease is more severe in males, and correlated with testosterone levels (Tian et al. 2012; White et al. 2012).
There is an effect of host sex in viral infections of the CNS. Female mice are more resistant than males to lethal VSV infections (Barna et al. 1996), and show improved clearance of MHV and Semliki Forest virus infection from oligodendrocytes (Fazakerley et al. 2006; Parra et al. 1999). However, female mice undergo more severe demyelination in TMEV infections (Fuller et al. 2005). HIV-E and HIV-dementia incidence may be lower in females due to estrogen effects on immune responses (Wilson et al. 2006). Thus, where inflammation is beneficial to clearing virus and resolving infection, females are at an advantage. In contrast, where inflammation contributes to viral disease pathology in the CNS , females are disproportionately affected.
Summary and Speculation
Innate immunity in the CNS is complex and includes protein effectors (IFNs, cytokines, chemokines, defensins, complement, lactoferrin, and other molecules), lipid mediators (PGs, LTs, Cannabinoids, etc.), small diffusible molecules (NO, ONOO−), and both parenchymal and inflammatory cells. These responses are highly regulated and are triggered, in part, by receptors that bind common pathogen-associated or damage-associated molecules. Combined, these responses provide a critical barrier, controlling viral replication until adaptive immune responses are marshalled. Inflammation is essential, but must be carefully controlled to prevent tissue damage and pathology. Virtually every pathway has regulation (e.g., kinases and phosphatases) that ultimately determines the magnitude and then the resolution of responses . We are still learning about the essential pathways and their controls, about how drugs may alter the dynamic equilibrium, and in which situations responses need to be ramped up or downregulated. Nonetheless, innate immune responses are critically essential in the central nervous system, to buy the host time for the adaptive immune response to mature and provide antigen-specific effector T cells and antibody to the viral infection. I predict we will make many future advances that will benefit the health of the populations who are infected with neurotropic viruses.
References
Aalaei-andabili SH, Rezaei N (2013) Toll like receptor (TLR)-induced differential expression of microRNAs (MiRs) promotes proper immune response against infections: a systematic review. J Infect 67(4):251–264. doi:10.1016/j.jinf.2013.07.016
Abbott NJ, Ronnback L, Hansson E (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7(1):41–53
Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, Hornung V (2013) Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503(7477):530–534. doi:10.1038/nature12640
Aboul-Enein F, Rauschka H, Kornek B, Stadelmann C, Stefferl A, Bruck W, Lucchinetti C, Schmidbauer M, Jellinger K, Lassmann H (2003) Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol 62(1):25–33
Abraham S, Manjunath R (2006) Induction of classical and nonclassical MHC-I on mouse brain astrocytes by Japanese encephalitis virus. Virus Res 119(2):216–220
Acheampong E, Parveen Z, Mengistu A, Ngoubilly N, Wigdahl B, Lossinsky AS, Pomerantz RJ, Mukhtar M (2007) Cholesterol-depleting statin drugs protect postmitotically differentiated human neurons against ethanol- and human immunodeficiency virus type 1-induced oxidative stress in vitro. J Virol 81(3):1492–1501
Adams O, Besken K, Oberdorfer C, MacKenzie CR, Takikawa O, Daubener W (2004) Role of indoleamine-2,3-dioxygenase in alpha/beta and gamma interferon-mediated antiviral effects against herpes simplex virus infections. J Virol 78(5):2632–2636
Agapkina J, Yanvarev D, Anisenko A, Korolev S, Vepsalainen J, Kochetkov S, Gottikh M (2014) Specific features of HIV-1 integrase inhibition by bisphosphonate derivatives. Eur J Med Chem 73:73–82. doi:10.1016/j.ejmech.2013.11.028
Aguirre K, Miller S (2002) MHC class II-positive perivascular microglial cells mediate resistance to Cryptococcus neoformans brain infection. Glia 39(2):184–188
Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, Maringer K, Bernal-Rubio D, Shabman RS, Simon V, Rodriguez-Madoz JR, Mulder LC, Barber GN, Fernandez-Sesma A (2012) DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog 8(10):e1002934. doi:10.1371/journal.ppat.1002934
Akira S, Hemmi H (2003) Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett 85(2):85–95
Albornoz A, Carletti T, Corazza G, Marcello A (2014) The stress granule component TIA-1 binds tick-borne encephalitis virus RNA and is recruited to perinuclear sites of viral replication to inhibit viral translation. J Virol 88(12):6611–6622. doi:10.1128/jvi.03736-13
Alldinger S, Wunschmann A, Baumgartner W, Voss C, Kremmer E (1996) Up-regulation of major histocompatibility complex class II antigen expression in the central nervous system of dogs with spontaneous canine distemper virus encephalitis. Acta Neuropathol (Berl) 92(3):273–280
Ambrosini E, Remoli ME, Giacomini E, Rosicarelli B, Serafini B, Lande R, Aloisi F, Coccia EM (2005) Astrocytes produce dendritic cell-attracting chemokines in vitro and in multiple sclerosis lesions. J Neuropathol Exp Neurol 64(8):706–715
Anastasiou CA, Yannakoulia M, Scarmeas N (2014) Vitamin D and cognition: an update of the current evidence. J Alzheimers Dis 42:S71–S80. doi:10.3233/jad-132636
Andrews DM, Matthews VB, Sammels LM, Carrello AC, McMinn PC (1999) The severity of Murray valley encephalitis in mice is linked to neutrophil infiltration and inducible nitric oxide synthase activity in the central nervous system. J Virol 73(10):8781–8790
Angeli A, Masera RG, Staurenghi AH, Solomon S, Bateman A, Sartori ML, Lazzero A, Griot G (1994) The expanding field of hypothalamic-pituitary-adrenal modulation of human natural killer cell activity. Ann N Y Acad Sci 719:328–342
Antoniou T, Zheng H, Singh S, Juurlink DN, Mamdani MM, Gomes T (2014) Statins and the risk of herpes zoster: a population-based cohort study. Clin Infect Dis 58(3):350–356. doi:10.1093/cid/cit745
Argaw AT, Zhang Y, Snyder BJ, Zhao ML, Kopp N, Lee SC, Raine CS, Brosnan CF, John GR (2006) IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J Immunol 177(8):5574–5584
Arias JF, Heyer LN, von Bredow B, Weisgrau KL, Moldt B, Burton DR, Rakasz EG, Evans DT (2014) Tetherin antagonism by Vpu protects HIV-infected cells from antibody-dependent cell-mediated cytotoxicity. Proc Natl Acad Sci U S A 111(17):6425–6430. doi:10.1073/pnas.1321507111
Arima Y, Kamimura D, Sabharwal L, Yamada M, Bando H, Ogura H, Atsumi T, Murakami M (2013) Regulation of immune cell infiltration into the CNS by regional neural inputs explained by the gate theory. Mediators Inflamm 2013:898165. doi:10.1155/2013/898165
Arnold R, Konig W (2006) Peroxisome proliferator-activated receptor-gamma agonists inhibit the replication of respiratory syncytial virus (RSV) in human lung epithelial cells. Virology 350(2):335–346
Asensio VC, Kincaid C, Campbell IL (1999) Chemokines and the inflammatory response to viral infection in the central nervous system with a focus on lymphocytic choriomeningitis virus. J Neurovirol 5(1):65–75
Atkin G, Paulson H (2014) Ubiquitin pathways in neurodegenerative disease. Front Mol Neurosci 7:63. doi:10.3389/fnmol.2014.00063
Auriel E, Regev K, Korczyn AD (2014) Nonsteroidal anti-inflammatory drugs exposure and the central nervous system. Handb Clin Neurol 119:577–584. doi:10.1016/b978-0-7020-4086-3.00038-2
Baccala R, Welch MJ, Gonzalez-Quintial R, Walsh KB, Teijaro JR, Nguyen A, Ng CT, Sullivan BM, Zarpellon A, Ruggeri ZM, de la Torre JC, Theofilopoulos AN, Oldstone MB (2014) Type I interferon is a therapeutic target for virus-induced lethal vascular damage. Proc Natl Acad Sci U S A 111(24):8925–8930. doi:10.1073/pnas.1408148111
Baltzis D, Qu LK, Papadopoulou S, Blais JD, Bell JC, Sonenberg N, Koromilas AE (2004) Resistance to vesicular stomatitis virus infection requires a functional cross talk between the eukaryotic translation initiation factor 2alpha kinases PERK and PKR. J Virol 78(23):12747–12761
Bampi C, Rasga L, Roux L (2013) Antagonism to human BST-2/tetherin by Sendai virus glycoproteins. J Gen Virol 94(Pt 6):1211–1219. doi:10.1099/vir.0.051771-0
Banach M, Piskorska B, Czuczwar SJ, Borowicz KK (2011) Nitric oxide, epileptic seizures, and action of antiepileptic drugs. CNS Neurol Disord Drug Targets 10(7):808–819
Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, Hong S, Serhan CN (2005) Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol 174(7):4345–4355
Barna M, Komatsu T, Bi Z, Reiss CS (1996) Sex differences in susceptibility to viral infection of the central nervous system. J Neuroimmunol 67(1):31–39, doi: S0165572896000227 [pii]
Barreto GE, Santos-Galindo M, Garcia-Segura LM (2014) Selective estrogen receptor modulators regulate reactive microglia after penetrating brain injury. Front Aging Neurosci 6:132. doi:10.3389/fnagi.2014.00132
Bauer S, Kerr BJ, Patterson PH (2007) The neuropoietic cytokine family in development, plasticity, disease and injury. Nat Rev Neurosci 8(3):221–232
Baynes RD, Bezwoda WR (1994) Lactoferrin and the inflammatory response. Adv Exp Med Biol 357:133–141
Bechmann I, Galea I, Perry VH (2007) What is the blood-brain barrier (not)? Trends Immunol 28(1):5–11
Belayev L, Busto R, Watson BD, Ginsberg MD (1995) Post-ischemic administration of HU-211, a novel non-competitive NMDA antagonist, protects against blood-brain barrier disruption in photochemical cortical infarction in rats: a quantitative study. Brain Res 702(1–2):266–270
Belfiore MC, Natoni A, Barzellotti R, Merendino N, Pessina G, Ghibelli L, Gualandi G (2007) Involvement of 5-lipoxygenase in survival of Epstein-Barr virus (EBV)-converted B lymphoma cells. Cancer Lett 254(2):236–243. doi:10.1016/j.canlet.2007.03.010
Bell MD, Taub DD, Kunkel SJ, Strieter RM, Foley R, Gauldie J, Perry VH (1996) Recombinant human adenovirus with rat MIP-2 gene insertion causes prolonged PMN recruitment to the murine brain. Eur J Neurosci 8(9):1803–1811
Berlanga JJ, Ventoso I, Harding HP, Deng J, Ron D, Sonenberg N, Carrasco L, de Haro C (2006) Antiviral effect of the mammalian translation initiation factor 2alpha kinase GCN2 against RNA viruses. EMBO J 25(8):1730–1740
Berman NE, Sheffield LG, Purcell J, Joag SV, Narayan O, Cheney P (1998) Gradient of microglial activation in the brain of SIV infected macaques. J Neuro AIDS 2(1):43–54
Bernstein HG, Keilhoff G, Steiner J, Dobrowolny H, Bogerts B (2011) Nitric oxide and schizophrenia: present knowledge and emerging concepts of therapy. CNS Neurol Disord Drug Targets 10(7):792–807
Bertin J, Barat C, Belanger D, Tremblay MJ (2012a) Leukotrienes inhibit early stages of HIV-1 infection in monocyte-derived microglia-like cells. J Neuroinflammation 9:55. doi:10.1186/1742-2094-9-55
Bertin J, Barat C, Methot S, Tremblay MJ (2012b) Interactions between prostaglandins, leukotrienes and HIV-1: possible implications for the central nervous system. Retrovirology 9:4. doi:10.1186/1742-4690-9-4
Bertin J, Jalaguier P, Barat C, Roy MA, Tremblay MJ (2014) Exposure of human astrocytes to leukotriene C4 promotes a CX3CL1/fractalkine-mediated transmigration of HIV-1-infected CD4(+) T cells across an in vitro blood-brain barrier model. Virology 454–455:128–138. doi:10.1016/j.virol.2014.02.007
Bhattacharyya S (2014) Can’t RIDD off viruses. Front Microbiol 5:292. doi:10.3389/fmicb.2014.00292
Billcliff PG, Rollason R, Prior I, Owen DM, Gaus K, Banting G (2013) CD317/tetherin is an organiser of membrane microdomains. J Cell Sci 126(Pt 7):1553–1564. doi:10.1242/jcs.112953
Binder GK, Griffin DE (2003) Immune-mediated clearance of virus from the central nervous system. Microbes Infect 5(5):439–448
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8(1):57–69
Brehin AC, Mouries J, Frenkiel MP, Dadaglio G, Despres P, Lafon M, Couderc T (2008) Dynamics of immune cell recruitment during West Nile encephalitis and identification of a new CD19+B220-BST-2+ leukocyte population. J Immunol 180(10):6760–6767
Broberg EK, Hukkanen V (2005) Immune response to herpes simplex virus and gamma134.5 deleted HSV vectors. Curr Gene Ther 5(5):523–530
Brodie C, Weizman N, Katzoff A, Lustig S, Kobiler D (1997) Astrocyte activation by Sindbis virus: expression of GFAP, cytokines, and adhesion molecules. Glia 19(4):275–285
Brogden KA, Guthmiller JM, Salzet M, Zasloff M (2005) The nervous system and innate immunity: the neuropeptide connection. Nat Immunol 6(6):558–564
Bruckdorfer R (2005) The basics about nitric oxide. Mol Aspects Med 26(1–2):3–31
Bruder C, Hagleitner M, Darlington G, Mohsenipour I, Wurzner R, Hollmuller I, Stoiber H, Lass-Florl C, Dierich MP, Speth C (2004) HIV-1 induces complement factor C3 synthesis in astrocytes and neurons by modulation of promoter activity. Mol Immunol 40(13):949–961
Bryan DL, Hart P, Forsyth K, Gibson R (2005) Modulation of respiratory syncytial virus-induced prostaglandin E2 production by n-3 long-chain polyunsaturated fatty acids in human respiratory epithelium. Lipids 40(10):1007–1011
Bryson KJ, Nash AA, Norval M (2014) Does vitamin D protect against respiratory viral infections? Epidemiol Infect 142(9):1789–1801. doi:10.1017/s0950268814000193
Burrer R, Buchmeier MJ, Wolfe T, Ting JP, Feuer R, Iglesias A, von Herrath MG (2007) Exacerbated pathology of viral encephalitis in mice with central nervous system-specific autoantibodies. Am J Pathol 170(2):557–566. doi:10.2353/ajpath.2007.060893
Butchi NB, Hinton DR, Stohlman SA, Kapil P, Fensterl V, Sen GC, Bergmann CC (2014) Ifit2 deficiency results in uncontrolled neurotropic coronavirus replication and enhanced encephalitis via impaired alpha/beta interferon induction in macrophages. J Virol 88(2):1051–1064. doi:10.1128/jvi.02272-13
Cabral GA, McNerney PJ, Mishkin EM (1987) Delta-9-tetrahydrocannabinol inhibits the splenocyte proliferative response to herpes simplex virus type 2. Immunopharmacol Immunotoxicol 9(2–3):361–370
Callahan TA, Moynihan JA, Piekut DT (1998) Central nervous system activation following peripheral chemical sympathectomy: implications for neural-immune interactions. Brain Behav Immun 12(3):230–241
Campbell IL (2005) Cytokine-mediated inflammation, tumorigenesis, and disease-associated JAK/STAT/SOCS signaling circuits in the CNS. Brain Res Brain Res Rev 48(2):166–177
Campbell JA, Lenschow DJ (2013) Emerging roles for immunomodulatory functions of free ISG15. J Interferon Cytokine Res 33(12):728–738. doi:10.1089/jir.2013.0064
Campbell T, Meagher MW, Sieve A, Scott B, Storts R, Welsh TH, Welsh CJ (2001) The effects of restraint stress on the neuropathogenesis of Theiler’s virus infection: I. Acute disease. Brain Behav Immun 15(3):235–254
Campbell BM, Charych E, Lee AW, Moller T (2014) Kynurenines in CNS disease: regulation by inflammatory cytokines. Front Neurosci 8:12. doi:10.3389/fnins.2014.00012
Cao L, Filipov NM, Lawrence DA (2002) Sympathetic nervous system plays a major role in acute cold/restraint stress inhibition of host resistance to Listeria monocytogenes. J Neuroimmunol 125(1–2):94–102
Caplazi P, Ehrensperger F (1998) Spontaneous Borna disease in sheep and horses: immunophenotyping of inflammatory cells and detection of MHC-I and MHC-II antigen expression in Borna encephalitis lesions. Vet Immunol Immunopathol 61(2–4):203–220
Carr JM, Mahalingam S, Bonder CS, Pitson SM (2013) Sphingosine kinase 1 in viral infections. Rev Med Virol 23(2):73–84. doi:10.1002/rmv.1718
Carrier EJ, Auchampach JA, Hillard CJ (2006) Inhibition of an equilibrative nucleoside transporter by cannabidiol: a mechanism of cannabinoid immunosuppression. Proc Natl Acad Sci U S A 103(20):7895–7900
Carty M, Reinert L, Paludan SR, Bowie AG (2014) Innate antiviral signalling in the central nervous system. Trends Immunol 35(2):79–87. doi:10.1016/j.it.2013.10.012
Castelli J, Wood KA, Youle RJ (1998) The 2-5A system in viral infection and apoptosis. Biomed Pharmacother 52(9):386–390
Cavanaugh SE, Holmgren AM, Rall GF (2015) Homeostatic interferon expression in neurons is sufficient for early control of viral infection. J Neuroimmunol 279:11–19. doi:10.1016/j.jneuroim.2014.12.012
Cheeran MC, Hu S, Gekker G, Lokensgard JR (2000) Decreased cytomegalovirus expression following proinflammatory cytokine treatment of primary human astrocytes. J Immunol 164(2):926–933
Chen BJ, Lamb RA (2008) Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology 372(2):221–232. doi:10.1016/j.virol.2007.11.008
Chen N, Reiss CS (2002a) Distinct roles of eicosanoids in the immune response to viral encephalitis: or why you should take NSAIDS. Viral Immunol 15(1):133–146
Chen N, Reiss CS (2002b) Innate immunity in viral encephalitis: role of C5. Viral Immunol 15(2):365–372. doi:10.1089/08828240260066288
Chen N, Warner JL, Reiss CS (2000) NSAID treatment suppresses VSV propagation in mouse CNS. Virology 276(1):44–51. doi:10.1006/viro.2000.0562, S0042-6822(00)90562-2 [pii]
Chen N, Restivo A, Reiss CS (2001) Leukotrienes play protective roles early during experimental VSV encephalitis. J Neuroimmunol 120(1–2):94–102, doi: S0165572801004155 [pii]
Chen N, Restivo A, Reiss CS (2002) Selective inhibition of COX-2 is beneficial to mice infected intranasally with VSV. Prostaglandins Other Lipid Mediat 67(2):143–155
Chen W, Han C, Xie B, Hu X, Yu Q, Shi L, Wang Q, Li D, Wang J, Zheng P, Liu Y, Cao X (2013) Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 152(3):467–478, http://dx.doi.org/10.1016/j.cell.2013.01.011
Cheng G, Feng Z, He B (2005) Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2alpha dephosphorylation by the gamma(1)34.5 protein. J Virol 79(3):1379–1388
Cherry JD, Olschowka JA, O’Banion MK (2014) Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation 11:98. doi:10.1186/1742-2094-11-98
Chesler DA, Reiss CS (2002) The role of IFN-gamma in immune responses to viral infections of the central nervous system. Cytokine Growth Factor Rev 13(6):441–454, doi:S1359610102000448 [pii]
Chesler DA, Munoz-Jordan JL, Donelan N, Garcia-Sastre A, Reiss CS (2003) PKR is not required for interferon-gamma inhibition of VSV replication in neurons. Viral Immunol 16(1):87–96. doi:10.1089/088282403763635474
Chesler DA, Dodard C, Lee GY, Levy DE, Reiss CS (2004a) Interferon-gamma-induced inhibition of neuronal vesicular stomatitis virus infection is STAT1 dependent. J Neurovirol 10(1):57–63, doi: D87PT7Y8MM5P8MR9 [pii]
Chesler DA, McCutcheon JA, Reiss CS (2004b) Posttranscriptional regulation of neuronal nitric oxide synthase expression by IFN-gamma. J Interferon Cytokine Res 24(2):141–149. doi:10.1089/107999004322813381
Cho H, Shrestha B, Sen GC, Diamond MS (2013) A role for Ifit2 in restricting West Nile virus infection in the brain. J Virol 87(15):8363–8371. doi:10.1128/JVI.01097-13
Christian AY, Barna M, Bi Z, Reiss CS (1996) Host immune response to vesicular stomatitis virus infection of the central nervous system in C57BL/6 mice. Viral Immunol 9(3):195–205
Christophi GP, Massa PT (2009) Central neuroinvasion and demyelination by inflammatory macrophages after peripheral virus infection is controlled by SHP-1. Viral Immunol 22(6):371–387. doi:10.1089/vim.2009.0052
Chu JJ, Ng ML (2003) The mechanism of cell death during West Nile virus infection is dependent on initial infectious dose. J Gen Virol 84(Pt 12):3305–3314
Chuah YK, Basir R, Talib H, Tie TH, Nordin N (2013) Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int J Inflamm 2013:403460. doi:10.1155/2013/403460
Claria J (2003) Cyclooxygenase-2 biology. Curr Pharm Des 9:2177–2190
Clarke P, Leser JS, Bowen RA, Tyler KL (2014) Virus-induced transcriptional changes in the brain include the differential expression of genes associated with interferon, apoptosis, interleukin 17 receptor A, and glutamate signaling as well as flavivirus-specific upregulation of tRNA synthetases. mBio 5(2):e00902–e00914. doi: 10.1128/mBio.00902-14
Cocuzza CE, Piazza F, Musumeci R, Oggioni D, Andreoni S, Gardinetti M, Fusco L, Frigo M, Banfi P, Rottoli MR, Confalonieri P, Rezzonico M, Ferro MT, Cavaletti G (2014) Quantitative detection of Epstein-Barr virus DNA in cerebrospinal fluid and blood samples of patients with relapsing-remitting multiple sclerosis. PLoS One 9(4):e94497. doi:10.1371/journal.pone.0094497
Cohn L, Delamarre L (2014) Dendritic cell-targeted vaccines. Front Immunol 5:255. doi:10.3389/fimmu.2014.00255
Cole AM, Ganz T, Liese AM, Burdick MD, Liu L, Strieter RM (2001) Cutting edge: IFN-inducible ELR-CXC chemokines display defensin-like antimicrobial activity. J Immunol 167(2):623–627
Cotter R, Williams C, Ryan L, Erichsen D, Lopez A, Peng H, Zheng J (2002) Fractalkine (CX3CL1) and brain inflammation: implications for HIV-1-associated dementia. J Neurovirol 8(6):585–598. doi:10.1080/13550280290100950
Crack LR, Jones L, Malavige GN, Patel V, Ogg GS (2012) Human antimicrobial peptides LL-37 and human beta-defensin-2 reduce viral replication in keratinocytes infected with varicella zoster virus. Clin Exp Dermatol 37(5):534–543. doi:10.1111/j.1365-2230.2012.04305.x
Cronstein BN, Levin RI, Philips M, Hirschhorn R, Abramson SB, Weissmann G (1992) Neutrophil adherence to endothelium is enhanced via adenosine A1 receptors and inhibited via adenosine A2 receptors. J Immunol 148(7):2201–2206
Croxford JL, Miller SD (2003) Immunoregulation of a viral model of multiple sclerosis using the synthetic cannabinoid R(+)WIN55,212. J Clin Investig 111(8):1231–1240
Cruz-Orengo L, Daniels BP, Dorsey D, Basak SA, Grajales-Reyes JG, McCandless EE, Piccio L, Schmidt RE, Cross AH, Crosby SD, Klein RS (2014) Enhanced sphingosine-1-phosphate receptor 2 expression underlies female CNS autoimmunity susceptibility. J Clin Invest 124(6):2571–2584. doi:10.1172/jci73408
Cue L, Diaz F, Briegel KJ, Patel HH, Raval AP (2015) Periodic estrogen receptor-beta activation: a novel approach to prevent ischemic brain damage. Neurochem Res 40(10):2009–2017. doi:10.1007/s11064-014-1346-7
Cuevas CD, Ross SR (2014) Toll-like receptor 2-mediated innate immune responses against Junin virus in mice lead to antiviral adaptive immune responses during systemic infection and do not affect viral replication in the brain. J Virol 88(14):7703–7714. doi:10.1128/jvi.00050-14
Curtin JF, King GD, Barcia C, Liu C, Hubert FX, Guillonneau C, Josien R, Anegon I, Lowenstein PR, Castro MG (2006) Fms-like tyrosine kinase 3 ligand recruits plasmacytoid dendritic cells to the brain. J Immunol 176(6):3566–3577
D’Agostino PM, Reiss CS (2010) A confocal and electron microscopic comparison of interferon beta-induced changes in vesicular stomatitis virus infection of neuroblastoma and nonneuronal cells. DNA Cell Biol 29(3):103–120. doi:10.1089/dna.2009.0963
D’Agostino PM, Amenta JJ, Reiss CS (2009a) IFN-beta-induced alteration of VSV protein phosphorylation in neuronal cells. Viral Immunol 22(6):353–369. doi:10.1089/vim.2009.0057
D’Agostino PM, Yang J, Reiss CS (2009b) Distinct mechanisms of inhibition of VSV replication in neurons mediated by type I and II IFN. Virus Rev Res 14(2):20–29
D’Agostino PM, Gottfried-Blackmore A, Anandasabapathy N, Bulloch K (2012a) Brain dendritic cells: biology and pathology. Acta Neuropathol 124(5):599–614. doi:10.1007/s00401-012-1018-0
D’Agostino PM, Kwak C, Vecchiarelli HA, Toth JG, Miller JM, Masheeb Z, McEwen BS, Bulloch K (2012b) Viral-induced encephalitis initiates distinct and functional CD103+ CD11b+ brain dendritic cell populations within the olfactory bulb. Proc Natl Acad Sci U S A 109(16):6175–6180. doi:10.1073/pnas.1203941109
Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin TY, Schneller S, Zust R, Dong H, Thiel V, Sen GC, Fensterl V, Klimstra WB, Pierson TC, Buller RM, Gale M Jr, Shi PY, Diamond MS (2010) 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468(7322):452–456. doi:10.1038/nature09489
Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116(2):205–219
Daniels BP, Holman DW, Cruz-Orengo L, Jujjavarapu H, Durrant DM, Klein RS (2014) Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. mBio 5(5). doi:10.1128/mBio.01476-14
Dantzer R (2004) Cytokine-induced sickness behaviour: a neuroimmune response to activation of innate immunity. Eur J Pharmacol 500(1–3):399–411
Dantzer R (2005) Somatization: a psychoneuroimmune perspective. Psychoneuroendocrinology 30(10):947–952
de Aquino MT, Kapil P, Hinton DR, Phares TW, Puntambekar SS, Savarin C, Bergmann CC, Stohlman SA (2014) IL-27 limits central nervous system viral clearance by promoting IL-10 and enhances demyelination. J Immunol 193(1):285–294. doi:10.4049/jimmunol.1400058
De Caterina R, Madonna R, Massaro M (2004) Effects of omega-3 fatty acids on cytokines and adhesion molecules. Curr Atheroscler Rep 6(6):485–491
de Souza KP, Silva EG, de Oliveira Rocha ES, Figueiredo LB, de Almeida-Leite CM, Arantes RM, de Assis Silva Gomes J, Ferreira GP, de Oliveira JG, Kroon EG, Campos MA (2013) Nitric oxide synthase expression correlates with death in an experimental mouse model of dengue with CNS involvement. Virol J 10:267. doi:10.1186/1743-422x-10-267
Degraeve F, Bolla M, Blaie S, Creminon C, Quere I, Boquet P, Levy-Toledano S, Bertoglio J, Habib A (2001) Modulation of COX-2 expression by statins in human aortic smooth muscle cells. Involvement of geranylgeranylated proteins. J Biol Chem 276(50):46849–46855
Delgado M, Gonzalez-Rey E, Ganea D (2006) Vasoactive intestinal peptide: the dendritic cell → regulatory T cell axis. Ann N Y Acad Sci 1070:233–238
Delhaye S, Paul S, Blakqori G, Minet M, Weber F, Staeheli P, Michiels T (2006) Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci U S A 103(20):7835–7840
Denizot M, Neal JW, Gasque P (2012) Encephalitis due to emerging viruses: CNS innate immunity and potential therapeutic targets. J Infect 65(1):1–16. doi:10.1016/j.jinf.2012.03.019
Deretic V (2005) Autophagy in innate and adaptive immunity. Trends Immunol 26(10):523–528
Deretic V, Saitoh T, Akira S (2013) Autophagy in infection, inflammation and immunity. Nat Rev Immunol 13(10):722–737. doi:10.1038/nri3532
Desforges M, Le Coupanec A, Brison E, Meessen-Pinard M, Talbot PJ (2014) Neuroinvasive and neurotropic human respiratory coronaviruses: potential neurovirulent agents in humans. Adv Exp Med Biol 807:75–96. doi:10.1007/978-81-322-1777-0_6
Detje CN, Lienenklaus S, Chhatbar C, Spanier J, Prajeeth CK, Soldner C, Tovey MG, Schluter D, Weiss S, Stangel M, Kalinke U (2015) Upon intranasal vesicular stomatitis virus infection, astrocytes in the olfactory bulb are important interferon Beta producers that protect from lethal encephalitis. J Virol 89(5):2731–2738. doi:10.1128/jvi.02044-14
Di Iorio G, Lupi M, Sarchione F, Matarazzo I, Santacroce R, Petruccelli F, Martinotti G, Di Giannantonio M (2013) The endocannabinoid system: a putative role in neurodegenerative diseases. Int J High Risk Behav Addict 2(3):100–106. doi:10.5812/ijhrba.9222
Diamond MS, Farzan M (2013) The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol 13(1):46–57. doi:10.1038/nri3344
Dietzschold B, Morimoto K (1997) Signaling pathways in virus-induced CNS inflammation. J Neurovirol 3(Suppl 1):S58–S59
Dietzschold B, Schwaeble W, Schafer MK, Hooper DC, Zehng YM, Petry F, Sheng H, Fink T, Loos M, Koprowski H (1995) Expression of C1q, a subcomponent of the rat complement system, is dramatically enhanced in brains of rats with either Borna disease or experimental allergic encephalomyelitis. J Neurol Sci 130(1):11–16
Ding Q, Cao X, Lu J, Huang B, Liu YJ, Kato N, Shu HB, Zhong J (2013) Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J Hepatol 59(1):52–58. doi:10.1016/j.jhep.2013.03.019
Dionne KR, Galvin JM, Schittone SA, Clarke P, Tyler KL (2011) Type I interferon signaling limits reoviral tropism within the brain and prevents lethal systemic infection. J Neurovirol 17(4):314–326. doi:10.1007/s13365-011-0038-1
Dionne KR, Zhuang Y, Leser JS, Tyler KL, Clarke P (2013) Daxx upregulation within the cytoplasm of reovirus-infected cells is mediated by interferon and contributes to apoptosis. J Virol 87(6):3447–3460. doi:10.1128/jvi.02324-12
Disanto G, Morahan JM, Ramagopalan SV (2012) Multiple sclerosis: risk factors and their interactions. CNS Neurol Disord Drug Targets 11(5):545–555
Dragone T, Cianciulli A, Calvello R, Porro C, Trotta T, Panaro MA (2014) Resveratrol counteracts lipopolysaccharide-mediated microglial inflammation by modulating a SOCS-1 dependent signaling pathway. Toxicol In Vitro 28(6):1126–1135. doi:10.1016/j.tiv.2014.05.005
Drew PD, Xu J, Storer PD, Chavis JA, Racke MK (2006) Peroxisome proliferator-activated receptor agonist regulation of glial activation: relevance to CNS inflammatory disorders. Neurochem Int 49(2):183–189
Drobni P, Naslund J, Evander M (2004) Lactoferrin inhibits human papillomavirus binding and uptake in vitro. Antiviral Res 64(1):63–68
Dubensky TW Jr, Kanne DB, Leong ML (2013) Rationale, progress and development of vaccines utilizing STING-activating cyclic dinucleotide adjuvants. Ther Adv Vaccines 1(4):131–143. doi:10.1177/2051013613501988
Dunford JE, Rogers MJ, Ebetino FH, Phipps RJ, Coxon FP (2006) Inhibition of protein prenylation by bisphosphonates causes sustained activation of Rac, Cdc42, and Rho GTPases. J Bone Miner Res 21(5):684–694
Dunn SE, Ousman SS, Sobel RA, Zuniga L, Baranzini SE, Youssef S, Crowell A, Loh J, Oksenberg J, Steinman L (2007) Peroxisome proliferator-activated receptor (PPAR){alpha} expression in T cells mediates gender differences in development of T cell-mediated autoimmunity. J Exp Med 204(2):321–330
Durrant DM, Robinette ML, Klein RS (2013) IL-1R1 is required for dendritic cell-mediated T cell reactivation within the CNS during West Nile virus encephalitis. J Exp Med 210(3):503–516. doi:10.1084/jem.20121897
Einav S, Glenn JS (2003) Prenylation inhibitors: a novel class of antiviral agents. J Antimicrob Chemother 52(6):883–886
El-Azami-El-Idrissi M, Franquin S, Day MJ, Mazza G, Elson CJ, Preat V, Pfau CJ, Coutelier JP (2005) Distinct host-dependent pathogenic mechanisms leading to a similar clinical anemia after infection with lymphocytic choriomeningitis virus. Exp Biol Med 230(11):865–871
El-Gindy EM, Ali-Eldin FA, Meguid MA (2012) Serum leptin level and its association with fatigue in patients with chronic hepatitis C virus infection. Arab J Gastroenterol 13(2):54–57. doi:10.1016/j.ajg.2012.05.001
El-Hage N, Podhaizer EM, Sturgill J, Hauser KF (2011) Toll-like receptor expression and activation in astroglia: differential regulation by HIV-1 Tat, gp120, and morphine. Immunol Invest 40(5):498–522. doi:10.3109/08820139.2011.561904
Errett JS, Suthar MS, McMillan A, Diamond MS, Gale M Jr (2013) The essential, nonredundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J Virol 87(21):11416–11425. doi:10.1128/jvi.01488-13
Eskandari F, Sternberg EM (2002) Neural-immune interactions in health and disease. Ann N Y Acad Sci 966:20–27
Esposito P, Gheorghe D, Kandere K, Pang X, Connolly R, Jacobson S, Theoharides TC (2001) Acute stress increases permeability of the blood-brain-barrier through activation of brain mast cells. Brain Res 888(1):117–127
Evans EW, Harmon BG (1995) A review of antimicrobial peptides: defensins and related cationic peptides. Vet Clin Pathol 24(4):109–116
Fadnis PR, Ravi V, Desai A, Turtle L, Solomon T (2013) Innate immune mechanisms in Japanese encephalitis virus infection: effect on transcription of pattern recognition receptors in mouse neuronal cells and brain tissue. Viral Immunol 26(6):366–377. doi:10.1089/vim.2013.0016
Fan Y, Mao R, Yu Y, Liu S, Shi Z, Cheng J, Zhang H, An L, Zhao Y, Xu X, Chen Z, Kogiso M, Zhang D, Zhang H, Zhang P, Jung JU, Li X, Xu G, Yang J (2014) USP21 negatively regulates antiviral response by acting as a RIG-I deubiquitinase. J Exp Med 211(2):313–328. doi:10.1084/jem.20122844
Faria PA, Chakraborty P, Levay A, Barber GN, Ezelle HJ, Enninga J, Arana C, van Deursen J, Fontoura BM (2005) VSV disrupts the Rae1/mrnp41 mRNA nuclear export pathway. Mol Cell 17(1):93–102
Fazakerley JK, Cotterill CL, Lee G, Graham A (2006) Virus tropism, distribution, persistence and pathology in the corpus callosum of the Semliki Forest virus-infected mouse brain: a novel system to study virus-oligodendrocyte interactions. Neuropathol Appl Neurobiol 32(4):397–409
Fensterl V, Sen GC (2015) Interferon-induced Ifit proteins: their role in viral pathogenesis. J Virol 89(5):2462–2468. doi:10.1128/jvi.02744-14
Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, Bergmann CC, Diamond MS, Virgin HW, Sen GC (2012) Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog 8(5):e1002712. doi:10.1371/journal.ppat.1002712
Fensterl V, Wetzel JL, Sen GC (2014) The interferon-induced protein, Ifit2, protects mice from infection of the peripheral nervous system by vesicular stomatitis virus. J Virol 88(18):10303–10311. doi:10.1128/jvi.01341-14
Fernandes ER, De Andrade HF Jr, Lancellotti CL, Quaresma JA, Demachki S, da Costa Vasconcelos PF, Duarte MI (2011) In situ apoptosis of adaptive immune cells and the cellular escape of rabies virus in CNS from patients with human rabies transmitted by Desmodus rotundus. Virus Res 156(1-2):121–126. doi:10.1016/j.virusres.2011.01.006
Fidaleo M, Fanelli F, Ceru MP, Moreno S (2014) Neuroprotective properties of peroxisome proliferator-activated receptor alpha (PPARalpha) and its lipid ligands. Curr Med Chem 21(24):2803–2821
Fiordalisi JJ, Keller PJ, Cox AD (2006) PRL tyrosine phosphatases regulate rho family GTPases to promote invasion and motility. Cancer Res 66(6):3153–3161
Fitzgerald KC, O’Reilly ÉJ, Falcone GJ et al (2014a) Dietary ω-3 polyunsaturated fatty acid intake and risk for amyotrophic lateral sclerosis. JAMA Neurol 71(9):1102–1110. doi:10.1001/jamaneurol.2014.1214
Fitzgerald ME, Rawling DC, Vela A, Pyle AM (2014b) An evolving arsenal: viral RNA detection by RIG-I-like receptors. Curr Opin Microbiol 20C:76–81. doi:10.1016/j.mib.2014.05.004
Flamand L, Borgeat P, Lalonde R, Gosselin J (2004) Release of anti-HIV mediators after administration of leukotriene B4 to humans. J Infect Dis 189(11):2001–2009
Foell D, Wittkowski H, Vogl T, Roth J (2007) S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol 81(1):28–37
Forger JM III, Bronson RT, Huang AS, Reiss CS (1991) Murine infection by vesicular stomatitis virus: initial characterization of the H-2d system. J Virol 65(9):4950–4958
Fox HS, Bond BL, Parslow TG (1991) Estrogen regulates the IFN-gamma promoter. J Immunol 146(12):4362–4367
Fu Z, Jackson AC (2005) Neuronal dysfunction and death in rabies virus infection. J Neurovirol 11(1):101–106
Fuchs A, Pinto AK, Schwaeble WJ, Diamond MS (2011) The lectin pathway of complement activation contributes to protection from West Nile virus infection. Virology 412(1):101–109. doi:10.1016/j.virol.2011.01.003
Fuller AC, Kang B, Kang HK, Yahikozowa H, Dal Canto MC, Kim BS (2005) Gender bias in Theiler’s virus-induced demyelinating disease correlates with the level of antiviral immune responses. J Immunol 175(6):3955–3963
Fullmer JJ, Khan AM, Elidemir O, Chiappetta C, Stark JM, Colasurdo GN (2005) Role of cysteinyl leukotrienes in airway inflammation and responsiveness following RSV infection in BALB/c mice. Pediatr Allergy Immunol 16(7):593–601
Furman D, Hejblum BP, Simon N, Jojic V, Dekker CL, Thiebaut R, Tibshirani RJ, Davis MM (2014) Systems analysis of sex differences reveals an immunosuppressive role for testosterone in the response to influenza vaccination. Proc Natl Acad Sci U S A 111(2):869–874. doi:10.1073/pnas.1321060111
Furr SR, Marriott I (2012) Viral CNS infections: role of glial pattern recognition receptors in neuroinflammation. Front Microbiol 3:201. doi:10.3389/fmicb.2012.00201
Gabriel C, Her Z, Ng LF (2013) Neutrophils: neglected players in viral diseases. DNA Cell Biol 32(12):665–675. doi:10.1089/dna.2013.2211
Gaddy DF, Lyles DS (2007) Oncolytic vesicular stomatitis virus induces apoptosis via signaling through PKR, Fas, and Daxx. J Virol 81(6):2792–2804
Gay NJ, Symmons MF, Gangloff M, Bryant CE (2014) Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol 14(8):546–558. doi:10.1038/nri3713, http://www.nature.com/nri/journal/v14/n8/abs/nri3713.html—supplementary-information
Gebremedhin D, Lange AR, Lowry TF, Taheri MR, Birks EK, Hudetz AG, Narayanan J, Falck JR, Okamoto H, Roman RJ, Nithipatikom K, Campbell WB, Harder DR (2000) Production of 20-HETE and its role in autoregulation of cerebral blood flow. Circ Res 87(1):60–65
Geffin R, McCarthy M (2013) Innate immune responses to HIV infection in the central nervous system. Immunol Res 57(1–3):292–302. doi:10.1007/s12026-013-8445-4
Geiss GK, Carter VS, He Y, Kwieciszewski BK, Holzman T, Korth MJ, Lazaro CA, Fausto N, Bumgarner RE, Katze MG (2003) Gene expression profiling of the cellular transcriptional network regulated by alpha/beta interferon and its partial attenuation by the hepatitis C virus nonstructural 5A protein. J Virol 77(11):6367–6375
Gendelman HE, Genis P, Jett M, Zhai QH, Nottet HS (1994) An experimental model system for HIV-1-induced brain injury. Adv Neuroimmunol 4(3):189–193
Gianforcaro A, Hamadeh MJ (2014) Vitamin D as a potential therapy in amyotrophic lateral sclerosis. CNS Neurosci Ther 20(2):101–111. doi:10.1111/cns.12204
Gilbert GL, Kim HJ, Waataja JJ, Thayer SA (2007) Delta(9)-tetrahydrocannabinol protects hippocampal neurons from excitotoxicity. Brain Res 1128(1):61–69
Glasker S, Toller M, Kummerer BM (2014) The alternate triad motif of the poly(ADP-ribose) polymerase-like domain of the human zinc finger antiviral protein is essential for its antiviral activity. J Gen Virol 95(Pt 4):816–822. doi:10.1099/vir.0.060988-0
Gnirss K, Fiedler M, Kramer-Kuhl A, Bolduan S, Mittler E, Becker S, Schindler M, Pohlmann S (2014) Analysis of determinants in filovirus glycoproteins required for tetherin antagonism. Viruses 6(4):1654–1671. doi:10.3390/v6041654
Gomez RM, Yep A, Schattner M, Berria MI (2003) Junin virus-induced astrocytosis is impaired by iNOS inhibition. J Med Virol 69(1):145–149
Gonzalez-Ariki S, Husband AJ (1999) Sympathectomy abrogates immunodeficiency associated with protein-energy malnutrition. J Neuroimmunol 99(1):97–104
Gonzalez-Rey E, Chorny A, Delgado M (2007) Regulation of immune tolerance by anti-inflammatory neuropeptides. Nat Rev Immunol 7(1):52–63
Goody RJ, Hoyt CC, Tyler KL (2005) Reovirus infection of the CNS enhances iNOS expression in areas of virus-induced injury. Exp Neurol 195(2):379–390
Gorchakov R, Frolova E, Williams BR, Rice CM, Frolov I (2004) PKR-dependent and -independent mechanisms are involved in translational shutoff during Sindbis virus infection. J Virol 78(16):8455–8467
Gosselin J, Borgeat P, Flamand L (2005) Leukotriene B4 protects latently infected mice against murine cytomegalovirus reactivation following allogeneic transplantation. J Immunol 174(3):1587–1593
Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen AG, Fujimura T, Rehwinkel J, Iskarpatyoti JA, Barchet W, Ludwig J, Dermody TS, Hartmann G, Reis ESC (2014) Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 514(7522):372–375. doi:10.1038/nature13590
Gower TL, Graham BS (2001) Antiviral activity of lovastatin against respiratory syncytial virus in vivo and in vitro. Antimicrob Agents Chemother 45(4):1231–1237
Grebe KM, Takeda K, Hickman HD, Bailey AL, Embry AC, Bennink JR, Yewdell JW (2010) Cutting edge: sympathetic nervous system increases proinflammatory cytokines and exacerbates influenza A virus pathogenesis. J Immunol 184(2):540–544. doi:10.4049/jimmunol.0903395
Green AM, Beatty PR, Hadjilaou A, Harris E (2014) Innate immunity to dengue virus infection and subversion of antiviral responses. J Mol Biol 426(6):1148–1160. doi:10.1016/j.jmb.2013.11.023
Gresser O, Hein A, Riese S, Regnier-Vigouroux A (2000) Tumor necrosis factor alpha and interleukin-1 alpha inhibit through different pathways interferon-gamma-induced antigen presentation, processing and MHC class II surface expression on astrocytes, but not on microglia. Cell Tissue Res 300(3):373–382
Griffin DE (2005) Neuronal cell death in alphavirus encephalomyelitis. Curr Top Microbiol Immunol 289:57–77
Griffin D, Levine B, Tyor W, Ubol S, Despres P (1997) The role of antibody in recovery from alphavirus encephalitis. Immunol Rev 159:155–161
Grishkan IV, Fairchild AN, Calabresi PA, Gocke AR (2013) 1,25-Dihydroxyvitamin D3 selectively and reversibly impairs T helper-cell CNS localization. Proc Natl Acad Sci U S A 110(52):21101–21106. doi:10.1073/pnas.1306072110
Grote E, Baba M, Ohsumi Y, Novick PJ (2000) Geranylgeranylated SNAREs are dominant inhibitors of membrane fusion. J Cell Biol 151(2):453–466
Grygiel-Gorniak B (2014) Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications—a review. Nutr J 13:17. doi:10.1186/1475-2891-13-17
Guayasamin RC, Reynolds TD, Wei X, Fujiwara M, Robek MD (2014) Type III interferon attenuates a vesicular stomatitis virus-based vaccine vector. J Virol 88(18):10909–10917. doi:10.1128/jvi.01910-14
Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, Yona S (2014) Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol 14(8):571–578. doi:10.1038/nri3712, http://www.nature.com/nri/journal/v14/n8/abs/nri3712.html—supplementary-information
Gustin JK, Douglas JL (2013) BST-2/tetherin: viral tether, viral sensor or both? Future Virol 8(11):1053–1060. doi:10.2217/fvl.13.96
Gwyer Findlay E, Currie SM, Davidson DJ (2013) Cationic host defence peptides: potential as antiviral therapeutics. BioDrugs 27(5):479–493. doi:10.1007/s40259-013-0039-0
Halmer R, Walter S, Fassbender K (2014) Sphingolipids: important players in multiple sclerosis. Cell Physiol Biochem 34(1):111–118. doi:10.1159/000362988
Han YW, Choi JY, Uyangaa E, Kim SB, Kim JH, Kim BS, Kim K, Eo SK (2014) Distinct dictation of Japanese encephalitis virus-induced neuroinflammation and lethality via triggering TLR3 and TLR4 signal pathways. PLoS Pathog 10(9):e1004319. doi:10.1371/journal.ppat.1004319
Harizi H, Gualde N (2006) Pivotal role of PGE2 and IL-10 in the cross-regulation of dendritic cell-derived inflammatory mediators. Cell Mol Immunol 3(4):271–277
Hatalski CG, Hickey WF, Lipkin WI (1998) Evolution of the immune response in the central nervous system following infection with Borna disease virus. J Neuroimmunol 90(2):137–142
Havers F, Smeaton L, Gupte N, Detrick B, Bollinger RC, Hakim J, Kumarasamy N, Andrade A, Christian P, Lama JR, Campbell TB, Gupta A (2014) 25-Hydroxyvitamin D insufficiency and deficiency is associated with HIV disease progression and virological failure post-antiretroviral therapy initiation in diverse multinational settings. J Infect Dis 210(2):244–253. doi:10.1093/infdis/jiu259
Hayashi T, Yamada K, Esaki T, Mutoh E, Iguchi A (1997) Effect of estrogen on isoforms of nitric oxide synthase: possible mechanism of anti-atherosclerotic effect of estrogen. Gerontology 43(Suppl 1):24–34
He C, Klionsky DJ (2006) Autophagy and neurodegeneration. ACS Chem Biol 1(4):211–213
Heilingloh CS, Muhl-Zurbes P, Steinkasserer A, Kummer M (2014) Herpes simplex virus type 1 ICP0 induces CD83 degradation in mature dendritic cells independent of its E3 ubiquitin ligase function. J Gen Virol 95(Pt 6):1366–1375. doi:10.1099/vir.0.062810-0
Helbig KJ, Beard MR (2014) The role of viperin in the innate antiviral response. J Mol Biol 426(6):1210–1219. doi:10.1016/j.jmb.2013.10.019
Hellwig-Burgel T, Stiehl DP, Wagner AE, Metzen E, Jelkmann W (2005) Review: hypoxia-inducible factor-1 (HIF-1): a novel transcription factor in immune reactions. J Interferon Cytokine Res 25(6):297–310
Hermant P, Michiels T (2014) Interferon-lambda in the context of viral infections: production, response and therapeutic implications. J Innate Immun 6(5):563–574. doi:10.1159/000360084
Herrera RA, Oved JH, Reiss CS (2008) Disruption of IFN-gamma-mediated antiviral activity in neurons: the role of cannabinoids. Viral Immunol 21(2):141–152. doi:10.1089/vim.2007.0109
Herrmann J, Lerman LO, Lerman A (2007) Ubiquitin and ubiquitin-like proteins in protein regulation. Circ Res 100(9):1276–1291. doi:10.1161/01.RES.0000264500.11888.f0
Hodges JL, Ireland DD, Reiss CS (2001) The role of interleukin-18 in vesicular stomatitis virus infection of the CNS. Viral Immunol 14(2):181–191. doi:10.1089/088282401750234556
Hoenen A, Gillespie L, Morgan G, van der Heide P, Khromykh A, Mackenzie J (2014) The West Nile virus assembly process evades the conserved antiviral mechanism of the interferon-induced MxA protein. Virology 448:104–116. doi:10.1016/j.virol.2013.10.005
Hooks JJ, Chin MS, Srinivasan K, Momma Y, Hooper LC, Nagineni CN, Chan CC, Detrick B (2006) Human cytomegalovirus induced cyclooxygenase-2 in human retinal pigment epithelial cells augments viral replication through a prostaglandin pathway. Microbes Infect 8(8):2236–2244
Hooper DC, Kean RB, Scott GS, Spitsin SV, Mikheeva T, Morimoto K, Bette M, Rohrenbeck AM, Dietzschold B, Weihe E (2001) The central nervous system inflammatory response to neurotropic virus infection is peroxynitrite dependent. J Immunol 167(6):3470–3477
Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G (2006) 5′-Triphosphate RNA is the ligand for RIG-I. Science 314(5801):994–997
Hornung V, Hartmann R, Ablasser A, Hopfner K-P (2014) OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat Rev Immunol 14(8):521–528. doi:10.1038/nri3719
Huang WH, Chen CW, Wu HL, Chen PJ (2006) Post-translational modification of delta antigen of hepatitis D virus. Curr Top Microbiol Immunol 307:91–112
Hudson CA, Christophi GP, Gruber RC, Wilmore JR, Lawrence DA, Massa PT (2008) Induction of IL-33 expression and activity in central nervous system glia. J Leukoc Biol 84(3):631–643. doi:10.1189/jlb.1207830
Hyde JL, Gardner CL, Kimura T, White JP, Liu G, Trobaugh DW, Huang C, Tonelli M, Paessler S, Takeda K, Klimstra WB, Amarasinghe GK, Diamond MS (2014) A viral RNA structural element alters host recognition of nonself RNA. Science 343(6172):783–787. doi:10.1126/science.1248465
Ibrahim MM, Rude ML, Stagg NJ, Mata HP, Lai J, Vanderah TW, Porreca F, Buckley NE, Makriyannis A, Malan TP Jr (2006) CB2 cannabinoid receptor mediation of antinociception. Pain 122(1–2):36–42
Ijaz MK, Dent D, Babiuk LA (1990) Neuroimmunomodulation of in vivo anti-rotavirus humoral immune response. J Neuroimmunol 26(2):159–171
Ireland DD, Reiss CS (2004) Expression of IL-12 receptor by neurons. Viral Immunol 17(3):411–422. doi:10.1089/0882824041856987
Ireland DD, Reiss CS (2006) Gene expression contributing to recruitment of circulating cells in response to vesicular stomatitis virus infection of the CNS. Viral Immunol 19(3):536–545. doi:10.1089/vim.2006.19.536
Ireland DD, Bang T, Komatsu T, Reiss CS (1999) Delayed administration of interleukin-12 is efficacious in promoting recovery from lethal viral encephalitis. Viral Immunol 12(1):35–40
Ireland DD, Palian BM, Reiss CS (2005) Interleukin (IL)-12 receptor beta1 or IL-12 receptor beta 2 deficiency in mice indicates that IL-12 and IL-23 are not essential for host recovery from viral encephalitis. Viral Immunol 18(2):397–402. doi:10.1089/vim.2005.18.397
Isaacs A, Lindenmann J (1957) Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 147(927):258–267
Ivashkiv LB, Donlin LT (2014) Regulation of type I interferon responses. Nat Rev Immunol 14(1):36–49. doi:10.1038/nri3581
Jackson WT, Giddings TH Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K (2005) Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 3(5):e156
Jakobsen MR, Bak RO, Andersen A, Berg RK, Jensen SB, Tengchuan J, Laustsen A, Hansen K, Ostergaard L, Fitzgerald KA, Xiao TS, Mikkelsen JG, Mogensen TH, Paludan SR (2013) IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc Natl Acad Sci U S A 110(48):E4571–E4580. doi:10.1073/pnas.1311669110
Jaulmes A, Thierry S, Janvier B, Raymondjean M, Marechal V (2006) Activation of sPLA2-IIA and PGE2 production by high mobility group protein B1 in vascular smooth muscle cells sensitized by IL-1beta. FASEB J 20(10):1727–1729
John L, Samuel CE (2014) Induction of stress granules by interferon and down-regulation by the cellular RNA adenosine deaminase ADAR1. Virology 454–455:299–310. doi:10.1016/j.virol.2014.02.025
Johnson HL, Jin F, Pirko I, Johnson AJ (2014) Theiler’s murine encephalomyelitis virus as an experimental model system to study the mechanism of blood-brain barrier disruption. J Neurovirol 20(2):107–112. doi:10.1007/s13365-013-0187-5
Johnston C, Jiang W, Chu T, Levine B (2001) Identification of genes involved in the host response to neurovirulent alphavirus infection. J Virol 75(21):10431–10445
Jurgens HA, Amancherla K, Johnson RW (2012) Influenza infection induces neuroinflammation, alters hippocampal neuron morphology, and impairs cognition in adult mice. J Neurosci 32(12):3958–3968. doi:10.1523/jneurosci.6389-11.2012
Kalamvoki M, Roizman B (2014) HSV-1 degrades, stabilizes, requires, or is stung by STING depending on ICP0, the US3 protein kinase, and cell derivation. Proc Natl Acad Sci U S A 111(5):E611–E617. doi:10.1073/pnas.1323414111
Kanakasabai S, Chearwae W, Walline CC, Iams W, Adams SM, Bright JJ (2010) Peroxisome proliferator-activated receptor delta agonists inhibit T helper type 1 (Th1) and Th17 responses in experimental allergic encephalomyelitis. Immunology 130(4):572–588. doi:10.1111/j.1365-2567.2010.03261.x
Kang SS, McGavern DB (2010) Microbial induction of vascular pathology in the CNS. J Neuroimmune Pharmacol 5(3):370–386. doi:10.1007/s11481-010-9208-9
Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, Huang J, Yu Y, Fan XG, Yan Z, Sun X, Wang H, Wang Q, Tsung A, Billiar TR, Zeh HJ III, Lotze MT, Tang D (2014) HMGB1 in health and disease. Mol Aspects Med 40:1–116. doi:10.1016/j.mam.2014.05.001
Kapadia SB, Chisari FV (2005) Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc Natl Acad Sci U S A 102(7):2561–2566
Kapadia SB, Levine B, Speck SH, Virgin HW (2002) Critical role of complement and viral evasion of complement in acute, persistent, and latent gamma-herpesvirus infection. Immunity 17(2):143–155
Kapil P, Stohlman SA, Hinton DR, Bergmann CC (2014) PKR mediated regulation of inflammation and IL-10 during viral encephalomyelitis. J Neuroimmunol 270(1–2):1–12. doi:10.1016/j.jneuroim.2014.02.012
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441(7089):101–105
Kawai T, Akira S (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34(5):637–650. doi:10.1016/j.immuni.2011.05.006
Kelley SP, Moynihan JA, Stevens SY, Grota LJ, Felten DL (2002) Chemical sympathectomy has no effect on the severity of murine AIDS: murine AIDS alone depletes norepinephrine levels in infected spleen. Brain Behav Immun 16(2):118–139
Khan MM, Wakade C, de Sevilla L, Brann DW (2015) Selective estrogen receptor modulators (SERMs) enhance neurogenesis and spine density following focal cerebral ischemia. J Steroid Biochem Mol Biol 146:38–47. doi:10.1016/j.jsbmb.2014.05.001
Kim JB, Sig CJ, Yu YM, Nam K, Piao CS, Kim SW, Lee MH, Han PL, Park JS, Lee JK (2006a) HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci 26(24):6413–6421
Kim SH, Won SJ, Mao XO, Jin K, Greenberg DA (2006b) Molecular mechanisms of cannabinoid protection from neuronal excitotoxicity. Mol Pharmacol 69(3):691–696
Kim SE, Lee EO, Yang JH, Kang JH, Suh YH, Chong YH (2012) 15-Deoxy-delta(1)(2), (1)(4)-prostaglandin J(2) inhibits human immunodeficiency virus-1 tat-induced monocyte chemoattractant protein-1/CCL2 production by blocking the extracellular signal-regulated kinase-1/2 signaling pathway independently of peroxisome proliferator-activated receptor-gamma and heme oxygenase-1 in rat hippocampal slices. J Neurosci Res 90(9):1732–1742. doi:10.1002/jnr.23051
Kimura R, Mori N (2014) Abundant expression of HMGB1 in human T-cell lymphotropic virus type I-infected T-cell lines and high plasma levels of HMGB1 in patients with adult T-cell leukemia. Oncol Lett 7(4):1239–1242. doi:10.3892/ol.2014.1851
Kimura T, Katoh H, Kayama H, Saiga H, Okuyama M, Okamoto T, Umemoto E, Matsuura Y, Yamamoto M, Takeda K (2013) Ifit1 inhibits Japanese encephalitis virus replication through binding to 5′ capped 2′-O unmethylated RNA. J Virol 87(18):9997–10003. doi:10.1128/jvi.00883-13
King VR, Huang WL, Dyall SC, Curran OE, Priestley JV, Michael-Titus AT (2006) Omega-3 fatty acids improve recovery, whereas omega-6 fatty acids worsen outcome, after spinal cord injury in the adult rat. J Neurosci 26(17):4672–4680
Kissick HT, Sanda MG, Dunn LK, Pellegrini KL, On ST, Noel JK, Arredouani MS (2014) Androgens alter T-cell immunity by inhibiting T-helper 1 differentiation. Proc Natl Acad Sci U S A 111(27):9887–9892. doi:10.1073/pnas.1402468111
Kitson MT, Sarrazin C, Toniutto P, Eslick GD, Roberts SK (2014) Vitamin D level and sustained virologic response to interferon-based antiviral therapy in chronic hepatitis C: a systematic review and meta-analysis. J Hepatol 61(6):1247–1252. doi:10.1016/j.jhep.2014.08.004
Klamp T, Boehm U, Schenk D, Pfeffer K, Howard JC (2003) A giant GTPase, very large inducible GTPase-1, is inducible by IFNs. J Immunol 171(3):1255–1265
Klotman ME, Chang TL (2006) Defensins in innate antiviral immunity. Nat Rev Immunol 6(6):447–456
Knopf PM, Cserr HF, Nolan SC, Wu TY, Harling-Berg CJ (1995) Physiology and immunology of lymphatic drainage of interstitial and cerebrospinal fluid from the brain. Neuropathol Appl Neurobiol 21(3):175–180
Kochs G, Trost M, Janzen C, Haller O (1998) MxA GTPase: oligomerization and GTP-dependent interaction with viral RNP target structures. Methods 15(3):255–263
Koeberle PD, Gauldie J, Ball AK (2004) Effects of adenoviral-mediated gene transfer of interleukin-10, interleukin-4, and transforming growth factor-beta on the survival of axotomized retinal ganglion cells. Neuroscience 125(4):903–920
Kolli D, Gupta MR, Sbrana E, Velayutham TS, Hong C, Casola A, Garofalo RP (2014) Alveolar macrophages contribute to the pathogenesis of hMPV infection while protecting against RSV infection. Am J Respir Cell Mol Biol 51(4):502–515. doi:10.1165/rcmb.2013-0414OC
Komatsu T, Bi Z, Reiss CS (1996) Interferon-gamma induced type I nitric oxide synthase activity inhibits viral replication in neurons. J Neuroimmunol 68(1–2):101–108, doi: 0165-5728(96)00083-5 [pii]
Komatsu T, Barna M, Reiss CS (1997) Interleukin-12 promotes recovery from viral encephalitis. Viral Immunol 10(1):35–47
Komatsu T, Ireland DD, Chen N, Reiss CS (1999a) Neuronal expression of NOS-1 is required for host recovery from viral encephalitis. Virology 258(2):389–395. doi:10.1006/viro.1999.9734, S0042-6822(99)99734-9 [pii]
Komatsu T, Ireland DD, Chung N, Dore A, Yoder M, Reiss CS (1999b) Regulation of the BBB during viral encephalitis: roles of IL-12 and NOS. Nitric Oxide 3(4):327–339. doi:10.1006/niox.1999.0237, S1089-8603(99)90237-9 [pii]
Kong T, Westerman KA, Faigle M, Eltzschig HK, Colgan SP (2006) HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEB J 20(13):2242–2250
Korom M, Wylie KM, Wang H, Davis KL, Sangabathula MS, Delassus GS, Morrison LA (2013) A proautophagic antiviral role for the cellular prion protein identified by infection with a herpes simplex virus 1 ICP34.5 mutant. J Virol 87(10):5882–5894. doi:10.1128/jvi.02559-12
Koustova E, Sei Y, McCarty T, Espey MG, Ming R, Morse HC III, Basile AS (2000) Accelerated development of neurochemical and behavioral deficits in LP-BM5 infected mice with targeted deletions of the IFN-gamma gene. J Neuroimmunol 108(1–2):112–121
Krenn BM, Holzer B, Gaudernak E, Triendl A, van Kuppeveld FJ, Seipelt J (2005) Inhibition of polyprotein processing and RNA replication of human rhinovirus by pyrrolidine dithiocarbamate involves metal ions. J Virol 79(22):13892–13899
Ku CC, Che XB, Reichelt M, Rajamani J, Schaap-Nutt A, Huang KJ, Sommer MH, Chen YS, Chen YY, Arvin AM (2011) Herpes simplex virus-1 induces expression of a novel MxA isoform that enhances viral replication. Immunol Cell Biol 89(2):173–182. doi:10.1038/icb.2010.83
Kulkarni AP, Kellaway LA, Kotwal GJ (2005) Herbal complement inhibitors in the treatment of neuroinflammation: future strategy for neuroprotection. Ann N Y Acad Sci 1056:413–429
Kundu K, Dutta K, Nazmi A, Basu A (2013) Japanese encephalitis virus infection modulates the expression of suppressors of cytokine signaling (SOCS) in macrophages: implications for the hosts’ innate immune response. Cell Immunol 285(1–2):100–110. doi:10.1016/j.cellimm.2013.09.005
Labrada L, Liang XH, Zheng W, Johnston C, Levine B (2002) Age-dependent resistance to lethal alphavirus encephalitis in mice: analysis of gene expression in the central nervous system and identification of a novel interferon-inducible protective gene, mouse ISG12. J Virol 76(22):11688–11703
Lallemand-Breitenbach V, de The H (2010) PML nuclear bodies. Cold Spring Harb Perspect Biol 2(5):a000661. doi:10.1101/cshperspect.a000661
Lane TE, Buchmeier MJ, Watry DD, Fox HS (1996) Expression of inflammatory cytokines and inducible nitric oxide synthase in brains of SIV-infected rhesus monkeys: applications to HIV-induced central nervous system disease. Mol Med 2(1):27–37
Lane TE, Fox HS, Buchmeier MJ (1999) Inhibition of nitric oxide synthase-2 reduces the severity of mouse hepatitis virus-induced demyelination: implications for NOS2/NO regulation of chemokine expression and inflammation. J Neurovirol 5(1):48–54
Lang PO, Samaras N, Samaras D, Aspinall R (2013) How important is vitamin D in preventing infections? Osteoporos Int 24(5):1537–1553. doi:10.1007/s00198-012-2204-6
Langhans W (2000) Anorexia of infection: current prospects. Nutrition 16(10):996–1005
Larena M, Regner M, Lobigs M (2013) Cytolytic effector pathways and IFN-gamma help protect against Japanese encephalitis. Eur J Immunol 43(7):1789–1798. doi:10.1002/eji.201243152
Latini S, Pazzagli M, Pepeu G, Pedata F (1996) A2 adenosine receptors: their presence and neuromodulatory role in the central nervous system. Gen Pharmacol 27(6):925–933
Lefebvre JS, Levesque T, Picard S, Pare G, Gravel A, Flamand L, Borgeat P (2011) Extra domain A of fibronectin primes leukotriene biosynthesis and stimulates neutrophil migration through activation of Toll-like receptor 4. Arthritis Rheum 63(6):1527–1533. doi:10.1002/art.30308
Lenschow DJ (2010) Antiviral properties of ISG15. Viruses 2(10):2154–2168. doi:10.3390/v2102154
Leo NA, Callahan TA, Bonneau RH (1998) Peripheral sympathetic denervation alters both the primary and memory cellular immune responses to herpes simplex virus infection. Neuroimmunomodulation 5(1–2):22–35
Lester SN, Li K (2014) Toll-like receptors in antiviral innate immunity. J Mol Biol 426(6):1246–1264. doi:10.1016/j.jmb.2013.11.024
Levine B, Sodora DL (2006) HIV and CXCR4 in a kiss of autophagic death. J Clin Investig 116(8):2078–2080
Li Y, Fan X, He X, Sun H, Zou Z, Yuan H, Xu H, Wang C, Shi X (2012) MicroRNA-466l inhibits antiviral innate immune response by targeting interferon-alpha. Cell Mol Immunol 9(6):497–502. doi:10.1038/cmi.2012.35
Li JM, Darlak KA, Southerland L, Hossain MS, Jaye DL, Josephson CD, Rosenthal H, Waller EK (2013a) VIPhyb, an antagonist of vasoactive intestinal peptide receptor, enhances cellular antiviral immunity in murine cytomegalovirus infected mice. PLoS One 8(5):e63381. doi:10.1371/journal.pone.0063381
Li Y, Xie J, Xu X, Wang J, Ao F, Wan Y, Zhu Y (2013b) MicroRNA-548 down-regulates host antiviral response via direct targeting of IFN-lambda1. Protein Cell 4(2):130–141. doi:10.1007/s13238-012-2081-y
Liao JK, Laufs U (2005) Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol 45:89–118
Liao PH, Hsu YH, Yang HH, Wang MH, Chen LK (2012) Involvement of extraneural tissues and upregulation of inducible nitric oxide synthase after experimental infection with rabies virus in BALB/c mice and LEW/SsN rats. Pathol Int 62(9):619–627. doi:10.1111/j.1440-1827.2012.02846.x
Libbey JE, Kirkman NJ, Wilcox KS, White HS, Fujinami RS (2010) Role for complement in the development of seizures following acute viral infection. J Virol 84(13):6452–6460. doi:10.1128/jvi.00422-10
Libbey JE, Kennett NJ, Wilcox KS, White HS, Fujinami RS (2011) Interleukin-6, produced by resident cells of the central nervous system and infiltrating cells, contributes to the development of seizures following viral infection. J Virol 85(14):6913–6922. doi:10.1128/jvi.00458-11
Libraty DH, Zhang L, Woda M, Giaya K, Kathivu CL, Acosta LP, Tallo V, Segubre-Mercado E, Bautista A, Obcena A, Brion JD, Capeding RZ (2014) Low adiposity during early infancy is associated with a low risk for developing dengue hemorrhagic fever: a preliminary model. PLoS One 9(2):e88944. doi:10.1371/journal.pone.0088944
Ligeti E, Settleman J (2006) Regulation of RhoGAP specificity by phospholipids and prenylation. Methods Enzymol 406:104–117
Lilley CE, Chaurushiya MS, Boutell C, Everett RD, Weitzman MD (2011) The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog 7(6):e1002084. doi:10.1371/journal.ppat.1002084
Lima RG, Moreira L, Paes-Leme J, Barreto-de-Souza V, Castro-Faria-Neto HC, Bozza PT, Bou-Habib DC (2006) Interaction of macrophages with apoptotic cells enhances HIV type 1 replication through PGE2, PAF, and vitronectin receptor. AIDS Res Hum Retroviruses 22(8):763–769
Lin MT, Hinton DR, Parra B, Stohlman SA, van der Veen RC (1998) The role of IL-10 in mouse hepatitis virus-induced demyelinating encephalomyelitis. Virology 245(2):270–280
Lindenmann J, Deuel E, Fanconi S, Haller O (1978) Inborn resistance of mice to myxoviruses: macrophages express phenotype in vitro. J Exp Med 147(2):531–540
Lindersson EK, Hojrup P, Gai WP, Locker D, Martin D, Jensen PH (2004) alpha-Synuclein filaments bind the transcriptional regulator HMGB-1. Neuroreport 15(18):2735–2739
Liu MT, Chen BP, Oertel P, Buchmeier MJ, Armstrong D, Hamilton TA, Lane TE (2000) The T cell chemoattractant IFN-inducible protein 10 is essential in host defense against viral-induced neurologic disease. J Immunol 165(5):2327–2330
Liu XY, Chen W, Wei B, Shan YF, Wang C (2011) IFN-induced TPR protein IFIT3 potentiates antiviral signaling by bridging MAVS and TBK1. J Immunol 187(5):2559–2568. doi:10.4049/jimmunol.1100963
Lopez-Ramirez MA, Wu D, Pryce G, Simpson JE, Reijerkerk A, King-Robson J, Kay O, de Vries HE, Hirst MC, Sharrack B, Baker D, Male DK, Michael GJ, Romero IA (2014) MicroRNA-155 negatively affects blood-brain barrier function during neuroinflammation. FASEB J 28(6):2551–2565. doi:10.1096/fj.13-248880
Lord GM (2006) Leptin as a proinflammatory cytokine. Contrib Nephrol 151:151–164
Loria F, Petrosino S, Mestre L, Spagnolo A, Correa F, Hernangomez M, Guaza C, Di MV, Docagne F (2008) Study of the regulation of the endocannabinoid system in a virus model of multiple sclerosis reveals a therapeutic effect of palmitoylethanolamide. Eur J Neurosci 28(4):633–641
Lubomski M, Louise Rushworth R, Lee W, Bertram KL, Williams DR (2014) Sex differences in Parkinson’s disease. J Clin Neurosci 35(3):370–384. doi:10.1016/j.jocn.2013.12.016
Lucas RM, Ponsonby AL, Dear K, Valery P, Pender MP, Burrows JM, Burrows SR, Chapman C, Coulthard A, Dwyer DE, Dwyer T, Kilpatrick T, Lay ML, McMichael AJ, Taylor BV, van der Mei IA, Williams D (2011) Current and past Epstein-Barr virus infection in risk of initial CNS demyelination. Neurology 77(4):371–379. doi:10.1212/WNL.0b013e318227062a
Luster AD, Alon R, von Andrian UH (2005) Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol 6(12):1182–1190
Machado FS, Johndrow JE, Esper L, Dias A, Bafica A, Serhan CN, Aliberti J (2006) Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nat Med 12(3):330–334
MacMicking JD (2004) IFN-inducible GTPases and immunity to intracellular pathogens. Trends Immunol 25(11):601–609
Maloney EM, St Claire MO, Widner B, Werner ER, Fuchs D (2000) Central nervous system activation of the indoleamine-2,3-dioxygenase pathway in human T cell lymphotropic virus type I-associated myelopathy/tropical spastic paraparesis. J Infect Dis 181(6):2037–2040
Mana P, Linares D, Fordham S, Staykova M, Willenborg D (2006) Deleterious role of IFN{gamma} in a toxic model of central nervous system demyelination. Am J Pathol 168(5):1464–1473
Mandey SH, Kuijk LM, Frenkel J, Waterham HR (2006) A role for geranylgeranylation in interleukin-1beta secretion. Arthritis Rheum 54(11):3690–3695
Mansour E, Pereira FG, Araujo EP, Amaral ME, Morari J, Ferraroni NR, Ferreira DS, Lorand-Metze I, Velloso LA (2006) Leptin inhibits apoptosis in thymus through a Janus kinase-2-independent, insulin receptor substrate-1/phosphatidylinositol-3 kinase-dependent pathway. Endocrinology 147(11):5470–5479
Mansouri M, Viswanathan K, Douglas JL, Hines J, Gustin J, Moses AV, Fruh K (2009) Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma herpesvirus. J Virol 83(19):9672–9681
Markle JG, Fish EN (2014) SeXX matters in immunity. Trends Immunol 35(3):97–104. doi:10.1016/j.it.2013.10.006
Marriott SJ, Semmes OJ (2005) Impact of HTLV-I Tax on cell cycle progression and the cellular DNA damage repair response. Oncogene 24(39):5986–5995
Martin R, Sospedra M (2014) Sphingosine-1 phosphate and central nervous system. Curr Top Microbiol Immunol 378:149–170. doi:10.1007/978-3-319-05879-5_7
Martinez NE, Karlsson F, Sato F, Kawai E, Omura S, Minagar A, Grisham MB, Tsunoda I (2014) Protective and detrimental roles for regulatory T cells in a viral model for multiple sclerosis. Brain Pathol 24(5):436–451. doi:10.1111/bpa.12119
Martinez-Gutierrez M, Castellanos JE, Gallego-Gomez JC (2011) Statins reduce dengue virus production via decreased virion assembly. Intervirology 54(4):202–216. doi:10.1159/000321892
Massad MG, Navarro RA, Rubeiz H, Kpodonu J, Karol J, Blacha M, Evans A (2004) Acute postoperative shingles after thoracic sympathectomy for hyperhidrosis. Ann Thorac Surg 78(6):2159–2161. doi:10.1016/s0003-4975(03)01499-1
Matarese G, La CA, Sanna V, Lord GM, Lechler RI, Fontana S, Zappacosta S (2002) Balancing susceptibility to infection and autoimmunity: a role for leptin? Trends Immunol 23(4):182–187
Matthews V, Robertson T, Kendrick T, Abdo M, Papadimitriou J, McMinn P (2000) Morphological features of Murray valley encephalitis virus infection in the central nervous system of Swiss mice. Int J Exp Pathol 81(1):31–40
Mattioli B, Straface E, Quaranta MG, Giordani L, Viora M (2005) Leptin promotes differentiation and survival of human dendritic cells and licenses them for Th1 priming. J Immunol 174(11):6820–6828
Mattoscio D, Segre CV, Chiocca S (2013) Viral manipulation of cellular protein conjugation pathways: the SUMO lesson. World J Virol 2(2):79–90. doi:10.5501/wjv.v2.i2.79
Matyszak MK, Perry VH (1997) Dendritic cells in inflammatory responses in the CNS. Adv Exp Med Biol 417:295–299
McCann KB, Lee A, Wan J, Roginski H, Coventry MJ (2003) The effect of bovine lactoferrin and lactoferricin B on the ability of feline calicivirus (a norovirus surrogate) and poliovirus to infect cell cultures. J Appl Microbiol 95(5):1026–1033
McKimmie CS, Johnson N, Fooks AR, Fazakerley JK (2005) Viruses selectively upregulate Toll-like receptors in the central nervous system. Biochem Biophys Res Commun 336(3):925–933
Mecha M, Feliu A, Inigo PM, Mestre L, Carrillo-Salinas FJ, Guaza C (2013) Cannabidiol provides long-lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: a role for A2A receptors. Neurobiol Dis 59:141–150. doi:10.1016/j.nbd.2013.06.016
Mehlhop E, Whitby K, Oliphant T, Marri A, Engle M, Diamond MS (2005) Complement activation is required for induction of a protective antibody response against West Nile virus infection. J Virol 79(12):7466–7477
Mehrbod P, Hair-Bejo M, Tengku Ibrahim TA, Omar AR, El Zowalaty M, Ajdari Z, Ideris A (2014) Simvastatin modulates cellular components in influenza A virus-infected cells. Int J Mol Med 34(1):61–73. doi:10.3892/ijmm.2014.1761
Melkonian KA, Ostermeyer AG, Chen JZ, Roth MG, Brown DA (1999) Role of lipid modifications in targeting proteins to detergent-resistant membrane rafts. Many raft proteins are acylated, while few are prenylated. J Biol Chem 274(6):3910–3917
Merabova N, Kaminski R, Krynska B, Amini S, Khalili K, Darbinyan A (2012) JCV agnoprotein-induced reduction in CXCL5/LIX secretion by oligodendrocytes is associated with activation of apoptotic signaling in neurons. J Cell Physiol 227(8):3119–3127. doi:10.1002/jcp.23065
Mestre L, Correa F, Arevalo-Martin A, Molina-Holgado E, Valenti M, Ortar G, Di Marzo V, Guaza C (2005) Pharmacological modulation of the endocannabinoid system in a viral model of multiple sclerosis. J Neurochem 92(6):1327–1339
Mestre L, Correa F, Docagne F, Clemente D, Guaza C (2006) The synthetic cannabinoid WIN 55,212-2 increases COX-2 expression and PGE2 release in murine brain-derived endothelial cells following Theiler’s virus infection. Biochem Pharmacol 72(7):869–880
Mestre L, Docagne F, Correa F, Loria F, Hernangomez M, Borrell J, Guaza C (2009) A cannabinoid agonist interferes with the progression of a chronic model of multiple sclerosis by downregulating adhesion molecules. Mol Cell Neurosci 40(2):258–266
Methe H, Kim JO, Kofler S, Nabauer M, Weis M (2005) Statins decrease Toll-like receptor 4 expression and downstream signaling in human CD14+ monocytes. Arterioscler Thromb Vasc Biol 25(7):1439–1445
Metz-Boutigue MH, Kieffer AE, Goumon Y, Aunis D (2003) Innate immunity: involvement of new neuropeptides. Trends Microbiol 11(12):585–592
Michel T, Hentges F, Zimmer J (2012) Consequences of the crosstalk between monocytes/macrophages and natural killer cells. Front Immunol 3:403. doi:10.3389/fimmu.2012.00403
Michlmayr D, McKimmie CS, Pingen M, Haxton B, Mansfield K, Johnson N, Fooks AR, Graham GJ (2014) Defining the chemokine basis for leukocyte recruitment during viral encephalitis. J Virol 88(17):9553–9567. doi:10.1128/jvi.03421-13
Miller E, Bhardwaj N (2013) Dendritic cell dysregulation during HIV-1 infection. Immunol Rev 254(1):170–189. doi:10.1111/imr.12082
Minagar A, Shapshak P, Fujimura R, Ownby R, Heyes M, Eisdorfer C (2002) The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J Neurol Sci 202(1–2):13–23
Mizrahi A, Molshanski-Mor S, Weinbaum C, Zheng Y, Hirshberg M, Pick E (2005) Activation of the phagocyte NADPH oxidase by Rac Guanine nucleotide exchange factors in conjunction with ATP and nucleoside diphosphate kinase. J Biol Chem 280(5):3802–3811
Modiano N, Lu YE, Cresswell P (2005) Golgi targeting of human guanylate-binding protein-1 requires nucleotide binding, isoprenylation, and an IFN-gamma-inducible cofactor. Proc Natl Acad Sci U S A 102(24):8680–8685
Molina-Holgado F, Hernanz A, De la FM, Guaza C (1999) N-Acetyl-cysteine inhibition of encephalomyelitis Theiler’s virus-induced nitric oxide and tumour necrosis factor-alpha production by murine astrocyte cultures. Biofactors 10(2–3):187–193
Moore PM (2000) Vasculitis of the central nervous system. Curr Rheumatol Rep 2(5):376–382
Morris T, Stables M, Gilroy DW (2006) New perspectives on aspirin and the endogenous control of acute inflammatory resolution. Scientific World Journal 6:1048–1065
Mott KR, Gate D, Zandian M, Allen SJ, Rajasagi NK, van Rooijen N, Chen S, Arditi M, Rouse BT, Flavell RA, Town T, Ghiasi H (2011) Macrophage IL-12p70 signaling prevents HSV-1-induced CNS autoimmunity triggered by autoaggressive CD4+ Tregs. Invest Ophthalmol Vis Sci 52(5):2321–2333. doi:10.1167/iovs.10-6536
Muller N, Myint AM, Krause D, Weidinger E, Schwarz MJ (2013) Anti-inflammatory treatment in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 42:146–153. doi:10.1016/j.pnpbp.2012.11.008
Murad F (2006) Shattuck lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N Engl J Med 355(19):2003–2011
Murphy S (2000) Production of nitric oxide by glial cells: regulation and potential roles in the CNS. Glia 29(1):1–13
Nallar SC, Kalvakolanu DV (2014) Interferons, signal transduction pathways, and the central nervous system. J Interferon Cytokine Res 34(8):559–576. doi:10.1089/jir.2014.0021
Navarra M, Baltrons MA, Sardon T, Pedraza CE, Garcia A (2004) HIV-1 coat protein gp120 decreases NO-dependent cyclic GMP accumulation in rat brain astroglia by increasing cyclic GMP phosphodiesterase activity. Neurochem Int 45(6):937–946
Nazmi A, Mukherjee S, Kundu K, Dutta K, Mahadevan A, Shankar SK, Basu A (2014) TLR7 is a key regulator of innate immunity against Japanese encephalitis virus infection. Neurobiol Dis 69:235–247. doi:10.1016/j.nbd.2014.05.036
Nchoutmboube JA, Viktorova EG, Scott AJ, Ford LA, Pei Z, Watkins PA, Ernst RK, Belov GA (2013) Increased long chain acyl-Coa synthetase activity and fatty acid import is linked to membrane synthesis for development of picornavirus replication organelles. PLoS Pathog 9(6):e1003401. doi:10.1371/journal.ppat.1003401
Neal JW (2014) Flaviviruses are neurotropic, but how do they invade the CNS? J Infect 69(3):203–215. doi:10.1016/j.jinf.2014.05.010
Neil SJ (2013) The antiviral activities of tetherin. Curr Top Microbiol Immunol 371:67–104. doi:10.1007/978-3-642-37765-5_3
Nikovics K, Dazza MC, Ekwalanga M, Mammano F, Clavel F, Saragosti S (2012) Counteraction of tetherin antiviral activity by two closely related SIVs differing by the presence of a Vpu gene. PLoS One 7(4):e35411. doi:10.1371/journal.pone.0035411
Noack J, Bernasconi R, Molinari M (2014) How viruses hijack the ERAD tuning machinery. J Virol 88(18):10272–10275. doi:10.1128/jvi.00801-14
Noch EK, Khalili K (2013) The role of AEG-1/MTDH/LYRIC in the pathogenesis of central nervous system disease. Adv Cancer Res 120:159–192. doi:10.1016/b978-0-12-401676-7.00006-1
Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK (1999) Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 285(5431):1276–1279
Noe SN, Nyland SB, Ugen K, Friedman H, Klein TW (1998) Cannabinoid receptor agonists enhance syncytia formation in MT-2 cells infected with cell free HIV-1MN. Adv Exp Med Biol 437:223–229
O’Connor KA, Hansen MK, Rachal PC, Deak MM, Biedenkapp JC, Milligan ED, Johnson JD, Wang H, Maier SF, Tracey KJ, Watkins LR (2003) Further characterization of high mobility group box 1 (HMGB1) as a proinflammatory cytokine: central nervous system effects. Cytokine 24(6):254–265
Oberdorfer C, Adams O, MacKenzie CR, De Groot CJ, Daubener W (2003) Role of IDO activation in anti-microbial defense in human native astrocytes. Adv Exp Med Biol 527:15–26
Ogawa Y, Calhoun WJ (2006) The role of leukotrienes in airway inflammation. J Allergy Clin Immunol 118(4):789–798
Ogura R, Aoki H, Natsumeda M, Shimizu H, Kobayashi T, Saito T, Takizawa J, Okamoto K, Hasegawa G, Umezu H, Ohshima K, Takahashi H, Fujii Y, Kakita A (2013) Epstein-Barr virus-associated primary central nervous system cytotoxic T-cell lymphoma. Neuropathology 33(4):436–441. doi:10.1111/neup.12005
Olson JK, Miller SD (2004) Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol 173(6):3916–3924
Olson JK, Miller SD (2009) The innate immune response affects the development of the autoimmune response in Theiler’s virus-induced demyelinating disease. J Immunol 182(9):5712–5722
Onomoto K, Yoneyama M, Fung G, Kato H, Fujita T (2014) Antiviral innate immunity and stress granule responses. Trends Immunol 35(9):420–428. doi:10.1016/j.it.2014.07.006
Owens T, Khorooshi R, Wlodarczyk A, Asgari N (2014) Interferons in the central nervous system: a few instruments play many tunes. Glia 62(3):339–355
Palma JP, Kim BS (2001) Induction of selected chemokines in glial cells infected with Theiler’s virus. J Neuroimmunol 117(1–2):166–170
Palma JP, Kwon D, Clipstone NA, Kim BS (2003) Infection with Theiler’s murine encephalomyelitis virus directly induces proinflammatory cytokines in primary astrocytes via NF-kappaB activation: potential role for the initiation of demyelinating disease. J Virol 77(11):6322–6331
Parra B, Hinton DR, Marten NW, Bergmann CC, Lin MT, Yang CS, Stohlman SA (1999) IFN-gamma is required for viral clearance from central nervous system oligodendroglia. J Immunol 162(3):1641–1647
Pasarica M, Shin AC, Yu M, Ou Yang HM, Rathod M, Jen KL, MohanKumar S, MohanKumar PS, Markward N, Dhurandhar NV (2006) Human adenovirus 36 induces adiposity, increases insulin sensitivity, and alters hypothalamic monoamines in rats. Obesity 14(11):1905–1913. doi:10.1038/oby.2006.222
Patel TR, Corbett SA (2004) Simvastatin suppresses LPS-induced Akt phosphorylation in the human monocyte cell line THP-1. J Surg Res 116(1):116–120
Pattnaik AK, Dinh PX (2013) Manipulation of cellular processing bodies and their constituents by viruses. DNA Cell Biol 32(6):286–291. doi:10.1089/dna.2013.2054
Pei R, Qin B, Zhang X, Zhu W, Kemper T, Ma Z, Trippler M, Schlaak J, Chen X, Lu M (2014) Interferon-induced proteins with tetratricopeptide repeats 1 and 2 are cellular factors that limit hepatitis B virus replication. J Innate Immun 6(2):182–191. doi:10.1159/000353220
Perigolo-Vicente R, Ritt K, Goncalves-de-Albuquerque CF, Castro-Faria-Neto HC, Paes-de-Carvalho R, Giestal-de-Araujo E (2014) IL-6, A1 and A2aR: a crosstalk that modulates BDNF and induces neuroprotection. Biochem Biophys Res Commun 449(4):477–482. doi:10.1016/j.bbrc.2014.05.036
Perkins D (2005) Virus signaling and apoptosis in the central nervous system infection. Front Biosci 10:2804–2819
Peter JB, Sevall JS (2001) Review of 3200 serially received CSF samples submitted for type-specific HSV detection by PCR in the reference laboratory setting. Mol Cell Probes 15(3):177–182. doi:10.1006/mcpr.2001.0356
Peyssonaux C, Johnson RS (2004) An unexpected role for hypoxic response: oxygenation and inflammation. Cell Cycle 3(2):168–171
Phares TW, Stohlman SA, Bergmann CC (2013) Intrathecal humoral immunity to encephalitic RNA viruses. Viruses 5(2):732–752. doi:10.3390/v5020732
Pillay NS, Kellaway LA, Kotwal GJ (2005) Administration of vaccinia virus complement control protein shows significant cognitive improvement in a mild injury model. Ann N Y Acad Sci 1056:450–461
Post DE, Devi NS, Li Z, Brat DJ, Kaur B, Nicholson A, Olson JJ, Zhang Z, Van Meir EG (2004) Cancer therapy with a replicating oncolytic adenovirus targeting the hypoxic microenvironment of tumors. Clin Cancer Res 10(24):8603–8612
Potworowski E, Bischoff P, Oth D (1992) Prolongation of survival in retrovirally induced T cell lymphoma by dietary omega 6 fatty acid. Nutr Cancer 17(3):217–221
Probasco JC, Spudich SS, Critchfield J, Lee E, Lollo N, Deeks SG, Price RW (2008) Failure of atorvastatin to modulate CSF HIV-1 infection: results of a pilot study. Neurology 71(7):521–524. doi:10.1212/01.wnl.0000325006.84658.e7
Prod’homme T, Weber MS, Steinman L, Zamvil SS (2006) A neuropeptide in immune-mediated inflammation, Y? Trends Immunol 27(4):164–167
Proescholdt MA, Heiss JD, Walbridge S, Muhlhauser J, Capogrossi MC, Oldfield EH, Merrill MJ (1999) Vascular endothelial growth factor (VEGF) modulates vascular permeability and inflammation in rat brain. J Neuropathol Exp Neurol 58(6):613–627
Pryce G, Baker D (2012) Potential control of multiple sclerosis by cannabis and the endocannabinoid system. CNS Neurol Disord Drug Targets 11(5):624–641
Qian F, Thakar J, Yuan X, Nolan M, Murray KO, Lee WT, Wong SJ, Meng H, Fikrig E, Kleinstein SH, Montgomery RR (2014) Immune markers associated with host susceptibility to infection with West Nile virus. Viral Immunol 27(2):39–47. doi:10.1089/vim.2013.0074
Qiu S, Liu N, Jia L, Yang G, Su W, Li J, Song L, Yang C, Wang J, Zhang C, Wang Z, Qiao F, Tomlinson S, Atkinson C, Sun Y, Huang L, Song H, Wang Y, Li Z (2012) A new treatment for neurogenic inflammation caused by EV71 with CR2-targeted complement inhibitor. Virol J 9:285. doi:10.1186/1743-422x-9-285
Rajasagi NK, Reddy PB, Mulik S, Gjorstrup P, Rouse BT (2013) Neuroprotectin D1 reduces the severity of herpes simplex virus-induced corneal immunopathology. Invest Ophthalmol Vis Sci 54(9):6269–6279. doi:10.1167/iovs.13-12152
Rakic B, Sagan SM, Noestheden M, Belanger S, Nan X, Evans CL, Xie XS, Pezacki JP (2006) Peroxisome proliferator-activated receptor alpha antagonism inhibits hepatitis C virus replication. Chem Biol 13(1):23–30
Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM (2005) Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 15(7):16R–28R
Ran Y, Shu HB, Wang YY (2014) MITA/STING: a central and multifaceted mediator in innate immune response. Cytokine Growth Factor Rev 25(6):631–639. doi:10.1016/j.cytogfr.2014.05.003
Ransohoff RM, Benveniste EN (2005) Cytokines and the CNS. vol second edition. Taylor & Francis. ISBN 0-8493-1622-7
Razzini E, Baronzio GF (1993) Omega-3 fatty acids as coadjuvant treatment in AIDS. Med Hypotheses 41(4):300–305
Recchiuti A, Serhan CN (2012) Pro-resolving lipid mediators (SPMs) and their actions in regulating miRNA in novel resolution circuits in inflammation. Front Immunol 3:298. doi:10.3389/fimmu.2012.00298
Reed DS, Larsen T, Sullivan LJ, Lind CM, Lackemeyer MG, Pratt WD, Parker MD (2005) Aerosol exposure to western equine encephalitis virus causes fever and encephalitis in cynomolgus macaques. J Infect Dis 192(7):1173–1182
Reid E, Charleston B (2014) Type I and III interferon production in response to RNA viruses. J Interferon Cytokine Res 34(9):649–658. doi:10.1089/jir.2014.0066
Reiss CS (2010) Cannabinoids and viral infections. Pharmaceuticals 3(6):1873–1886. doi:10.3390/ph3061873
Reiss CS, Komatsu T (1998) Does nitric oxide play a critical role in viral infections? J Virol 72(6):4547–4551
Rempel JD, Murray SJ, Meisner J, Buchmeier MJ (2004) Differential regulation of innate and adaptive immune responses in viral encephalitis. Virology 318(1):381–392
Reynolds AE, Enquist LW (2006) Biological interactions between herpesviruses and cyclooxygenase enzymes. Rev Med Virol 16(6):393–403
Rikitake Y, Liao JK (2005) Rho GTPases, statins, and nitric oxide. Circ Res 97(12):1232–1235
Rink L, Haase H (2007) Zinc homeostasis and immunity. Trends Immunol 28(1):1–4
Robertson SJ, Lubick KJ, Freedman BA, Carmody AB, Best SM (2014) Tick-borne flaviviruses antagonize both IRF-1 and type I IFN signaling to inhibit dendritic cell function. J Immunol 192(6):2744–2755. doi:10.4049/jimmunol.1302110
Rohrenbeck AM, Bette M, Hooper DC, Nyberg F, Eiden LE, Dietzschold B, Weihe E (1999) Upregulation of COX-2 and CGRP expression in resident cells of the Borna disease virus-infected brain is dependent upon inflammation. Neurobiol Dis 6(1):15–34
Rong LL, Gooch C, Szabolcs M, Herold KC, Lalla E, Hays AP, Yan SF, Yan SS, Schmidt AM (2005) RAGE: a journey from the complications of diabetes to disorders of the nervous system—striking a fine balance between injury and repair. Restor Neurol Neurosci 23(5–6):355–365
Rosato PC, Leib DA (2014) Intrinsic innate immunity fails to control herpes simplex virus and vesicular stomatitis virus replication in sensory neurons and fibroblasts. J Virol 88(17):9991–10001. doi:10.1128/jvi.01462-14
Rothan HA, Han HC, Ramasamy TS, Othman S, Rahman NA, Yusof R (2012) Inhibition of dengue NS2B-NS3 protease and viral replication in Vero cells by recombinant retrocyclin-1. BMC Infect Dis 12:314. doi:10.1186/1471-2334-12-314
Rubio I, Wittig U, Meyer C, Heinze R, Kadereit D, Waldmann H, Downward J, Wetzker R (1999) Farnesylation of Ras is important for the interaction with phosphoinositide 3-kinase gamma. Eur J Biochem 266(1):70–82
Rubio N, Sanz-Rodriguez F, Lipton HL (2006) Theiler’s virus induces the MIP-2 chemokine (CXCL2) in astrocytes from genetically susceptible but not from resistant mouse strains. Cell Immunol 239(1):31–40. doi:10.1016/j.cellimm.2006.03.003
Ruller CM, Tabor-Godwin JM, Van Deren DA Jr, Robinson SM, Maciejewski S, Gluhm S, Gilbert PE, An N, Gude NA, Sussman MA, Whitton JL, Feuer R (2012) Neural stem cell depletion and CNS developmental defects after enteroviral infection. Am J Pathol 180(3):1107–1120. doi:10.1016/j.ajpath.2011.11.016
Rushing EJ, Liappis A, Smirniotopoulos JD, Smith AB, Henry JM, Man YG, Nelson AM (2008) Immune reconstitution inflammatory syndrome of the brain: case illustrations of a challenging entity. J Neuropathol Exp Neurol 67(8):819–827. doi:10.1097/NEN.0b013e318181b4da
Russell CD, Schwarze J (2014) The role of pro-resolution lipid mediators in infectious disease. Immunology 141(2):166–173. doi:10.1111/imm.12206
Ryman KD, Meier KC, Nangle EM, Ragsdale SL, Korneeva NL, Rhoads RE, MacDonald MR, Klimstra WB (2005) Sindbis virus translation is inhibited by a PKR/RNase L-independent effector induced by alpha/beta interferon priming of dendritic cells. J Virol 79(3):1487–1499
Sabouri AH, Marcondes MC, Flynn C, Berger M, Xiao N, Fox HS, Sarvetnick NE (2014) TLR signaling controls lethal encephalitis in WNV-infected brain. Brain Res 1574:84–95. doi:10.1016/j.brainres.2014.05.049
Sagredo O, Pazos MR, Valdeolivas S, Fernandez-Ruiz J (2012) Cannabinoids: novel medicines for the treatment of Huntington’s disease. Recent Pat CNS Drug Discov 7(1):41–48
Sahin U, Ferhi O, Carnec X, Zamborlini A, Peres L, Jollivet F, Vitaliano-Prunier A, de The H, Lallemand-Breitenbach V (2014a) Interferon controls SUMO availability via the Lin28 and let-7 axis to impede virus replication. Nat Commun 5:4187. doi:10.1038/ncomms5187
Sahin U, Ferhi O, Jeanne M, Benhenda S, Berthier C, Jollivet F, Niwa-Kawakita M, Faklaris O, Setterblad N, de The H, Lallemand-Breitenbach V (2014b) Oxidative stress-induced assembly of PML nuclear bodies controls sumoylation of partner proteins. J Cell Biol 204(6):931–945. doi:10.1083/jcb.201305148
Sakurai K, Zou JP, Tschetter JR, Ward JM, Shearer GM (2002) Effect of indoleamine 2,3-dioxygenase on induction of experimental autoimmune encephalomyelitis. J Neuroimmunol 129(1–2):186–196
Santoro F, Greenstone HL, Insinga A, Liszewski MK, Atkinson JP, Lusso P, Berger EA (2003) Interaction of glycoprotein H of human herpesvirus 6 with the cellular receptor CD46. J Biol Chem 278(28):25964–25969
Santos CM (2012) New agents promote neuroprotection in Parkinson’s disease models. CNS Neurol Disord Drug Targets 11(4):410–418
Sarojini S, Theofanis T, Reiss CS (2011) Interferon-induced tetherin restricts vesicular stomatitis virus release in neurons. DNA Cell Biol 30(12):965–974. doi:10.1089/dna.2011.1384
Sauter D (2014) Counteraction of the multifunctional restriction factor tetherin. Front Microbiol 5:163. doi:10.3389/fmicb.2014.00163
Sawynok J, Liu XJ (2003) Adenosine in the spinal cord and periphery: release and regulation of pain. Prog Neurobiol 69(5):313–340
Schluesener H, Meyermann R (1995) Neutrophilic defensins penetrate the blood-brain barrier. J Neurosci Res 42(5):718–723
Schmidt S, Fritz JV, Bitzegeio J, Fackler OT, Keppler OT (2011) HIV-1 Vpu blocks recycling and biosynthetic transport of the intrinsic immunity factor CD317/tetherin to overcome the virion release restriction. mBio 2(3):e00036–e00011. doi:10.1128/mBio.00036-11
Schneider WM, Chevillotte MD, Rice CM (2014) Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. doi:10.1146/annurev-immunol-032713-120231
Schneider-Schaulies J, ter Meulen V, Schneider-Schaulies S (2001) Measles virus interactions with cellular receptors: consequences for viral pathogenesis. J Neurovirol 7(5):391–399
Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM (2014) Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505(7485):691–695. doi:10.1038/nature12862
Schoneboom BA, Fultz MJ, Miller TH, McKinney LC, Grieder FB (1999) Astrocytes as targets for Venezuelan equine encephalitis virus infection. J Neurovirol 5(4):342–354
Seeler JS, Marchio A, Losson R, Desterro JM, Hay RT, Chambon P, Dejean A (2001) Common properties of nuclear body protein SP100 and TIF1alpha chromatin factor: role of SUMO modification. Mol Cell Biol 21(10):3314–3324
Seet RC, Lee CY, Lim EC, Quek AM, Yeo LL, Huang SH, Halliwell B (2009) Oxidative damage in dengue fever. Free Radic Biol Med 47(4):375–380. doi:10.1016/j.freeradbiomed.2009.04.035
Seganti L, Di Biase AM, Rega B, De GB, Nicoletti M, Antonini G, Valenti P (2001) Involvement of bovine lactoferrin moieties in the inhibition of herpes simplex virus type 1 infection. Int J Immunopathol Pharmacol 14(2):71–79
Serhan CN (2005a) Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids 73(3–4):141–162
Serhan CN (2005b) Novel omega—3-derived local mediators in anti-inflammation and resolution. Pharmacol Ther 105(1):7–21
Serra-Moreno R, Evans DT (2012) Adaptation of human and simian immunodeficiency viruses for resistance to tetherin/BST-2. Curr HIV Res 10(4):277–282
Shanks N, Harbuz MS, Jessop DS, Perks P, Moore PM, Lightman SL (1998) Inflammatory disease as chronic stress. Ann N Y Acad Sci 840:599–607
Shen F, Su H, Fan Y, Chen Y, Zhu Y, Liu W, Young WL, Yang GY (2006) Adeno-associated viral-vector-mediated hypoxia-inducible vascular endothelial growth factor gene expression attenuates ischemic brain injury after focal cerebral ischemia in mice. Stroke 37(10):2601–2606
Shirey KA, Lai W, Pletneva LM, Karp CL, Divanovic S, Blanco JC, Vogel SN (2014) Role of the lipoxygenase pathway in RSV-induced alternatively activated macrophages leading to resolution of lung pathology. Mucosal Immunol 7(3):549–557. doi:10.1038/mi.2013.71
Shukla DK, Kaiser CC, Stebbins GT, Feinstein DL (2010) Effects of pioglitazone on diffusion tensor imaging indices in multiple sclerosis patients. Neurosci Lett 472(3):153–156. doi:10.1016/j.neulet.2010.01.046
Shusta EV, Zhu C, Boado RJ, Pardridge WM (2002) Subtractive expression cloning reveals high expression of CD46 at the blood-brain barrier. J Neuropathol Exp Neurol 61(7):597–604
Siciliano R, Barone E, Calabrese V, Rispoli V, Butterfield DA, Mancuso C (2011) Experimental research on nitric oxide and the therapy of Alzheimer disease: a challenging bridge. CNS Neurol Disord Drug Targets 10(7):766–776
Silverman MN, Pearce BD, Biron CA, Miller AH (2005) Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol 18(1):41–78
Singh R, Patel V, Mureithi MW, Naranbhai V, Ramsuran D, Tulsi S, Hiramen K, Werner L, Mlisana K, Altfeld M, Luban J, Kasprowicz V, Dheda K, Abdool Karim SS, Ndung’u T (2014) TRIM5alpha and TRIM22 are differentially regulated according to HIV-1 infection phase and compartment. J Virol 88(8):4291–4303. doi:10.1128/jvi.03603-13
Skaper SD, Facci L, Giusti P (2014) Mast cells, glia and neuroinflammation: partners in crime? Immunology 141(3):314–327. doi:10.1111/imm.12170
Smith JA (2014) A new paradigm: innate immune sensing of viruses via the unfolded protein response. Front Microbiol 5:222. doi:10.3389/fmicb.2014.00222
So EY, Kang MH, Kim BS (2006) Induction of chemokine and cytokine genes in astrocytes following infection with Theiler’s murine encephalomyelitis virus is mediated by the Toll-like receptor 3. Glia 53(8):858–867
Solbrig MV, Fan Y, Hazelton P (2013) Prospects for cannabinoid therapies in viral encephalitis. Brain Res 1537:273–282. doi:10.1016/j.brainres.2013.08.032
Son KN, Pugazhenthi S, Lipton HL (2009) Activation of tumor suppressor protein p53 is required for Theiler’s murine encephalomyelitis virus-induced apoptosis in M1-D macrophages. J Virol 83(20):10770–10777. doi:10.1128/jvi.01030-09
Souza TM, Temerozo JR, Giestal-de-Araujo E, Bou-Habib DC (2014) The effects of neurotrophins and the neuropeptides VIP and PACAP on HIV-1 infection: histories with opposite ends. Neuroimmunomodulation 21(5):268–282. doi:10.1159/000357434
Speth C, Williams K, Hagleitner M, Westmoreland S, Rambach G, Mohsenipour I, Schmitz J, Wurzner R, Lass-Florl C, Stoiber H, Dierich MP, Maier H (2004) Complement synthesis and activation in the brain of SIV-infected monkeys. J Neuroimmunol 151(1–2):45–54
Sporbert A, Mertsch K, Smolenski A, Haseloff RF, Schonfelder G, Paul M, Ruth P, Walter U, Blasig IE (1999) Phosphorylation of vasodilator-stimulated phosphoprotein: a consequence of nitric oxide- and cGMP-mediated signal transduction in brain capillary endothelial cells and astrocytes. Mol Brain Res 67(2):258–266
Steel CD, Breving K, Tavakoli S, Kim WK, Sanford LD, Ciavarra RP (2014) Role of peripheral immune response in microglia activation and regulation of brain chemokine and proinflammatory cytokine responses induced during VSV encephalitis. J Neuroimmunol 267(1–2):50–60. doi:10.1016/j.jneuroim.2013.12.002
Steer SA, Corbett JA (2003) The role and regulation of COX-2 during viral infection. Viral Immunol 16(4):447–460
Su KP, Lai HC, Yang HT, Su WP, Peng CY, Chang JP, Chang HC, Pariante CM (2014) Omega-3 fatty acids in the prevention of interferon-alpha-induced depression: results from a randomized, controlled trial. Biol Psychiatry 76(7):559–566. doi:10.1016/j.biopsych.2014.01.008
Suh HS, Brosnan CF, Lee SC (2009) Toll-like receptors in CNS viral infections. Curr Top Microbiol Immunol 336:63–81. doi:10.1007/978-3-642-00549-7_4
Suhy DA, Giddings TH Jr, Kirkegaard K (2000) Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J Virol 74(19):8953–8965
Sumanasekera WK, Tien ES, Turpey R, Vanden Heuvel JP, Perdew GH (2003) Evidence that peroxisome proliferator-activated receptor alpha is complexed with the 90-kDa heat shock protein and the hepatitis virus B X-associated protein 2. J Biol Chem 278(7):4467–4473
Sumpter R Jr, Levine B (2011) Selective autophagy and viruses. Autophagy 7(3):260–265
Sun P, Zhou K, Wang S, Li P, Chen S, Lin G, Zhao Y, Wang T (2013) Involvement of MAPK/NF-kappaB signaling in the activation of the cholinergic anti-inflammatory pathway in experimental colitis by chronic vagus nerve stimulation. PLoS One 8(8):e69424. doi:10.1371/journal.pone.0069424
Sun T, Vasek MJ, Klein RS (2014) Congenitally acquired persistent lymphocytic choriomeningitis viral infection reduces neuronal progenitor pools in the adult hippocampus and subventricular zone. PLoS One 9(5):e96442. doi:10.1371/journal.pone.0096442
Sze A, Belgnaoui SM, Olagnier D, Lin R, Hiscott J, van Grevenynghe J (2013) Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe 14(4):422–434. doi:10.1016/j.chom.2013.09.009
Szretter KJ, Samuel MA, Gilfillan S, Fuchs A, Colonna M, Diamond MS (2009) The immune adaptor molecule SARM modulates tumor necrosis factor alpha production and microglia activation in the brainstem and restricts West Nile virus pathogenesis. J Virol 83(18):9329–9338. doi:10.1128/jvi.00836-09
Szretter KJ, Brien JD, Thackray LB, Virgin HW, Cresswell P, Diamond MS (2011) The interferon-inducible gene viperin restricts West Nile virus pathogenesis. J Virol 85(22):11557–11566. doi:10.1128/jvi.05519-11
Tabor-Godwin JM, Tsueng G, Sayen MR, Gottlieb RA, Feuer R (2012) The role of autophagy during coxsackievirus infection of neural progenitor and stem cells. Autophagy 8(6):938–953. doi:10.4161/auto.19781
Tam Vincent C, Quehenberger O, Oshansky Christine M, Suen R, Armando Aaron M, Treuting Piper M, Thomas Paul G, Dennis Edward A, Aderem A (2013) Lipidomic profiling of influenza infection identifies mediators that induce and resolve inflammation. Cell 154(1):213–227, http://dx.doi.org/10.1016/j.cell.2013.05.052
Taylor JM, Barry M (2006) Near death experiences: poxvirus regulation of apoptotic death. Virology 344(1):139–150
Taylor KE, Mossman KL (2013) Recent advances in understanding viral evasion of type I interferon. Immunology 138(3):190–197. doi:10.1111/imm.12038
Taylor DR, Puig M, Darnell ME, Mihalik K, Feinstone SM (2005) New antiviral pathway that mediates hepatitis C virus replicon interferon sensitivity through ADAR1. J Virol 79(10):6291–6298
Taylor RT, Lubick KJ, Robertson SJ, Broughton JP, Bloom ME, Bresnahan WA, Best SM (2011) TRIM79alpha, an interferon-stimulated gene product, restricts tick-borne encephalitis virus replication by degrading the viral RNA polymerase. Cell Host Microbe 10(3):185–196. doi:10.1016/j.chom.2011.08.004
Taylor KG, Woods TA, Winkler CW, Carmody AB, Peterson KE (2014) Age-dependent myeloid dendritic cell responses mediate resistance to La Crosse virus induced neurological disease. J Virol 88(19):11070–11079. doi:10.1128/jvi.01866-14
Templeton A, Nguyen G, Ash JD, Straub RH, Carr DJ (2008) Chemical sympathectomy increases susceptibility to ocular herpes simplex virus type 1 infection. J Neuroimmunol 197(1):37–46. doi:10.1016/j.jneuroim.2008.03.011
Tey SK, Khanna R (2012) Autophagy mediates transporter associated with antigen processing-independent presentation of viral epitopes through MHC class I pathway. Blood 120(5):994–1004. doi:10.1182/blood-2012-01-402404
Thomas AG, O'Driscoll CM, Bressler J, Kaufmann W, Rojas CJ, Slusher BS (2014) Small molecule glutaminase inhibitors block glutamate release from stimulated microglia. Biochem Biophys Res Commun 443(1):32–36. doi:10.1016/j.bbrc.2013.11.043
Thompson MR, Sharma S, Atianand M, Jensen SB, Carpenter S, Knipe DM, Fitzgerald KA, Kurt-Jones EA (2014) Interferon Gamma Inducible protein (IFI)16 transcriptionally regulates type I interferons and other interferon stimulated genes and controls the interferon response to both DNA and RNA viruses. J Biol Chem 289(34):23568–23581. doi:10.1074/jbc.M114.554147
Thongtan T, Cheepsunthorn P, Chaiworakul V, Rattanarungsan C, Wikan N, Smith DR (2010) Highly permissive infection of microglial cells by Japanese encephalitis virus: a possible role as a viral reservoir. Microbes Infect 12(1):37–45. doi:10.1016/j.micinf.2009.09.013
Thounaojam MC, Kaushik DK, Kundu K, Basu A (2014a) MicroRNA-29b modulates Japanese encephalitis virus-induced microglia activation by targeting tumor necrosis factor alpha-induced protein 3. J Neurochem 129(1):143–154. doi:10.1111/jnc.12609
Thounaojam MC, Kundu K, Kaushik DK, Swaroop S, Mahadevan A, Shankar SK, Basu A (2014b) MicroRNA 155 regulates Japanese encephalitis virus-induced inflammatory response by targeting Src homology 2-containing inositol phosphatase 1. J Virol 88(9):4798–4810. doi:10.1128/jvi.02979-13
Tian Y, Kuo CF, Chen WL, Ou JH (2012) Enhancement of hepatitis B virus replication by androgen and its receptor in mice. J Virol 86(4):1904–1910. doi:10.1128/JVI.06707-11
Tobon-Velasco JC, Cuevas E, Torres-Ramos MA (2014) Receptor for AGEs (RAGE) as mediator of NF-kB pathway activation in neuroinflammation and oxidative stress. CNS Neurol Disord Drug Targets 13(9):1615–1626
Tracey KJ (2007) Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Investig 117(2):289–296
Tran Thi Duc T, Desmecht D, Cornet A (2013) Functional characterization of new allelic polymorphisms identified in the promoter region of the human MxA gene. Int J Immunogenet 40(4):316–319. doi:10.1111/j.1744-313X.2012.01153.x
Troseid M, Lind A, Nowak P, Barqasho B, Heger B, Lygren I, Pedersen KK, Kanda T, Funaoka H, Damas JK, Kvale D (2013) Circulating levels of HMGB1 are correlated strongly with MD2 in HIV-infection: possible implication for TLR4-signalling and chronic immune activation. Innate Immun 19(3):290–297. doi:10.1177/1753425912461042
Trotta T, Porro C, Calvello R, Panaro MA (2014) Biological role of Toll-like receptor-4 in the brain. J Neuroimmunol 268(1–2):1–12. doi:10.1016/j.jneuroim.2014.01.014
Trottier MD Jr, Palian BM, Reiss CS (2005) VSV replication in neurons is inhibited by type I IFN at multiple stages of infection. Virology 333(2):215–225. doi:10.1016/j.virol.2005.01.009, S0042-6822(05)00016-4 [pii]
Trottier MD, Lyles DS, Reiss CS (2007) Peripheral, but not central nervous system, type I interferon expression in mice in response to intranasal vesicular stomatitis virus infection. J Neurovirol 13(5):433–445. doi:10.1080/13550280701460565
Tung WH, Lee IT, Hsieh HL, Yang CM (2010) EV71 induces COX-2 expression via c-Src/PDGFR/PI3K/Akt/p42/p44 MAPK/AP-1 and NF-kappaB in rat brain astrocytes. J Cell Physiol 224(2):376–386. doi:10.1002/jcp.22133
Ueno N, Takegoshi Y, Kamei D, Kudo I, Murakami M (2005) Coupling between cyclooxygenases and terminal prostanoid synthases. Biochem Biophys Res Commun 338(1):70–76
Uhrin P, Perkmann T, Binder B, Schabbauer G (2013) ISG12 is a critical modulator of innate immune responses in murine models of sepsis. Immunobiology 218(9):1207–1216. doi:10.1016/j.imbio.2013.04.009
Ullrich O, Merker K, Timm J, Tauber S (2007) Immune control by endocannabinoids—new mechanisms of neuroprotection? J Neuroimmunol 184(1–2):127–135
Ushikubi F, Sugimoto Y, Ichikawa A, Narumiya S (2000) Roles of prostanoids revealed from studies using mice lacking specific prostanoid receptors. Jpn J Pharmacol 83(4):279–285
Valimaa H, Tenovuo J, Waris M, Hukkanen V (2009) Human lactoferrin but not lysozyme neutralizes HSV-1 and inhibits HSV-1 replication and cell-to-cell spread. Virol J 6:53. doi:10.1186/1743-422x-6-53
van Doorn R, Lopes Pinheiro MA, Kooij G, Lakeman K, van het Hof B, van der Pol SM, Geerts D, van Horssen J, van der Valk P, van der Kam E, Ronken E, Reijerkerk A, de Vries HE (2012) Sphingosine 1-phosphate receptor 5 mediates the immune quiescence of the human brain endothelial barrier. J Neuroinflammation 9:133. doi:10.1186/1742-2094-9-133
Veillard NR, Braunersreuther V, Arnaud C, Burger F, Pelli G, Steffens S, Mach F (2006) Simvastatin modulates chemokine and chemokine receptor expression by geranylgeranyl isoprenoid pathway in human endothelial cells and macrophages. Atherosclerosis 188(1):51–58
Veloso S, Escote X, Ceperuelo-Mallafre V, Lopez-Dupla M, Peraire J, Vilades C, Domingo P, Castro A, Olona M, Sirvent JJ, Leal M, Vendrell J, Richart C, Vidal F (2012) Leptin and adiponectin, but not IL18, are related with insulin resistance in treated HIV-1-infected patients with lipodystrophy. Cytokine 58(2):253–260. doi:10.1016/j.cyto.2012.01.013
Ventoso I, Sanz MA, Molina S, Berlanga JJ, Carrasco L, Esteban M (2006) Translational resistance of late alphavirus mRNA to eIF2alpha phosphorylation: a strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev 20(1):87–100
Vigorito M, Cao J, Li MD, Chang SL (2013) Acquisition and long-term retention of spatial learning in the human immunodeficiency virus-1 transgenic rat: effects of repeated nicotine treatment. J Neurovirol 19(2):157–165. doi:10.1007/s13365-013-0154-1
Votteler J, Sundquist Wesley I (2013) Virus budding and the ESCRT pathway. Cell Host Microbe 14(3):232–241, http://dx.doi.org/10.1016/j.chom.2013.08.012
Waarts BL, Aneke OJ, Smit JM, Kimata K, Bittman R, Meijer DK, Wilschut J (2005) Antiviral activity of human lactoferrin: inhibition of alphavirus interaction with heparan sulfate. Virology 333(2):284–292
Wada K, Arita M, Nakajima A, Katayama K, Kudo C, Kamisaki Y, Serhan CN (2006) Leukotriene B4 and lipoxin A4 are regulatory signals for neural stem cell proliferation and differentiation. FASEB J 20(11):1785–1792
Wakimoto H, Johnson PR, Knipe DM, Chiocca EA (2003) Effects of innate immunity on herpes simplex virus and its ability to kill tumor cells. Gene Ther 10(11):983–990
Walsh JG, Muruve DA, Power C (2014) Inflammasomes in the CNS. Nat Rev Neurosci 15(2):84–97. doi:10.1038/nrn3638
Walters CE, Pryce G, Hankey DJ, Sebti SM, Hamilton AD, Baker D, Greenwood J, Adamson P (2002) Inhibition of Rho GTPases with protein prenyltransferase inhibitors prevents leukocyte recruitment to the central nervous system and attenuates clinical signs of disease in an animal model of multiple sclerosis. J Immunol 168(8):4087–4094
Wang G (2013) Database-guided discovery of potent peptides to combat HIV-1 or superbugs. Pharmaceuticals 6(6):728–758. doi:10.3390/ph6060728
Wang G (2014) Human antimicrobial peptides and proteins. Pharmaceuticals 7(5):545–594. doi:10.3390/ph7050545
Wang H, Ward MF, Fan XG, Sama AE, Li W (2006) Potential role of high mobility group box 1 in viral infectious diseases. Viral Immunol 19(1):3–9
Wang L, Cao Y, Tang Q, Liang G (2013) Role of the blood-brain barrier in rabies virus infection and protection. Protein Cell 4(12):901–903. doi:10.1007/s13238-013-3918-8
Wang Y, Marling SJ, Beaver EF, Severson KS, Deluca HF (2015) UV light selectively inhibits spinal cord inflammation and demyelination in experimental autoimmune encephalomyelitis. Arch Biochem Biophys 567:75–82. doi:10.1016/j.abb.2014.12.017
Ward SV, George CX, Welch MJ, Liou LY, Hahm B, Lewicki H, de la Torre JC, Samuel CE, Oldstone MB (2011) RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc Natl Acad Sci U S A 108(1):331–336. doi:10.1073/pnas.1017241108
Watkins LR, Maier SF (2000) The pain of being sick: implications of immune-to-brain communication for understanding pain. Annu Rev Psychol 51:29–57
Weber E, Finsterbusch K, Lindquist R, Nair S, Lienenklaus S, Gekara NO, Janik D, Weiss S, Kalinke U, Overby AK, Kroger A (2014a) Type I interferon protects mice from fatal neurotropic infection with Langat virus by systemic and local anti-viral response. J Virol 88(21):12202–12212. doi:10.1128/jvi.01215-14
Weber MS, Prod'homme T, Youssef S, Dunn SE, Steinman L, Zamvil SS (2014b) Neither T-helper type 2 nor Foxp3+ regulatory T cells are necessary for therapeutic benefit of atorvastatin in treatment of central nervous system autoimmunity. J Neuroinflammation 11:29. doi:10.1186/1742-2094-11-29
Weinberg JB, Jensen DR, Gralinski LE, Lake AR, Stempfle GS, Spindler KR (2007) Contributions of E1A to mouse adenovirus type 1 pathogenesis following intranasal inoculation. Virology 357(1):54–67
Weiss JM, Subleski JJ, Wigginton JM, Wiltrout RH (2007) Immunotherapy of cancer by IL-12-based cytokine combinations. Expert Opin Biol Ther 7(11):1705–1721
Welsh CJ, Bustamante L, Nayak M, Welsh TH, Dean DD, Meagher MW (2004) The effects of restraint stress on the neuropathogenesis of Theiler’s virus infection II: NK cell function and cytokine levels in acute disease. Brain Behav Immun 18(2):166–174
Wensman JJ, Ilback C, Hjertstrom E, Blomstrom AL, Gustavsson MH, Jaderlund KH, Strom-Holst B, Belak S, Berg AL, Berg M (2011) Expression of interferon gamma in the brain of cats with natural Borna disease virus infection. Vet Immunol Immunopathol 141(1–2):162–167. doi:10.1016/j.vetimm.2011.02.014
Whitacre CC, Reingold SC, O’Looney PA (1999) A gender gap in autoimmunity. Science 283(5406):1277–1278
White DL, Tavakoli-Tabasi S, Kuzniarek J, Pascua R, Ramsey DJ, El-Serag HB (2012) Higher serum testosterone is associated with increased risk of advanced hepatitis C-related liver disease in males. Hepatology 55(3):759–768. doi:10.1002/hep.24618
Wiens ME, Wilson SS, Lucero CM, Smith JG (2014) Defensins and viral infection: dispelling common misconceptions. PLoS Pathog 10(7):e1004186. doi:10.1371/journal.ppat.1004186
Wierucka-Rybak M, Bojanowska E (2014) Bacteria, viruses, and hypothalamic inflammation: potential new players in obesity. Postepy Hig Med Dosw (Online) 68:271–279
Wilkerson BA, Argraves KM (2014) The role of sphingosine-1-phosphate in endothelial barrier function. Biochim Biophys Acta 1841(10):1403–1412. doi:10.1016/j.bbalip.2014.06.012
Williams KC, Hickey WF (2002) Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. Annu Rev Neurosci 25:537–562
Williams K, Alvarez X, Lackner AA (2001) Central nervous system perivascular cells are immunoregulatory cells that connect the CNS with the peripheral immune system. Glia 36(2):156–164
Williams WM, Castellani RJ, Weinberg A, Perry G, Smith MA (2012) Do beta-defensins and other antimicrobial peptides play a role in neuroimmune function and neurodegeneration? Scientific World Journal 2012:905785. doi:10.1100/2012/905785
Williams DW, Veenstra M, Gaskill PJ, Morgello S, Calderon TM, Berman JW (2014a) Monocytes mediate HIV neuropathogenesis: mechanisms that contribute to HIV associated neurocognitive disorders. Curr HIV Res 12(2):85–96
Williams JL, Holman DW, Klein RS (2014b) Chemokines in the balance: maintenance of homeostasis and protection at CNS barriers. Front Cell Neurosci 8:154. doi:10.3389/fncel.2014.00154
Wilson ME, Dimayuga FO, Reed JL, Curry TE, Anderson CF, Nath A, Bruce-Keller AJ (2006) Immune modulation by estrogens: role in CNS HIV-1 infection. Endocrine 29(2):289–297. doi:10.1385/endo:29:2:289
Wilson SS, Wiens ME, Smith JG (2013) Antiviral mechanisms of human defensins. J Mol Biol 425(24):4965–4980. doi:10.1016/j.jmb.2013.09.038
Wiltzer L, Okada K, Yamaoka S, Larrous F, Kuusisto HV, Sugiyama M, Blondel D, Bourhy H, Jans DA, Ito N, Moseley GW (2014) Interaction of rabies virus P-protein with STAT proteins is critical to lethal rabies disease. J Infect Dis 209(11):1744–1753. doi:10.1093/infdis/jit829
Wobke TK, Sorg BL, Steinhilber D (2014) Vitamin D in inflammatory diseases. Front Physiol 5:244. doi:10.3389/fphys.2014.00244
Wollish AC, Ferris MT, Blevins LK, Loo YM, Gale M Jr, Heise MT (2013) An attenuating mutation in a neurovirulent Sindbis virus strain interacts with the IPS-1 signaling pathway in vivo. Virology 435(2):269–280. doi:10.1016/j.virol.2012.09.008
Wu Z, Qin J, Pu L (2012) Omega-3 fatty acid improves the clinical outcome of hepatectomized patients with hepatitis B virus (HBV)-associated hepatocellular carcinoma. J Biomed Res 26(6):395–399. doi:10.7555/jbr.26.20120058
Wu B, Peisley A, Richards C, Yao H, Zeng X, Lin C, Chu F, Walz T, Hur S (2013) Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 152(1–2):276–289. doi:10.1016/j.cell.2012.11.048
Xiao S, Li D, Zhu HQ, Song MG, Pan XR, Jia PM, Peng LL, Dou AX, Chen GQ, Chen SJ, Chen Z, Tong JH (2006) RIG-G as a key mediator of the antiproliferative activity of interferon-related pathways through enhancing p21 and p27 proteins. Proc Natl Acad Sci U S A 103(44):16448–16453
Yang J, Dennison NN, Reiss CS (2007) PIN: a novel protein involved in IFN-gamma accumulation of NOS-1 in neurons. DNA Cell Biol 29(1):9–17
Yang J, Dennison NN, Reiss CS (2008) PIN: a novel protein involved in IFN-gamma accumulation of NOS-1 in neurons. DNA Cell Biol 27(1):9–17. doi:10.1089/dna.2007.0673
Yu L, Yan K, Liu P, Li N, Liu Z, Zhu W, Chen Y, Han D (2014) Pattern recognition receptor-initiated innate antiviral response in mouse adipose cells. Immunol Cell Biol 92(2):105–115. doi:10.1038/icb.2013.66
Zaritsky LA, Gama L, Clements JE (2012) Canonical type I IFN signaling in simian immunodeficiency virus-infected macrophages is disrupted by astrocyte-secreted CCL2. J Immunol 188(8):3876–3885. doi:10.4049/jimmunol.1103024
Zenner HL, Mauricio R, Banting G, Crump CM (2013) Herpes simplex virus 1 counteracts tetherin restriction via its virion host shutoff activity. J Virol 87(24):13115–13123. doi:10.1128/jvi.02167-13
Zhang W, Du J, Evans SL, Yu Y, Yu XF (2012) T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature 481(7381):376–379. doi:10.1038/nature10718
Zhang AJ, To KK, Li C, Lau CC, Poon VK, Chan CC, Zheng BJ, Hung IF, Lam KS, Xu A, Yuen KY (2013a) Leptin mediates the pathogenesis of severe 2009 pandemic influenza A(H1N1) infection associated with cytokine dysregulation in mice with diet-induced obesity. J Infect Dis 207(8):1270–1280. doi:10.1093/infdis/jit031
Zhang B, Liu X, Chen W, Chen L (2013b) IFIT5 potentiates anti-viral response through enhancing innate immune signaling pathways. Acta Biochim Biophys Sin 45(10):867–874. doi:10.1093/abbs/gmt088
Zhang L, Mo J, Swanson KV, Wen H, Petrucelli A, Gregory SM, Zhang Z, Schneider M, Jiang Y, Fitzgerald KA, Ouyang S, Liu ZJ, Damania B, Shu HB, Duncan JA, Ting JP (2014) NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity 40(3):329–341. doi:10.1016/j.immuni.2014.01.010
Zhao LB, Li YJ, Zhang XD, Xie P (2006) [Progress and investigation of neuronal plasticity interfered by Borna disease virus infection]. Wei Sheng Wu Xue Bao 46(4):676–679
Zhao P, Zhao L, Zhang T, Qi Y, Wang T, Liu K, Wang H, Feng H, Jin H, Qin C, Yang S, Xia X (2011) Innate immune response gene expression profiles in central nervous system of mice infected with rabies virus. Comp Immunol Microbiol Infect Dis 34(6):503–512. doi:10.1016/j.cimid.2011.09.003
Zheng Y, Zhang W, Ye Q, Zhou Y, Xiong W, He W, Deng M, Zhou M, Guo X, Chen P, Fan S, Liu X, Wang Z, Li X, Ma J, Li G (2012) Inhibition of Epstein-Barr virus infection by lactoferrin. J Innate Immun 4(4):387–398. doi:10.1159/000336178
Zhou J, Stohlman SA, Hinton DR, Marten NW (2003) Neutrophils promote mononuclear cell infiltration during viral-induced encephalitis. J Immunol 170(6):3331–3336
Zhou S, Halle A, Kurt-Jones EA, Cerny AM, Porpiglia E, Rogers M, Golenbock DT, Finberg RW (2008) Lymphocytic choriomeningitis virus (LCMV) infection of CNS glial cells results in TLR2-MyD88/Mal-dependent inflammatory responses. J Neuroimmunol 194(1–2):70–82
Zhu H, Zheng C, Xing J, Wang S, Li S, Lin R, Mossman KL (2011) Varicella-zoster virus immediate-early protein ORF61 abrogates the IRF3-mediated innate immune response through degradation of activated IRF3. J Virol 85(21):11079–11089. doi:10.1128/jvi.05098-11
Zins SR, Nelson CD, Maginnis MS, Banerjee R, O'Hara BA, Atwood WJ (2014) The human alpha defensin HD5 neutralizes JC polyomavirus infection by reducing endoplasmic reticulum traffic and stabilizing the viral capsid. J Virol 88(2):948–960. doi:10.1128/jvi.02766-13
Zolini GP, Lima GK, Lucinda N, Silva MA, Dias MF, Pessoa NL, Coura BP, Cartelle CT, Arantes RM, Kroon EG, Campos MA (2014) Defense against HSV-1 in a murine model is mediated by iNOS and orchestrated by the activation of TLR2 and TLR9 in trigeminal ganglia. J Neuroinflammation 11:20. doi:10.1186/1742-2094-11-20
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Reiss, C.S. (2016). Innate Immunity in Viral Encephalitis. In: Reiss, C. (eds) Neurotropic Viral Infections. Springer, Cham. https://doi.org/10.1007/978-3-319-33189-8_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-33189-8_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-33188-1
Online ISBN: 978-3-319-33189-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)