
Abstract
Transcriptional enhancers play a key role in the initiation and maintenance of gene expression programmes, particularly in metazoa. How these elements control their target genes in the right place and time is one of the most pertinent questions in functional genomics, with wide implications for most areas of biology. Here, we synthesise classic and recent evidence on the regulatory logic of enhancers, including the principles of enhancer organisation, factors that facilitate and delimit enhancer–promoter communication, and the joint effects of multiple enhancers. We show how modern approaches building on classic insights have begun to unravel the complexity of enhancer–promoter relationships, paving the way towards a quantitative understanding of gene control.
Similar content being viewed by others

Introduction
In complex organisms, the vast majority of genes are not controlled by promoters alone, but additionally receive input from one or more non-coding DNA cis-regulatory elements, the best-characterised of which are enhancers. The first transcriptional enhancers were identified about 40 years ago, and their critical role in development has been clear for at least two decades. Advances in recent years have led to the identification of millions of enhancers active in an ever-expanding array of cell types. Meanwhile, population genetics studies have revealed the enrichment of these elements for genetic variants associated with common diseases. Most recently, targeted perturbation and imaging techniques have taken functional analyses of enhancers to a new level of scale and resolution. As the quest for a mechanistic understanding of enhancers continues, the function of these elements emerges as a complex phenomenon that integrates multiple levels of nuclear organisation, from primary DNA sequence and sequence-specific transcription factors to higher-order chromatin architecture through chromatin remodelling complexes, chromosomal loops and potentially phase-separated condensates. In this review, we synthesise classic and recent evidence on the organisation and function of enhancers, focusing in particular on the principles governing their communication with target gene promoters.
Organisation and function of enhancers in cis
Classic definition of enhancers
Enhancers were first described around 20 years after the discovery of the gene promoter [1,2,3,4,5,6,7]. In 1980, the first evidence for enhancers arose when short DNA sequences were discovered within the simian virus 40 (SV40) [2] and the sea urchin genome [3] that were remote from a gene promoter, yet seemed to stimulate gene expression by an unknown mechanism. Subsequent landmark experiments by the groups of Walter Schaffner [4] and Pierre Chambon [5] confirmed that a 72-bp repeat sequence element in SV40 [8, 9] was an “enhancing” sequence, capable of vastly upregulating gene expression from a plasmid upon transfection into mammalian cells. Moreover, this enhancer could activate rabbit or human β-globin genes from varying distances from their promoters, regardless of its orientation [4, 5]. At around the same time, similar observations were made for an unrelated enhancer sequence within the polyoma virus [10]. We now know that the ability to function remotely and independently of orientation are classic features of enhancers. The SV40 discovery was useful for identifying further enhancers via the “enhancer trap” method: an SV40 genome lacking its known enhancer was combined with fragments of other viruses and transfected into mammalian cells. DNA combinations with the ability to replicate must have gained an enhancer from the new fragment [11, 12].
Enhancers were soon discovered in the genomes of other model organisms in a range of complexities from yeast [13] to Drosophila [14] and mouse [15,16,17]; enhancer-like sequences were also detected in bacteria [18,19,20,21,22]. In Drosophila, newly discovered enhancers orchestrated the regulation of developmental genes that were crucial for the correct formation of the embryo [23, 24]. It soon became clear that enhancers also played vital roles in humans, with non-coding enhancer deletions starting to be linked to severe disease phenotypes such as beta thalassaemia and polydactyly [25,26,27,28].
Enhancers as transcription factor recruitment units
At the DNA level, enhancers represent clusters of binding sites for sequence-specific transcription factors (TFs) (Fig. 1A) (reviewed in [29, 30]). However, the ability of enhancers to recruit their cognate TFs may be constrained by inaccessible (“closed”) chromatin conformation, whereby enhancer regions are tightly packaged in nucleosomes. A subset of TFs known as pioneer factors are capable of overcoming this constraint through chromatin remodelling (reviewed in [31, 32]). Pioneer factors play particularly important roles in priming enhancers in order to program gene expression patterns during early development. In Drosophila, the Zinc Finger Early Drosophila Activator (Zelda) is critical for lowering the nucleosome barrier at enhancers for genes driving zygotic gene activation [33,34,35]. In mice and humans, the pioneer factors OCT4 and SOX2 form a core part of the regulatory network controlling stem cell pluripotency [36]. The mechanisms of pioneer factor action are still being established [37], but at least some of them (such as FOXA, OCT4 and SOX2/SOX11) have been shown to initiate chromatin opening by direct displacement of histones [38,39,40,41], whilst many others recruit chromatin remodelers [42,43,44,45,46,47,48,49,50]. Often, pioneer factors must recruit additional non-pioneer sequence-specific TFs for efficient enhancer activation [30, 31]. For instance, Zelda promotes the formation of local TF hubs involving important transcription factors required for developmental patterning, such as Bicoid and Dorsal [51,52,53,54]. Synthetic enhancers recruiting multiple heterotypic TFs showed stronger transcriptional effects compared with those recruiting high amounts of a single TF, providing a functional rationale for combinatorial TF recruitment to natural enhancers [55].

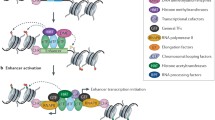
Organisation of active enhancers in cis. A Enhancers become activated by pioneer factors that increase chromatin accessibility, perhaps through histone displacement, allowing for binding of cell-specific TFs and cofactors. Cofactors such as P300 and MLL perform chromatin remodelling via acetylation or methylation of histone tails. TFs and cofactors recruit BRD4/Mediator and further transcriptional machinery to begin active transcription at the enhancer and the distal gene promoter. B Models of enhancer grammar include the rigid Enhanceosome model [88], in which TFs bind in a particular order and spacing; the Billboard model, in which TFs bind in a very flexible arrangement [90], and the TF Collective model, in which a full set of particular TFs is required for enhancer activation, but not all TFs must bind directly to the DNA and instead are recruited via protein–protein interactions [91]
Mechanistically, a key function of sequence-specific TFs is the recruitment of “workhorse” cofactors that facilitate enzymatic chromatin remodelling, histone modification and act as scaffolds for recruitment of additional factors. These cofactors include, but are not limited to, SWI/SNF and FACT chromatin remodelling complexes, P300 and CBP chromatin activators, Bromodomain-containing (BRD) proteins and the Mediator complex [56, 57]. The SWI/SNF complex is involved in regulating occupancy and spacing of nucleosomes at promoters and enhancers [58], while the most established role of FACT is in facilitating transcriptional elongation [57]. SWI/SNF recruits the chromatin activators P300 and CBP, which mediate the acetylation of histone H3 lysine 27 through their intrinsic histone acetyltransferase (HAT) activity [59,60,61,62]. This P300/CBP-induced acetylation has recently been implicated in the release of “paused” RNAP at both enhancers and promoters to promote active transcription [63]. P300 and CBP also serve as scaffolds that connect RNA polymerase II (RNAP) and RNAP-associated “general transcription factors” to the chromatin [64, 65]. At active enhancers, the BRD4 protein co-localises with the Mediator complex, which in turn assembles the pre-initiation complex (PIC) and RNAP to initiate transcription [66,67,68].
Recent experiments degrading Mediator and BRD4 have shown that these proteins are critical for gene expression [69]. In particular, Mediator and BRD4 co-localise at very high levels at “super-enhancers” (SEs): long stretches of sequences with gene regulatory activity that generally drive greater levels of gene expression than regular enhancers [69,70,71,72]. We will return to Mediator and BRD4 later in the review to discuss their possible roles in enhancer–promoter communication.
The majority of active enhancers are themselves transcribed, producing enhancer RNAs (eRNAs) in a bi-directional manner [73,74,75,76,77]. These eRNAs are rapidly degraded and might be merely by-products of RNAP recruitment to enhancers [78]. However, studies that knock down selected eRNA have found reduced expression of the corresponding gene targets, suggesting functionality [79,80,81]. The exact functions of eRNAs are as yet unclear but could include, among others, maintaining enhancer chromatin accessibility [82, 83] and interacting with TFs or cofactors such as BRD4 [84, 85] (reviewed in [86]).
The chain of molecular events following the binding of a pioneer TF to its target sequence (in this case, a promoter-proximal region) has been elegantly documented in a fine-grained time-course experiment following oestrogen receptor activation in a cancer cell line [87]. This study detected multiple “waves” of a progressive recruitment of at least 46 sequence-specific TFs and cofactors, including chromatin remodelling complexes and histone acetyl-transferases, eventually resulting in a derepression of an oestrogen-response gene pS2 [87].
Flexibility of enhancer organisation
Given that multiple TF binding sites are the essential building blocks of enhancers, the key question is whether their positioning within enhancers needs to follow any kind of “grammar”, with respect to their particular order and orientation, for an enhancer to function properly (Fig. 1B). The various proposed models for enhancer grammar range from very rigid to flexible, largely dependent on the species, cell types and loci in which they were formulated (reviewed in [29]). At the rigid end, the “enhanceosome” model suggests that the TF binding sites must remain in the same order and orientation for the enhancer to work. This model was based on careful dissection of the virus-responsive interferon beta enhancer, which has eight TF binding sites [88]. The model dictates a set organisation of motifs and spacers consistent with the formation of a heteromultimer of cooperatively bound TFs, each of which directly binds the DNA. Virtually every nucleotide of the interferon beta enhancer is evolutionarily conserved; this is likely because the eight TFs cooperate to form a composite surface that recognises the entire sequence of the enhancer as one effective binding site [88, 89]. In contrast, later studies of Drosophila developmental enhancers found evidence for much less rigid motif organisation [90, 91]. At the most flexible end, the loosely organised “billboard” model developed in Drosophila implies no cooperativity between TFs for DNA binding and, therefore, the location and spacing between binding sites is not crucial for conserved enhancer function [90, 92]. Enhancers consistent with the billboard model are likely also widespread in vertebrate genomes [55, 93]. An intermediate model for enhancer grammar known as the “TF collective” proposes that a set of the same TFs binds to multiple enhancers but an individual enhancer does not necessarily have the full set of TF binding sites [30, 91]. TF recruitment is therefore facilitated by a combination of DNA binding affinity and protein–protein interactions between TFs. The TF collective model was originally formulated in Drosophila, where a set of five TFs bound multitudes of enhancers required for heart development that showed no detectable similarity in their sequence organisation, beyond the presence of binding motifs for some of these TFs [91]. Later studies provided further examples of “TF collective” enhancers. For example, dissection of an enhancer controlling the specification of Drosophila leg precursor cells showed that it was robust against motif disruption, but relied on DNA–protein and protein–protein interactions between a particular set of TFs [94]. Similarly, TFs governing serotonergic neuron differentiation showed synergy despite flexible sequence organisation of their cognate enhancers [95]. Flexibility of enhancer organisation with respect to binding site order and orientation was also observed in a massively parallel synthetic enhancer experiment [55]. Biophysical modelling and machine learning approaches have provided more evidence for flexible enhancer grammar [96,97,98]. For example, modelling the effects of targeting engineered transcription activators and repressors to enhancers suggested that TFs can contribute additively to enhancer function, regardless of their identity [96]. Additionally, a deep learning algorithm applied to binding of pluripotency TFs at base resolution in mouse embryonic stem cells (ESCs) suggested a “soft motif syntax” whereby TFs help each other to bind directly to the enhancer with a lenient distance-dependency between binding sites [98].
The prevalence of flexible enhancer organisation is also supported by rapid evolutionary turnover of enhancers, which exceeds that of proximal promoter regions [29, 99, 100]. In terms of their sequence, there are no known enhancers that are completely conserved at the base level across the animal kingdom [101]. Instead, enhancer conservation is typically “modular”, commonly preserving the sets of binding sites for the required TFs, but often not their exact sequence and orientation [101,102,103,104,105,106,107,108,109]. Consistent with this, homologous enhancers in a pair of distantly related species can retain their function in the non-host organism despite differences in DNA sequence [101, 110, 111]. Furthermore, even enhancers with extremely high evolutionary conservation may not require the exact preservation of their sequence for their correct function, as was recently shown using CRISPR-mediated mutagenesis [112].
While it is clear that enhancers typically favour the more flexible models of sequence organisation, the degree of necessary cooperation between TFs is less obvious, particularly since TF binding to DNA appears to be very transient [113,114,115,116,117]. Recent technological advances such as single molecule footprinting (SMF) have begun to unpick how TFs work with each other at enhancers [118, 119]. First developed in Drosophila, SMF can detect the binding of multiple TFs on a single DNA molecule [116]. SMF showed high TF cooperativity at active enhancers and identified cases where TFs bind independently, sequentially or simultaneously [118]. SMF has also recently been adapted to mammalian cells, detecting widespread co-occupancy of cooperative TFs at enhancers [119]. Notably, at dimeric motif sites, TFs tended to bind initially as monomers with subsequent dimerisation presumably stabilising the DNA–protein complex [119]. Interestingly, at non-dimeric sites, physical interactions between co-bound TFs were not necessary [119], consistent with evidence from Drosophila where co-occupied DNA-bound TFs were often spaced quite widely apart (> 50 bp) [118]. Jointly, these results point to synergistic mechanisms between different TFs that do not require direct protein–protein interaction.
How does the flexibility in enhancer organisation result in the robust control of gene expression? In part, this is achieved through tight regulation of the expression of the TFs themselves. One of the best-characterised examples of this is the development of gene expression “stripes” in Drosophila embryonic development, which is driven by the expression of pair-rule genes such as even-skipped (eve) [120]. The stripe 2 enhancer controls eve expression in early embryogenesis via binding of tissue-specific TFs [121,122,123]. This enhancer contains clusters of TF binding sites, with the sites for transcriptional repressors overlapping those for the activators. Activating TFs Bicoid and Hunchback are expressed only in the anterior section of the embryo, while the repressors Giant and Kruppel are expressed in anterior and posterior regions on either side of stripe 2, such that the stripe 2 enhancer is only activated within a 2–3 cell-wide section of the embryo [108, 121, 124]. Another means by which flexible enhancer grammar ensures robust gene expression control is through “suboptimal” TF binding motifs, which reinforce the requirement for TF cooperativity and create opportunities for fine-tuning enhancer activity levels [125,126,127,128]. Consistent with this, “strengthening” TF binding sites at developmental enhancers results in aberrant gene expression patterns [125]. Finally, gene control is maintained by the multiplicity of enhancer inputs to the same gene and its regulation by the broader chromatin context, as will be reviewed below.
The chromatin states of enhancers
Active enhancers reside in open chromatin and occupy regions away from gene TSSs. However, open chromatin regions are not limited to active enhancers, and also harbour other regions including active gene promoters, regulatory elements in a ‘poised’ chromatin configuration (discussed below), as well as some structural regions such as tissue-invariant binding sites for the architectural protein CCCTC-binding factor (CTCF) [129,130,131,132]. Post-translational modifications of histone tails provide a more specific readout of enhancer activity. Currently mono-methylation of histone H3 lysine 4 (H3K4me1), associated with the binding of Trithorax/MLL complexes, is taken to be the hallmark of both ‘primed’ and active enhancers, whilst active enhancers additionally tend to associate with the acetylation of histone H3 lysine 27 (H3K27ac) [133,134,135,136,137]. In contrast, the trimethylation of histone H3 lysine 4 (H3K4me3) is typically considered a promoter-associated mark and, in regions proximal to a known TSS, has been used to distinguish the enhancer from a promoter [131]. In addition to the ‘primed’ and active states, enhancers can also reside in a ‘poised’ state exhibiting both H3K4me1 and H3 lysine 27 trimethylation (H3K27me3) that is associated with Polycomb repressive complexes [131, 135, 136, 138]. Finally, some enhancers are marked with H3K27me3 alone and are considered to be “Polycomb-repressed” [139,140,141]. Many developmental enhancers are found in the poised or Polycomb-repressed state in early development [139, 142, 143], and the functional role of these states remains an area of active research. One possibility is that poised enhancers may be ‘held in check’ for rapid activation [144]. However, enhancer association with Polycomb repressors may also merely serve to suppress enhancer activity [145] or potentially convert these regions to active ‘silencers’ [139].
Enhancer detection in high throughput
The epigenetic hallmarks of enhancers, including histone modifications and the binding of cofactors such as P300, have enabled their detection at a global scale. Following pioneering studies in a small number of cell types [146,147,148], large-scale consortia such as ENCODE, Roadmap Epigenomics and BLUEPRINT [131, 149,150,151] have profiled the epigenetic hallmarks of DNA regulatory elements across cell types in human and mouse cells, as well as in multiple Drosophila and Caenorhabditis species (through modENCODE) [152, 153]. While the initial efforts focused on cell lines, the analyses have eventually been expanded to multitudes of primary samples, including blood cells and solid tissues. The most recent (phase 3) release of the ENCODE project has compiled an updated registry of predicted cis regulatory elements in more than 500 cell or tissue types in human and mouse [131]. Finally, the EpiMap project has combined direct epigenetic mapping with imputation to generate a compendium of 833 reference epigenomes across 18 epigenetic assays [154]. The recently developed CUT&RUN [155] and CUT&TAG [156] methods that require fewer cells and less sequencing compared with ChIP are enabling enhancer identification in an ever-expanding array of cell types and tissues, as well as in single cells [157, 158]. Complementary to this, bespoke methods have been developed to partition the genome into distinct regulatory ‘states’ by integrating information across multiple histone marks accounting for spatial dependency [134, 159,160,161] to provide an easily interpretable and visualisable readout of enhancer activity in a given tissue.
While enhancer detection based on chromatin profiling has become standard, this approach may falsely detect regions without appreciable regulatory activity, as well as miss some functional elements [146, 162]. For example, enhancers devoid of the classic H3K27ac mark have recently been described [163]; consistent with this, it was shown that this mark is not required for enhancer function, at least in mouse ESCs [164]. These limitations can be partially mitigated by high-throughput techniques that obtain a readout of enhancer activity as opposed to the markers of their chromatin state. For example, methods such as Cap Analysis of Gene Expression (CAGE) can be used to detect eRNA transcripts generated from enhancers, including in single cells [74, 165,166,167]. In addition, massively parallel reporter assays such as MPFD [168] and STARR-seq [169] make it possible to assess huge libraries of enhancer sequences for transcriptional regulatory activity in vitro [168,169,170,171,172,173,174,175]. Finally, the advent of CRISPR-based techniques streamlines the perturbation analysis of enhancers in vivo. Most relevantly, using the fusions of the ‘dead’ (endonuclease-deficient) Cas9 protein (dCas9) with either transcriptional repressors such as KRAB, or activators such as P300 or tandems of herpesvirus VP16 transactivation domain, enables a guide RNA (gRNA)-targeted inhibition (CRISPRi) or activation (CRISPRa) of theoretically any regulatory region in the genome [176,177,178]. A recent modification of this approach (known as enCRISPRi/a) combines multiple effector proteins with dCas9 to modulate enhancer (rather than promoter) activity more specifically [179]. To enable CRISPR-mediated enhancer targeting in high throughput, populations of cells are transduced with pooled gRNA libraries, followed by single-cell readouts of enhancer or transcriptional activity from scRNA-seq [180, 181], scATAC-seq [182] and flow cytometry-based RNA FISH [183, 184] to obtain tiled maps of functional regulatory regions for multiple genomic loci in several cell types. In addition to targeting chromatin modifiers using dCas9, knockout screens using wild type Cas9 can also be useful for identifying regulatory elements in high throughput [185,186,187]. Most of these studies are performed in cell lines; however, CRISPR screening in primary cells is starting to gain momentum [188,189,190]. Genome editing of enhancers has even been conducted in situ, by injecting guide RNA combinations along with Cas9 mRNA into fertilised mouse oocytes [111]. These novel techniques hold promise for the functional identification of enhancers on a global scale in vivo.
Natural variation at enhancers and its consequences
While much of the evidence for enhancer function was obtained through perturbing these regions artificially, natural genetic variation at enhancers is commonly observed in human populations, consistent with the relatively low sequence constraint of these regions [191, 192]. Much of this variation has apparently little phenotypic effect due to redundancy both within enhancers (such as through multiple homotypic TF binding sites [191, 193]) and across them (such as through multiple enhancers regulating the same gene, as will be discussed below). Nonetheless, enhancer variation is also known to underlie significant pathologies in both model organisms and humans. In Drosophila, for example, point mutations in the binding site for the Kruppel TF within an enhancer controlling the abdominal fate specifier abd-A results in its misexpression in a thoracic segment, leading to its conversion to an abdominal one [194]. Likewise, mutations in the enhancers controlling the expression of Drosophila homolog of Pax2 affected mechanosensory bristle development [195]. In humans, a point mutation within a downstream enhancer controlling PAX6 expression causes the congenital eye malformation aniridia [196]. This mutation was found to prevent PAX6 itself from binding to the enhancer [196]. Point mutations in the ZRS enhancer of the sonic hedgehog (SHH) gene are associated with several types of congenital limb malformation and skeletal abnormalities [27, 197, 198] (notably this enhancer is located ~ 1 megabases away from SHH and within an intron of another gene, as will be further discussed below). Functional analysis of the ZRS enhancer revealed that these mutations either create gain-of-function binding sites for activating TFs (ETS1 and GABPα) [199], or abolish a repressive TF site [200], leading to ectopic SHH expression. Other examples of genes mis-regulated by point mutations in enhancers leading to human disease include TBX5 (congenital heart disease) [201], PTF1A (pancreatic agenesis) [202] and IRF6 (Van der Woude syndrome) [203]. Capitalising on these observations, sequences predicted to affect TF binding have been used to prioritise causal expression quantitative trait locus (eQTL) variants at enhancers [204,205,206,207].
The importance of enhancer variation in human pathology has been further highlighted by genome-wide association studies (GWAS), since GWAS-detected genetic variants associated with complex traits and diseases are typically non-coding, non-promoter associated and highly enriched in enhancers [184, 208, 209]. A classic example is in obesity susceptibility, where trait-associated SNPs are located within the intron of the FTO gene, but lead to the dis-regulation of the distal IRX3 gene [210]. In another example, a systemic lupus erythematosus-associated variant (termed TT > A) situated within an enhancer downstream of TNFAIP3 has been shown to abrogate NF-kB binding, affecting TNFAIP3 expression [211, 212]. Subsequent genome-wide TF binding analyses in population cohorts have identified multiple loci of differential TF binding (tfQTLs) across individuals that overlap GWAS variants. For example, genetic variants associated with differential binding of PU.1 were found to underpin GWAS variants associated with blood cell count and autoimmune diseases [213,214,215]. Beyond differential TF binding, population variation in readouts such as chromatin accessibility [216,217,218,219], DNA methylation, histone modifications [220,221,222,223] and chromatin looping [215, 224, 225] can provide insights into the genetic determinants of enhancer activity and a functional interpretation of disease associations.
Notably, some enhancer variants that appear functionally neutral when tested under normal steady-state conditions may show “cryptic” effects under stress or upon stimulus response [107, 226, 227]. For instance, eQTL analysis in monocytes stimulated with interferon-γ or lipopolysaccharide has revealed thousands of new variants affecting gene expression, which overlapped nearly 250 variants associated with response to infection and susceptibility to renal and immunological disorders [228]. Likewise, variants associated with type 1 diabetes were strongly enriched at promoter-connected active enhancers in activated, compared with resting, CD4+ T cells [229]. Condition-specific eQTLs were also detected in the cardiomyocytes of patients given the chemotherapy drug doxorubicin, with these loci potentially explaining the differential risks of heart failure upon doxorubicin treatment [230]. The emerging tools for QTL analysis in the single-cell setting [231, 232] have the potential to streamline the identification of regulatory genetic variants with transient effects.
Similarities and differences between enhancers and promoters
Contrary to the initial assumption that enhancers and promoters were biologically distinguishable elements, it is becoming clear that they share many properties (reviewed in [233]). The ENCODE project, for instance, found that an element might be classified as a promoter in the cell type-agnostic setting, but as a proximal enhancer-like element in a specific cell type [131]. Likewise, proximal and distal enhancer-like elements sometimes display elevated promoter-associated H3K4me3 [131, 234, 235]. Both active enhancers and promoters occupy nucleosome-free chromatin, bind RNAP and can be divergently transcribed [73, 236, 237]. In addition, enhancers within genes are sometimes capable of acting as alternative promoters [238]. Finally, evidence from multiple sources including reporter assays [169, 175, 239, 240], CRISPR [240,241,242], chromatin mapping [243, 244] and population genetics analysis [207, 245] suggests that certain promoters, termed ‘epromoters’, can also function as enhancers (reviewed in [246]). Notably, while many such elements control house-keeping genes in cis (as promoters), they may regulate lineage-specific or inducible genes distally (as enhancers) [240, 247]. In addition, whilst genetic variants at epromoters may affect the expression of both the proximal and distal gene, a large proportion are associated with only the distal one [207]. Therefore, the mechanisms by which epromoters control their target genes in cis and in trans may not be fully equivalent.
Recently, it has been proposed that cis-regulatory elements exist on a spectrum of enhancer/promoter ability [233, 248]. One key feature determining the position of a regulatory element on this spectrum is likely to be the directionality of its transcriptional output. In a Drosophila assay, unidirectionally transcribing elements were more likely to drive strong promoter and limited enhancer activity, whilst bi-directionally transcribing elements had strong potential for enhancer activity and could also function as weak promoters [248]. The “grey area” between enhancers and promoters is supported by evolutionary evidence that some enhancers have undergone genetic mutations allowing them to be repurposed into gene promoters in mammals [100]. Promoters are also sometimes repurposed into enhancers, but at a 13-fold lower rate than enhancer-to-promoter [100]. In terms of their sequence properties, enhancers and promoters tend to differ in GC content, with many promoters and only a subset of enhancers containing CpG islands [74, 249]. Therefore, enhancers predisposed to convert into promoters may have a particular GC sequence composition and presence of 5′ splicing regulatory motif patterns [100].
The potential interchangeability of enhancers and promoters is relevant for functionally interpreting promoter–promoter contacts that are abundantly detected in the genome, as will be discussed later in the review.
Enhancer–promoter communication in three dimensions
Whilst enhancers were classically tested by cloning them upstream of a core promoter, it quickly became clear that in their native context they are often located large distances away from the genes they control. The classic example of this is the Shh ZRS enhancer, which is located within the intron of the Lmbr1 gene approximately 1 megabase distant from Shh [27]. In this chapter, we discuss classic models of how enhancers and promoters can communicate over large distances, focusing on the evidence for the currently prevalent model of direct enhancer–promoter looping and factors thought to mediate it. We then move on to review the emerging exceptions from this model, with potential implications for non-looping mechanisms such as liquid–liquid phase separation. We have left the discussions on the dynamics of enhancer–promoter looping in specific biological settings outside the scope of this review. Several recent articles have provided up-to-date perspectives in this regard for various systems, including lymphocyte development [250, 251], heart development and disease [252], and cancer [253].
Evidence for enhancer–promoter looping
Historically, several conceptual models were proposed for enhancer–promoter communication. These models were initially applied to the mammalian β-globin locus, in which the developmental β-globin genes are regulated by a powerful tissue-specific enhancer known as a locus control region (LCR) [254,255,256,257]. It was unclear how the LCR could faithfully control the expression of the appropriate set of genes in each stage of erythroid cell development (embryonic, foetal and then adult β-type globin genes). In the “looping” model, the chromatin fibre between the LCR and the gene promoter could loop out until the LCR, as a single unit, is brought close to the promoter in 3D space [257, 258]. Looping between the LCR and gene promoters one by one would mean that gene promoters compete for LCR activation. It was unclear whether the amount of cellular energy required for such dynamic looping was achievable, particularly over increasingly large distances [259]. Therefore, in the alternative “linking” model, TFs binding to the enhancer begin a chain of events whereby multiple proteins bind along the intervening chromatin between the enhancer and the promoter, creating a bridge-like complex [259,260,261]. In a similar vein, the “scanning” model suggests that proteins recruited by the enhancer slide along the chromatin towards the promoter [262]. According to the linking and scanning models, the protein complexes would eventually become impeded by the presence of transcriptional machinery at the appropriate gene promoter, presumably to prevent spurious gene transcription [263, 264]. At the time, each of these proposed models were equally plausible as there were no appropriate experimental techniques to distinguish between them [259].
The development of novel techniques provided evidence that looping mechanisms are in fact widespread, even though the picture that is currently emerging is more complex, as will be discussed later. Two papers published in 2002 used different novel biochemical techniques to demonstrate that the β-globin LCR forms chromosomal loops with the active β-globin genes in vivo. Peter Fraser’s team used a method called RNA TRAP (“Tagging and Recovery of Associated Proteins”), in which horseradish peroxidase (HRP)-labelled probes bind to mRNA of specific genes and catalyse the deposition of biotin on chromatin proteins in close proximity with the transcript [265]. Loci that interact with the gene of interest can then be detected by confocal microscopy using fluorescently labelled avidin [265]. In parallel, Wouter de Laat’s team adapted the emerging chromosome conformation capture (3C) technology to explore the β-globin locus [266], after it was pioneered in yeast [267]. In 3C, chromatin is fixed by formaldehyde crosslinking and then digested into smaller fragments with a restriction enzyme. Fragments held together in 3D by interacting proteins are ligated (using DNA ligase), with the relative concentration of re-ligated pairs reflecting the frequency (and potentially strength) of their 3D contacts in vivo [267]. 3C confirmed that the LCR comes into close contact with active β-globin genes, whilst the intervening inactive genes are looped out [266]. Enhancer–promoter looping occurred in erythroid cells, in which the β-globin genes are expressed, but not in brain cells, in which they are not [266]. Further 3C experiments over a developmental time course revealed that the LCR interacts with different β-globin genes depending on the stage of erythroid cell development [268]. These findings supported a dynamic looping model rather than a linking or scanning model; neither of which predicted physical proximity between the LCR and the distal gene promoters.
3C technology, which probes the interactions between a single “anchor” with multiple “other end” fragments, was soon expanded to increase throughput. For example, 4C (“Circularized Chromosome Conformation Capture” or “Chromosome Conformation Capture-on-Chip”) employs a secondary restriction digest and inverse PCR to profile all loci interacting with the anchor fragment [269, 270]. In 4C experiments, the LCR was found to interact with multiple highly transcribed genes in the corresponding active cell type (foetal liver) but with different, transcriptionally silent genes in an inactive cell type (brain tissue) [269]. Further modifications of the 3C technology included 5C (few anchors vs few other ends, [271]) and, most significantly, Hi-C that uses biotin pulldown to enrich for ligation junctions, and is thereby capable of detecting theoretically all pairwise interactions between chromatin fragments in the nucleus [272]. The newest modification, Micro-C, replaces restriction enzyme digestion with micrococcal nuclease (MNase) treatment, resulting in smaller fragments and thereby increased resolution of the assay [273, 274]. Originally applied in yeast, it was then adapted to mammalian cells, including human ESCs and fibroblasts [275] and mouse ESCs [276]. Herein, we shall generally refer to 3C/Hi-C/Micro-C methods and their modifications as “3C-derived” methods.
The high complexity of libraries generated by 3C-derived methods necessitates very deep sequencing for identification of individual enhancer–promoter loops. This has nevertheless been achieved in a number of cell lines, providing further evidence for looping in transcriptional regulation [277]. Technologies that combine 3C, Hi-C or Micro-C with sequence capture to enrich the libraries for contacts involving (at least on one end) promoters or enhancers prior to sequencing have made this more achievable. Capture Hi-C (CHi-C) enriches Hi-C libraries (generated using biotin pulldown of proximity ligation junctions) [278,279,280,281], while Capture-C instead enriches 3C libraries obtained without biotin pulldown [282,283,284]. Most recently reported Micro-Capture-C (Micro-C coupled with sequence capture) enables the profiling of pairwise contacts involving selected regions of interest, at up to single-base pair resolution [285]. These enrichment methods have detected multitudes of enhancer–promoter loops in many human and mouse cell types [243, 286,287,288,289,290,291]. Alternatively, methods such as HiChIP [292] and PLAC-seq [293] (based on Hi-C), or ChIA-PET [294] (based on 3C), use immunoprecipitation to enrich for contacts that are bound to a protein of interest. Using these techniques with antibodies to enhancer- and promoter-associated histone modifications or those to RNA polymerase has also revealed large numbers of 3D enhancer–promoter contacts [244, 295,296,297,298,299,300,301].
Imaging techniques provide a complementary way to ascertain the spatial relationships in the genome. The ‘classic’ DNA fluorescence in situ hybridisation (FISH) [302] enables measuring the distance between candidate genomic loci at a single-cell level. DNA FISH has been used to characterise enhancer–promoter loops for several developmental genes such as Shh and HoxD [303, 304]. Unlike standard 3C-derived methods, imaging techniques obtain a direct distance measurement between the loci of interest at a single-cell level. This gives an opportunity to distinguish between interacting loci that either (a) are moderately proximal in the majority of cells (unimodal behaviour) or (b) have great cell-to-cell variability i.e. are very close in some cells and far apart in other cells (bimodal behaviour) [305]. Single-cell Hi-C can also be used to assess the heterogeneity of chromosomal topologies across cells (reviewed in [306]). However, along with other 3C-derived methods, it can only detect contacts between loci in those cells in which they localise at a close enough proximity to be ligated together (the exact such distance is not fully established). In contrast, DNA FISH gives access to the full distribution of inter-locus distances across all analysed cells [307]. Therefore, 3C-derived methods and FISH cannot be considered merely as “mutually validating” techniques, and they may produce genuinely divergent results, which need to be interpreted carefully [307]. At present, standard DNA FISH cannot reliably detect contacts between loci that are located short genomic distances away (dozens of kilobases) from each other, and have a limited throughput that may miss rare but functionally significant genomic contacts. However, advances in super-resolution microscopy are beginning to mitigate these limitations [308, 309]. Finally, high-resolution live imaging has also paved the way for a real-time visualisation of enhancer–promoter dynamics in vivo using genetic tools and CRISPR-mediated targeting to label these loci with fluorophores [310,310,312].
The recently developed “forced looping” methodologies have made it possible to probe the causal relationship between chromatin looping and gene expression [313, 314]. The CLOuD9 approach is based on the fusions of two dCas9 variants with the plant molecules ABI1 and PYL1, which dimerise in the presence of a plant phytohormone [313]. The two fusion proteins are directed to the chosen genomic anchors via complementary gRNAs, and phytohormone-induced dimerisation then drives the formation of an artificial chromatin loop. Meanwhile, LADL uses dCas9 fusions of plant proteins that form a heterodimer in response to blue light [314]. Forced looping between β-globin and its LCR was shown to rescue β-globin expression in K562 erythroid cells (which instead aberrantly express γ-globin) in a reversible manner [313]. Forced looping between these loci could also be achieved in HEK293 cells, in which they are heterochromatic, but in this case β-globin derepression was not achieved [313]. Hence, as expected, looping alone is insufficient for gene activation by enhancers.
Factors mediating enhancer–promoter looping
Following the discovery of chromatin looping in the β-globin locus, it was ascertained that several TFs were required for looping between the LCR and the distal genes, including GATA-1, FOG-1, EKLF and NLI/Ldb1 [315,315,317]. However, whether these factors directly facilitated chromatin looping remained unclear. A direct role in looping in this locus was eventually found for CTCF, whose role we will review in this section, alongside other looping factors (Fig. 2). Some of these proteins, including CTCF, also play a role in organising the global 3D chromosomal architecture, which will be discussed later in the text.

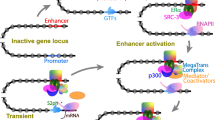
Effect of depletion of selected proteins on enhancer–promoter looping and gene expression. A In the wild type, enhancer–promoter contacts (arcs) occur in the context of large-scale contact domains such as TADs (red triangles) facilitated by the joint action of cohesin and CTCF (see “Large-scale chromosomal architecture”). Other cofactors relevant for enhancer–promoter communication (represented in various colours and shapes) can bind independently or in association with CTCF/cohesin at enhancers. Many of these factors are also found at promoters (not shown for simplicity). B Removal of CTCF or cohesin abolishes TADs. Long-range enhancer–promoter contacts proximal to TAD boundaries are disrupted, although some short-range contacts remain. In contrast, some long-range Polycomb-associated contacts (arc connecting red squares) and short-range enhancer–promoter contacts appear that spread across the native TAD boundaries. The transcriptional effects of this perturbation, however, remain relatively mild. C Removal of LDB1 abolishes an enhancer–promoter loop and leads to decreased gene expression for the affected gene. D Removal of BRD4 and/or Mediator does not disrupt enhancer–promoter contacts, but decreases gene expression. See section “Factors mediating enhancer–promoter looping” for details on individual factors and references to primary studies
CTCF and cohesin
CTCF is a remarkably versatile protein that contains eleven highly conserved zinc fingers with both DNA-binding and protein-binding capacity [318]. Reporter gene assays in selected loci identified context-dependent capabilities of this factor as a transcriptional activator and repressor [319,319,320,322]. More unusually, it emerged that CTCF binding has an insulating capacity, effectively blocking signals between enhancers and their target genes in the β-globin and H19/Igf2 loci [323,323,324,326]. Dozens of thousands of CTCF binding sites were then identified genome-wide in silico [327] and in vivo [328,328,330], and it was found that divergent gene pairs separated by CTCF sites had lower than expected correlation of gene expression genome-wide, confirming the insulatory role of CTCF on the global scale [327]. According to the linking or scanning model of enhancer communication, an insulatory role for CTCF could be imagined as a “roadblock” preventing movement of proteins along the chromatin fibre. However, its mechanistic action in the looping model was initially unclear [318]. This was partially reconciled by the finding that CTCF itself can mediate chromatin loop formation at the nucleolar surface [331]. Transgene assays of the human β-globin locus then showed that two insulator sites binding CTCF could form a chromatin loop [332]. Indeed, CTCF binding sites flanking the β-globin locus were found to physically interact in erythroid progenitor cells, forming a large domain encompassing the LCR and its target genes [268, 333].
A key insight into how CTCF mediates chromosomal looping was the discovery of its association with the cohesin complex [132, 334, 335]. Cohesin had long been known for its role in holding sister chromatids together from DNA replication to chromosomal segregation [336]. The cohesin complex has four core subunits: SMC1, SMC3, SCC1 and SCC3 that join to form a ring-like structure [337,337,339]. It is currently accepted that this “ring” holds sections of chromatin together, both in trans (in sister chromatid cohesion) and in cis (when mediating chromomal loops). CTCF is not directly required for cohesin loading and unloading from chromatin, which is mediated by other factors (reviewed in [340]). Instead, it stabilises cohesin loops by blocking the movement of the cohesin ring [341, 342]. Consistent with this, cohesin recruitment to regulatory elements may also occur independently of CTCF, typically in a cell type-specific manner [343].
Beyond mediating specific enhancer–promoter loops, the cooperative action of cohesin and CTCF has emerged as a central organiser of the global chromosomal architecture through a mechanism termed loop extrusion, which will be discussed later in the text (chapter “Determinants of enhancer–promoter selectivity”, section “Large-scale chromosomal architecture”). Surprisingly however, deletion or degradation of cohesin or CTCF in differentiated cells has only mild effects on gene expression [344,344,345,346,347,349]. Consistent with this, significant numbers of enhancer–promoter contacts remain unaffected upon rapid degradation of these proteins, and some contacts even appear de novo [349, 350]. Mechanisms underlying this phenomenon are still not fully understood, but recent studies suggest that loops that require CTCF might be preferentially long-range (> 100 kb) and situated in loci where enhancers are generally sparse [301, 349]. For example, the contact between versican (Vcan) promoter and Xrcc4 epromoter spanning over 350 kb is significantly reduced, and Vcan expression diminished, upon deletion of the CTCF motif at the Vcan promoter. Artificial tethering of CTCF to the Vcan promoter using a dCas9-CTCF construct effectively rescues the loop and, partially, the expression of Vcan [301].
Polycomb repressive complexes
Polycomb group (PcG) complexes have long been known to orchestrate the epigenetic silencing of chromatin throughout development. Initially discovered in Drosophila, PcG contain two main families: Polycomb Repressive Complex 1 (PRC1), which has E3 ligase activity causing the monoubiquitination of histone H2A lysine 118 (Drosophila) or 119 (vertebrates), and Polycomb Repressive Complex 2 (PRC2), which catalyses the di- or tri- methylation of histone H3 lysine 27 [351]. These histone modifications are thought to directly block transcription (reviewed in [352]). In Drosophila, PcG complexes aggregate into foci known as Polycomb bodies, which contain co-repressed genes such as Hox gene clusters (reviewed in [353]). Within Polycomb bodies, PcG complexes were discovered to mediate chromatin loops between distal loci. For example, 4C and FISH revealed that genes within the Hox loci Antennapedia and Bithorax, which are separated by ~ 10 megabases, are looped together with their associated regulatory elements, ensuring coordinated epigenetic silencing [354]. These findings were corroborated by genome-wide evidence of PcG-mediated looping in embryonic Drosophila cells [355, 356]. In mammals, PcG-associated promoters and enhancers also engage in multitudes of looping contacts, particularly in early development [139, 141, 276, 357]. Many of these contacts are undetectable in human naive ESCs where PcG proteins are dispersed throughout chromatin and emerge in primed ESCs [358].
Polycomb-mediated looping is likely to work largely independently of CTCF and cohesin and is potentially even counteracted by the looping activities of these factors in ESCs [357]. Whilst the exact mechanisms of how Polycomb organises the 3D genome are not fully understood, formation of bonds between the PHC1/2 SAM domains of PRC1 complexes was proposed as one potential mechanism [359]. However, knockouts of both PRC1 and PRC2 components affect chromosomal contacts between Polycomb-bound loci [280, 360].
Notably, some contacts between PcG-associated regions, including poised promoters and enhancers in ESCs, are retained after these regions are activated during cell differentiation [141, 143]. Moreover, PRC1 was recently found to mediate chromatin interactions between a subset of active developmental enhancers and promoters in Drosophila [361]. Jointly, these results raise the possibility that at least in some cases, Polycomb-mediated chromatin loops may “prime” the connections between regulatory elements in early embryonic cells to ensure their appropriate pairing upon activation later in development.
YY1 and ZNF143
YY1 and ZNF143 are both zinc finger TFs with DNA binding domains that have recently been implicated as looping factors mediating interactions between active regulatory elements [295, 362,361,362,365]. YY1 was initially proposed to establish loops in the Ig heavy chain locus in pro-B cells [366]. Recent studies have confirmed its global role in connecting cell type-specific active enhancers and promoters in a range of mammalian cell types including mESCs [295] and neural progenitor cells [365]. Meanwhile, ZNF143 binds to promoters globally [367] and was found at the anchors of many lineage-specific enhancer–promoter loops in human and mouse cells [362,361,364, 368]. Unlike CTCF and cohesin, long-term depletion of YY1 or ZNF143 in mammalian cells causes a significant reduction of enhancer–promoter loops, coupled with substantial changes in gene expression [295, 368, 369] (although short-term YY1 depletion has only a modest effect on both [350]).
The mechanisms by which YY1 or ZNF143 mediate regulatory chromatin contacts are still under investigation. YY1 does not seem to co-localise with CTCF [365] and instead has been proposed to associate with cohesin independently, in a manner analogous to CTCF itself [295]. In contrast, ZNF143 co-localises with both CTCF and cohesin [369], and it has recently been shown that depletion of ZNF143 disrupts CTCF-mediated enhancer–promoter looping [368].
LDB1
LIM domain-binding protein 1 (LDB1) is a dimeric cofactor that does not have direct DNA-binding capacity, but interacts with the LIM domain of other TFs to form multi-protein complexes [370]. LDB1 was first identified as a looping factor in the mouse β-globin locus [317, 371]. Later studies identified a role for LDB1 in facilitating intra- and inter-chromosomal interactions upon mouse ESC differentiation to myogenic progenitors [372, 373] and in mouse olfactory sensory neurons [373]. In these two studies, deletion of LDB1 significantly disrupted the expression of the gene programs underpinning appropriate cell differentiation [372, 373]. Ectopic tethering of LDB1 to chromatin is sufficient for loop formation, increased deposition of the enhancer mark H3K4me1 and transcriptional activation [372, 374]. Jointly, these results implicate LDB1 in connecting lineage-specific enhancers with their distal target genes by chromosomal looping. In contrast to YY1 and ZNF143, LDB1 has been shown to form loops independently of both cohesin and CTCF [371, 375]. Consistent with this, the binding of sequence-specific TF PAX3 associates with either LDB1 or CTCF but not both [372]. Likewise, binding sites for Lhx2, another TF that recruits LDB1, are devoid of CTCF and cohesin binding [373].
The mediator complex and BRD4
The multimeric Mediator complex is recruited to active enhancers, where it initiates the assembly of the pre-initiation complex and recruitment of RNAP [376]. Mediator was implicated as a looping factor, potentially in association with CTCF and cohesin [377,376,379]. However, it was shown recently that rapid removal of chromatin-bound Mediator and RNAP does not significantly affect chromosomal architecture, including enhancer–promoter interactions, in spite of triggering widespread transcriptional changes and cell cycle arrest [380].
Mediator recruitment to active enhancers is facilitated by the bromodomain and extraterminal domain (BET) protein BRD4 [70, 71] that plays crucial roles in pluripotency and cancer [71, 381, 382]. Loss of BRD4 leads to global changes in transcription and widespread chromatin decompaction [383, 384], and BRD4 is enriched at enhancers engaged in cohesin-independent promoter contacts in HeLa cells [349]. Nonetheless, removing BRD4 (and, consequently, Mediator) from chromatin using chemical inhibitors did not perturb the majority of enhancer–promoter contacts, despite major changes in transcription [69].
Thus, despite their importance for transcription, Mediator and BRD4 are likely not essential for looping between regulatory elements. Instead, it was suggested that Mediator can aid transcription by supplying TFs and transcriptional machinery to the promoter by diffusion over a small distance, which would not require a direct “bridging” interaction [380]. Whilst these activities might occur in association with the looping contacts facilitated by cohesin and CTCF [380], Mediator and BRD4 may also facilitate liquid–liquid phase separation of enhancers and their target promoters that does not require direct looping [69, 385], as discussed below.
RNA polymerase and transcription
The transcriptional process and associated RNA molecules can also influence DNA architecture. Whilst it is established that transcription helps shape up higher-level genome conformation (reviewed in [386]), there is also evidence that it can either disrupt or stabilise specific enhancer–promoter loops. For example, transcription at stimulus–response genes was shown to disrupt chromatin loops [387, 388], potentially through destabilising cohesin association with the chromatin fibre by RNAP machinery [388]. However, a study in bacteria suggests that a related chromatin “ring” (condensin II complex) recovers quickly from the encounters with the elongating RNAP machinery [389].
There is also evidence suggesting that transcription stabilises enhancer–promoter loops [276, 311]. Live cell imaging experiments using a fluorescent reporter gene in the eve locus in Drosophila detected a causal association between transcription and the enhanced compaction of the locus, bringing the eve enhancers into close proximity with the promoter [311]. Moreover, enhancer–promoter loops dissociated tenfold faster in the absence of transcription than when the reporter gene was being transcribed, suggesting that transcription can stabilise contact between these elements [311]. One possible mechanism by which transcription may stabilise loops is through eRNAs (reviewed in [86]), potentially through their interaction with cohesin [81, 378, 390]. Knockdown of eRNAs resulted in decreased enhancer–promoter proximity in human cells [81, 390]. Other proposed mechanisms implicate the transcriptional machinery itself. Transcription by RNAP was found to support some short-range enhancer–promoter and promoter–promoter interactions in fine-scale Micro-C data [276]. However, Promoter Capture Hi-C-based studies did not observe a significant change in enhancer–promoter contacts upon RNAP inhibition, including in cells rapidly depleted of cohesin or CTCF [349, 380].
In conclusion, while cohesin and CTCF remain the established key organisers of chromosomal loops, there is evidence for loops independent of these proteins and of additional looping factors, including potentially RNAP and/or transcription itself. To what extent this explains cohesin-independent contacts and the small effects of cohesin/CTCF deletion on gene expression still remains unclear, prompting the question of whether mechanisms other than direct looping can mediate enhancer–promoter communication, as discussed below.
Permissive and instructive enhancer–promoter contacts
A long-standing question in the field has been whether enhancer–promoter contacts form concomitantly with the activation of these regions (“instructive”) or, alternatively, precede it (“permissive”) [391]. There are currently ample examples in support of both phenomena. In Drosophila, pronounced changes in enhancer activity during embryonic development seem to occur without prominent changes in their 3D conformation [392, 393]. In mammalian differentiation models, in contrast, extensive remodelling of enhancer–promoter contacts is typically observed consistently with enhancer activation [141, 394,393,394,397]. In addition, the patterns of promoter contacts in human primary blood cells recapitulate the hematopoietic lineage tree, likely reflecting their gradual remodelling during lineage specification [243]. One possibility is that the pace of development underlies the observed differences in interaction dynamics between Drosophila and mammalian models. Nonetheless, a significant minority of enhancers in human ESCs also show pre-existing “permissive” promoter contacts prior to activation [141, 394]. In addition, permissive contacts were also observed in mammalian cells upon TNF-induced enhancer activation [398].
The presence of both instructive and permissive contacts reflects the likely diversity of mechanisms facilitating enhancer–promoter connectivity. One proposed mechanism linking enhancer activity with connectivity is based on the observation that cohesins can be recruited to H3K4me1-marked chromatin, which is associated with the “primed” state of enhancers and the binding of MLL/Trithorax transcriptional coactivators [399]. Additionally, depletion of H3K27ac was shown to result in weaker enhancer–promoter looping in human lymphoma cells [400]. However, the exact determinants of enhancer–promoter loop formation at a given point of time, either prior to or upon the activation of these elements, still remain incompletely understood.
Enhancer–promoter communication beyond direct looping
Whilst there is now a definitive bulk of evidence in support of physical looping between enhancers and promoters, several recently described phenomena suggest that looping is not the only mechanism of how these regions can communicate, as will be discussed in this section.
Deviations from the looping model
The Sox2 gene has an essential enhancer, the Sox2 Control Region (SCR), which was found by 3C-derived techniques to loop to the Sox2 promoter (for example, [379, 401]). However, live cell imaging over a ~ 25 min interval could not detect close proximity between the SLC and the Sox2 promoter, even during the bursts of Sox2 transcription [402]. Moreover, there was no correlation between Sox2 transcriptional level and enhancer–promoter distance, or any obvious reduction in distance prior to transcription initiation, suggesting that this enhancer may activate Sox2 transcription via a looping-independent mechanism [402]. While this discrepancy between 3C-derived and imaging-based results is puzzling, the findings are not necessarily contradictory, and may be jointly consistent with either the presence or lack of direct looping. For example, the proximity signal between Sox2 promoter and enhancer in 3C/Hi-C might be detected if these regions become connected by protein bridges without getting physically closer to each other [403], as would be expected under the linking or scanning models. In addition, 3C-derived methods can pick up rare events that imaging is underpowered to identify [404]. Finally, recent biophysical modelling suggests that functionally causal enhancer–promoter contacts do not have to be temporally concomitant with transcriptional bursts [405, 406]. The exact mechanisms underlying the observed phenomenon, however, remain to be elucidated.
In contrast, a proximal enhancer of the Shh gene moves further away from the Shh promoter upon activation, according to both 3C-derived methods and imaging [407]. Recruiting activators such as VP64, Mediator and SIX3 to this enhancer also increased the enhancer–promoter distance whilst upregulating Shh expression [407]. It has been argued that this might be due to increased enhancer mobility [408] whereby non-thermal energy produced during transcription can “stir” the chromatin [312]. However, whilst this stirring model predicts that the enhancer explores a larger nuclear space, it also moves faster, which would potentially increase the frequency of contacts with the gene promoter [312]. Notably, synthetic activation of the Shh promoter, bypassing the need for enhancer activation and causing increased transcription, did not lead to increased distance [407]. Combined, these results would argue against the classical looping model for this enhancer. Given that the looping between the distal ZRS enhancer and Shh promoter is well-established, this suggests that multiple enhancers may control the expression of their shared target gene through a combination of looping and non-looping mechanisms.
Finally, multiple lines of recent evidence suggest that many functional enhancers likely do not come into direct contact with their target promoters upon Drosophila development (although some “permissive” enhancer–promoter loops are detectable as discussed above) [392]. For example, neither Hi-C nor Micro-C detected a widespread enrichment for enhancer–promoter interactions in early Drosophila embryos irrespective of the activity of these elements [393]. In addition, using high-resolution live imaging, it was observed that genes regulated by a single enhancer can localise surprisingly large distances away from each other in 3D (at least 100–200 nm), and yet show coordinated bursts in transcription, which is inconsistent with their direct looping to the shared enhancer [409].
Liquid–liquid phase separation
It has been proposed that chromatin can compartmentalise into membrane-less biomolecular condensates in the phenomenon known as liquid–liquid phase separation (LLPS) [410]. Several recent studies argue the case for LLPS as a key mechanism underlying transcriptional control by enhancers [385, 411, 412]. Mechanistically, LLPS occurs when a particular protein increases in quantity to a critically high point, whereupon it spontaneously separates into two phases that contain either a high or a low concentration of the molecule [413]. LLPS is thought to be mediated by weak protein–protein interactions involving intrinsically disordered regions (IDRs) of the protein [412, 414]. Histone proteins themselves might have the ability to form LLPS via intrinsically disordered nucleosome tails [415,413,417]. In addition, many sequence-specific TFs also have intrinsically disordered low complexity domains within their activation domains that are capable of forming LLPS [412]. In line with this, DNA-bound TFs may have the ability to drive condensate formation via weak, multivalent interactions with recruited cofactors, such as Mediator and BRD4 [69, 385, 414]. The number and density of TF binding sites within enhancers potentially facilitates this by crowding DNA-bound TFs beyond the required threshold required for LLPS formation [418, 410].
The engagement of cofactors Mediator and BRD4 in LLPS could explain why their depletion results in transcriptional changes without significantly affecting chromatin loops [69, 380]. However, looping and LLPS may not be fully orthogonal phenomena, as it was recently proposed that cohesin may induce LLPS of chromatin through forming protein bridges [419]. Likewise, a recent biophysical model could explain the absence of contacts between the SCR and the Sox2 promoter observed in live imaging data by a combination of looping and phase separation [420].
Whilst LLPS in general has been demonstrated both in vitro and in vivo [421, 422], evidence for chromatin LLPS and its functional significance in gene control is still incomplete. For instance, whilst TFs were shown to form high-concentration stochastic hubs with chromatin in living human cells, overexpression of their low complexity domains beyond endogenous levels was required for LLPS to occur [412]. Furthermore, since high-concentration hubs were capable of transactivation and recruitment of RNAP even without LLPS, the question remains if LLPS is necessary for any genomic locus [412]. It is possible, for example, that LLPS is a distinctive feature of super-enhancers and/or is a consequence, rather than cause, of enhancer–promoter association [411, 412].
The recent interest in LLPS has prompted calls for more rigorous experimental guidelines on its identification and characterisation in the cellular environment [423, 424]. For example, commonly used in vivo approaches like fast fluorescence recovery after photobleaching (FRAP) and sensitivity to treatment with 1,6-hexanediol are not always indicative of LLPS [423]. Nonetheless, it is compelling to consider if LLPS could be a mechanism of enhancer–promoter communication that is complementary to chromosomal looping. First, LLPS could underlie gene control by “contactless” enhancers, uniting them with their target promoters and supplying them with the required proteins without the need for physical proximity [52]. LLPS could also provide a mechanistic explanation to the classic “transcription factory” model [425], whereby specific activators or repressors are assembled in high concentration at appropriate genomic loci [426, 427]. For example, it has been suggested that Mediator and RNAP molecules may accumulate in condensates at super-enhancers, and these condensates could facilitate efficient loading of the transcriptional machinery to active gene promoters [426].
Notably, phase-separated condensates might form and dissolve in accordance with local chromatin features, such as histone tail acetylation, the presence of linker histone H1 presence and inter-nucleosome spacing, as well as the concentrations of recruited TFs [52, 415]. Molecules such as RNAP might also be passed between different condensates based on their post-translational modifications [426, 428]. Therefore, such “factories” formed by LLPS may be extremely transient [52], consistent with the dynamic and “bursty” nature of transcription. Finally, an important feature of LLPS is that it has been modelled quantitatively, at least in simpler systems, building on the foundations of soft matter physics [429]. These models facilitate formal testing of the involvement of LLPS in chromatin organisation and prediction of its implications in gene control.
Determinants of enhancer–promoter selectivity
It is clear from both genome-wide profiling and perturbation studies that, of the multitudes of enhancers in a locus, not all of them provide input to every promoter [430, 431]. The underpinning global genomic context, discussed immediately below, has emerged as a key factor determining enhancer–promoter selectivity, providing a means to mitigate and modify the effects of linear genomic distance. However, it still does not explain all observed effects, suggesting the involvement of other phenomena, such as the mutual compatibility of specific promoters and enhancers that is discussed later in this chapter.
Large-scale chromosomal architecture
Products of loop extrusion: TADs and stripe domains
It has become evident from early analyses of Hi-C data that metazoan genomes are organised into megabase-scale segregated domains that are often invariant between cell types, termed topologically associating domains (TADs) [432,431,434]. While initially, the mechanisms underlying TAD formation and maintenance were unclear, an explanation has eventually been provided by the model of chromosomal loop extrusion [342, 435]. Loop extrusion was initially proposed theoretically based on molecular dynamics simulations [342, 435] and then validated in single-molecule assays [436, 437]. Under this model, cohesin in interphase nuclei continuously extrudes chromosomal loops until it encounters a boundary, which prevents further extrusion. In the classic scenario, this boundary is presented by CTCF binding to its sequence motifs located in a divergent orientation at either end of the loop [277, 438,437,440], consistent with CTCF’s established role as an insulator-associated protein. Loops stalled by CTCF or other boundaries constrain chromosomal contacts, giving rise to “loop domains” detectable in Hi-C data. Notably, TADs correspond to only one level of a hierarchy of such “loop domains”, with super-/sub-TADs and insulated neighbourhoods potentially corresponding to other levels [434, 441, 442]. It has been suggested, however, that the level of the hierarchy corresponding to TADs is “functionally privileged”, as their boundaries show the highest enrichment in CTCF binding and conservation across cell types [443].
TAD boundaries are classically defined through a change in the predominant direction of chromosomal contacts observed in Hi-C data [444]. The majority of specific enhancer–promoter loops follow this pattern and are highly enriched within, compared with across, TAD boundaries [277, 401, 405]. Consistent with this, disruption of TADs can cause promiscuous enhancer–promoter contacts and ectopic gene expression leading to developmental aberrations and disease (reviewed in [445]). For example, loss of TAD insulation between limb-specific enhancers associated with EPHA4 expression and a set of adjacent genes, including WNT6 and PAX3, results in their misexpression and abnormal limb development in both mice and humans [446]. More recently, TAD fusion was also proposed as a mechanism leading to several disease phenotypes upon genomic duplications in the SOX9 locus [447]. However, TAD structure in general may play less definitive a role in gene regulation than originally thought. Most strikingly, rapid depletion of cohesin or CTCF quickly abolishes TADs but, as mentioned above, has only relatively mild effects on gene expression [301, 345,345,347, 448]. Consistent with this, the considerable TAD re-arrangements observed in Drosophila balancer chromosomes compared with their wild-type counterparts result in changes in the expression of only ~ 10% of genes [449]. Likewise, targeted deletions of TAD boundary-forming CTCF sites in the Shh locus did not entirely disrupt the developmental regulation of Shh expression [450, 451].
The less pronounced than expected role of TADs in gene control can be partially explained by the ability of some enhancer–promoter contacts to form and/or be maintained independently of TAD constraints. For example, Capture Hi-C analyses in multiple cell types consistently detect enhancer–promoter contacts that cross TAD boundaries [141, 243]. Recent mechanistic experiments suggest that the insulating effect of TAD boundaries may be inversely dependent on enhancer strength [405]. In addition, single-cell studies show both a substantial cell-to-cell heterogeneity of TADs and large numbers of inter-TAD chromosomal contacts [448, 452]. Furthermore, in the developing Drosophila embryo, enhancer–promoter loops are formed before TADs, and likely also precede the onset of zygotic transcription [453, 454]. Cis-regulatory contacts were also shown to emerge prior to the formation of TADs in murine cells exiting mitosis [455].
Another layer of complexity in the relationship between chromosomal domains and specific enhancer–promoter contacts arises from the fact that active promoters and enhancers can likely give rise to TAD boundaries even in the absence of convergent CTCF sites [401]. Disentangling the cause-and-effect relationship is particularly challenging in the case of a flavour of TAD-like structures known as stripe domains (from the way they appear on Hi-C maps) [276, 456, 457]. Stripe domains tend to anchor at a single CTCF binding site (as opposed to two divergent CTCF sites in the classic case for TADs), which points towards a nearby site for the cohesin-loading factor NIPBL [456, 457]. It has been suggested that they arise from one-sided loop extrusion, facilitated by the loading of cohesin to lineage-specific enhancers [457]. However, experiments using finer-scale mapping with Micro-C have revealed even smaller, nested stripes that may occur independently of loop extrusion [276]. These 10–50 kb pair stripes are enriched in promoter–promoter and enhancer–promoter interactions and are largely unassociated with CTCF/cohesin binding [276].
In conclusion, whilst there is clear evidence for a role of some TADs in gene control, the emerging view is more nuanced than originally envisaged. Rather than being fully insulated structures that predetermine enhancer–promoter contacts, TADs and related loop domains can be seen as flexible hierarchical features of genomic organisation that loosely contain, but also can potentially be generated by, enhancer–promoter contacts.
A/B compartments and LADs
Beyond TADs, other aspects of the global chromatin structure may also play a role in enhancer–promoter communication. Most prominently, contacts from active chromatin and heterochromatin regions appear largely segregated into separate compartments (referred to as A and B, respectively) [272]. These “A/B” compartments are dynamic across cell types and conditions. Most strikingly, around 1/6th of the genome undergoes compartment changes during the 24 h circadian cycle in mouse adult liver [458]. The A/B compartment structure does not disappear (and is in fact even strengthened) upon cohesin/CTCF depletion [344, 346,346,348], but, like TADs, dissolves in mitosis [459]. It has been suggested that A/B compartments may be facilitated by affinity of heterochromatin regions to each other [460, 461]. Whether contacts between active cis-regulatory elements (particularly those independent of cohesin/CTCF) also participate in compartment formation currently remains an open question.
A subset of B compartments corresponds to lamina-associated domains (LADs) [462]. LADs represent ~ 500 kb-long chromatin regions localised at the nuclear periphery. These regions are generally gene-poor regions with low transcriptional activity. Mechanisms by which LADs promote transcriptional repression are not fully understood, but may include sequestration of genes and their regulatory elements in heterochromatin foci [462]. Whilst many LADs remain generally invariant across cell types and conditions, others can move away from the nuclear periphery, for example, upon T cell differentiation [463]. This relocation is associated with the activation of the respective regulatory elements and their increased proximity [463]. Recently, some A compartment regions consisting of H3K9me2-marked chromatin were also found to localise at the nuclear periphery [464]. These domains (dubbed H3K9me2-Only Domains; KODs) are distinct from LADs and have been suggested to represent an “intermediate” chromatin state with a role in regulating tissue-specific gene expression [464].
Genomic distance
Chromosomal contacts follow the law of constrained Brownian motion (reviewed in [465]), with the contact probabilities showing a strong power–law dependence on the “linear” genomic distance [272, 466]. In mitosis, where TADs and compartments dissolve, genomic distance becomes the predominant factor governing the frequency of chromosomal contacts across the genome [459]. However, even in interphase nuclei, genomic distance continues to be a reasonable predictor of functional enhancer–promoter contacts, particularly in the short to medium range (< 100 kb), as evidenced by high-throughput enhancer perturbation data [183, 184]. Recently, Zuin et al. investigated the positional effects of enhancer–promoter contacts by embedding an enhancer into a PiggyBac transposon and integrating it ectopically next to a reporter gene [405]. PiggyBac “jumps” resulted in the enhancer relocating away from the promoter over a range of distances, but mostly within the same 560 kb TAD as the reporter gene, enabling a comparative analysis between different clones. In this systematic study, the probabilities of enhancer–promoter contacts showed a strong dependence on linear distance within the TAD and fell sharply near and across the TAD boundaries [405].
The effects of distance, however, are likely not absolute, even within the same TAD. For example, analysis in the Shh locus suggests that within-TAD chromosomal contacts there do not show a pronounced distance dependence [467]. In addition, many computational algorithms for the analysis of data from Hi-C and related techniques explicitly correct for distance decay, identifying “significant interactions” based on how unusual their contact frequency is at a given distance [277, 468,467,468,471]. Promoter-interacting regions identified using these approaches typically show enrichment for enhancer-associated chromatin marks and disease-associated variants, and in a number of cases their target genes have been validated directly (reviewed in [431]). In contrast, the model of Zuin et al. [405] discussed above was based on the potentially “contactless” SCR and its target Sox2 promoter. Consistent with this, in the ectopic context this enhancer–promoter pair did not show prominent specific interactions, despite the clearly detectable transcriptional effects of SCR [405]. Therefore, the extent of distance dependence of enhancer–promoter contacts likely depends on the underlying genomic context and potentially also on the properties of specific enhancers and promoters.
Enhancer–promoter compatibility
Even within the same TAD, enhancers are not always fully promiscuous and can sometimes “skip” nearby genes, in favour of more distal targets [472, 473]. Sequence compatibility between enhancers and promoters has been proposed as one of the factors underlying their pairing [474]. For example, enhancer trap experiments in Drosophila that used randomly integrated reporter genes driven by a TATA- and a DPE-containing promoter, respectively, showed that some endogenous enhancers had a preference for either of the two promoters [475]. More recently, a massively parallel reporter assay in Drosophila cells revealed distinct groups of enhancers that preferred one of two types of core promoter, derived from either a housekeeping or a developmental gene and enriched for different TF binding motifs [247]. Notably, this preference was consistent with the tissue-invariant or tissue-specific activity of the enhancers themselves [247].
Beyond the Drosophila model system, there is currently little evidence for inherent enhancer–promoter compatibility. Further insights on potential selectivity may yet come from machine learning methods that are gaining the ability to predict enhancer–promoter pairs from sequence alone in mammalian cells [476, 477]. While currently these are generally “black box” methods, such as deep neural networks, advances in interpretable machine learning may provide a handle on the specific sequence features of these elements that are predictive of their compatibility. In addition, at least some of the perceived selectivity of enhancer–promoter contacts could be due to other factors, such as enhancer competition for promoters. For example, it was shown that deleting or inactivating the target promoter of a particular enhancer releases the enhancer to activate other local promoters [478]. In addition, biophysical modelling suggests that the same enhancer may have vastly unequal effects on different promoters depending on their intrinsic sensitivity to enhancer–promoter contacts (irrespectively of the properties of a given enhancer) [406].
The findings discussed in this chapter show a diversity of the aspects of nuclear organisation that regulate enhancer–promoter communication. The outstanding question is how exactly these multiple aspects work together to establish appropriate enhancer–promoter relationships. One way to conceptualise this complexity at the current level of understanding is to assume enhancer–promoter pairing to be a largely stepwise process, as proposed recently by Schoenfelder and Fraser [431]. In this view, enhancers are “selected” for activation in cis by TFs, following which their contacts with target promoters are then “facilitated” by the underlying chromosomal topology and then further “specified” by parameters of enhancer–promoter compatibility [431]. Further functional analyses and quantitative modelling of enhancer–promoter communication will help to validate and improve our understanding of these phenomena.
Beyond pairwise enhancer–promoter relationships
It follows from the rather flexible requirements for enhancer–promoter selectivity (discussed above) and the very large number of enhancers active in a given cell at a time that genes often receive output from multiple enhancers. It is also currently clear that the same enhancer can control multiple genes. Here, we discuss the functional and structural considerations around this multiplicity of enhancer–promoter relationships.
Evidence for multiplicity of enhancer–promoter connections
One promoter—many enhancers
Enhancers vastly outnumber promoters in human and mouse genomes [131], and consistently promoters often contact more than one enhancer in the same cell type [141, 243, 278, 279]. For example, PCHi-C showed that active promoters contacted a median of two active enhancers in human primary blood cells [243]. CRISPR-based approaches have confirmed that promoters often receive signals from multiple functional enhancers [479, 480]. In developing mouse limb tissue, for instance, genes were regulated by a median of three functional enhancers and some genes, which coded for limb-specific TFs, had more than ten associated enhancers [480]. Promoters can also engage multiple enhancers in different cell types or at different developmental stages. For example, in the well-characterised mammalian Hox gene cluster, activation of genes with crucial roles in development is tightly controlled at each developmental stage by complex enhancer networks [481, 482]. In Drosophila, eve expression is controlled by five separate enhancers to produce stripe patterning in the embryo [483].
Multiple enhancers associated with a single promoter in a given cell type may be conceptualised as cis-regulatory units (CRUs) [141]. Analyses in human ESCs and early ES-derived neural progenitors found that, as expected, enhancers within a CRU tended to have the same activity state in a given cell type [141]. However, this effect was not absolute, with 20% of CRUs detected in these cells being “dual-state”, i.e. containing a mixture of active and poised or Polycomb-repressed enhancers [141]. Upon early neural differentiation, about a third of single-state CRUs in human ESCs became dual-state, while around half of all dual-state CRUs became single-state [141]. Changes in CRU state were predominantly driven by coordinated alterations in the chromatin state of enhancers and their promoter contacts, and were associated with significant changes in gene expression [141]. Notably, the chromatin state of the promoter at dual-state CRUs typically corresponded to the majority state of its enhancers, consistent with a competition of opposing regulatory effects at the promoters.
Many promoters—one enhancer
The same enhancer can also regulate more than one target gene—either in different spatiotemporal settings or in the same one. An example of the former is found in the Drosophila Hox locus, where one enhancer regulates either pb or zen2 depending on the developmental stage via distinct chromatin loops [484]. Examples of enhancers contacting multiple genes in the same cell type are also abundant in data from Hi-C and related technologies. While these data cannot ascertain whether these contacts occur in the same cell, recent evidence suggests that this may indeed be the case. For example, the LCR in the β-globin locus can simultaneously engage two adult globin genes, Hbb-b1 and Hbb-b2, while skipping over embryonic globin genes, in mouse fetal liver cells [485]. Likewise, evidence from live imaging shows that an enhancer located in-between two reporter genes can initiate simultaneous transcriptional bursting from both gene promoters [310].
Enhancer chains and networks
In addition to multiple direct enhancer–promoter contacts, there is also evidence for enhancer–enhancer interactions that create an enhancer “chain” or network [486,485,486,489]. In this mechanism, not all of the enhancers need to contact the gene promoter directly; rather, the signal could be passed along the chain. Evidence from Hi-C data analysis has suggested that the “first” enhancer in the chain, i.e. the enhancer with direct looping to the gene promoter, is often more distal than the “second” enhancer, attracts more tissue-specific factors and is more enriched for eQTLs than other enhancers within the chain [490]. Consistent with this, recent dissection of an enhancer chain in the INK4a/ARF locus revealed that perturbing any promoter-interacting enhancer within the chain abolished the functionality of the entire network, but deleting the enhancers further down the chain had more moderate effects [491]. Furthermore, deletion or inhibition of any one of the promoter-interacting enhancers caused disruption of H3K27ac and eRNA levels on the other enhancers in the network [491]. Combined, these data suggest that the “first” enhancer is responsible for connecting with the promoter and initiating gene transcription, but also relies on signals from further down the enhancer chain.
Promoter–promoter contacts
3C-derived methods commonly detect multitudes of promoter-promoter contacts, typically in addition to promoter-enhancer contacts of the same genes. In some well-characterised cases, multiple promoters engage in combinations of pairwise interactions with each other, suggestive of promoter-promoter “networks”. These include contacts between the promoters of Hox genes in ESCs (in which they are inactive) that were shown to depend on Polycomb Repressive complex 1 [280]. A similar network of contacts is also detectable between the active promoters of multiple histone genes in many mammalian cell types [243, 279, 471, 492]. These two examples may reflect co-regulatory relationships between the interacting promoters, consistent with the classic “transcription factory” model [425]. In addition, recent evidence that promoter-promoter contacts within nested stripe domains preferentially involve transcriptionally active, rather than inactive, promoters [276], as well as the existence of epromoters (discussed above), suggest that some promoter-promoter contacts likely have a direct cis-regulatory role similarly to enhancer–promoter contacts. This concept has also recently been supported by computational analyses based on Hi-C data [493].
Transient contacts or enhancer hubs?
The subsections immediately above provide an empirical view on the multiplicity of enhancer–promoter and promoter-promoter relationships. How can such configurations be achieved structurally? Whilst 3C-derived data have indicated that interactomes comprising multiple interacting elements may be common in mammalian genomes [360, 488, 494,493,496], these data alone cannot prove the simultaneous association of multiple loci. This is because in their standard form, 3C-derived methods detect pairwise ligation events. Therefore, whilst these findings might represent multi-way “hubs”, they might equally represent a superposition of multiple independent contacts between enhancers and promoters. In order to explore this question further, several techniques have been developed that circumvent the limitations of standard 3C-derived methods.
Genome Architecture Mapping (GAM) is one technique that enables probing multi-way chromosomal contacts [497]. GAM works by laser microdissection of cryopreserved nuclei, followed by DNA sequencing within a given slice, to find all loci that were nearby in 3D space [497]. Another inherently non-pairwise method is Split Pool Recognition of Interactions by Tag Extension (SPRITE), which uses chromatin crosslinking but no proximity ligation [403]. Instead, barcode signatures are created for restriction fragments that are then sequenced and clustered to identify chromatin complexes [403]. While both methods could successfully reveal multi-way contacts across TADs and chromosomes, their resolution currently limits robust detection of individual CRUs. Multi-way relationships can also be inferred using 3C-derived methods, provided that multiple pairwise ligation events are detectable in the same sequencing read. This was achieved with very short (~ 200 bp) restriction fragments (Tri-C [498, 499]), or by using very long (~2 kb) reads generated with Oxford Nanopore seqeuencing (MC-4C [485]). Both technologies, applied in the α- and β- globin loci respectively, provided evidence for higher-order hubs, containing three and more regulatory elements [499, 485]. Finally, advances in super-resolution imaging (reviewed in [500]) now enable the visualisation of multiple interacting loci to the resolution of a few kilobases in single cells [448, 501,500,503]. Hi-M is one such technique that combines visualisation of chromatin interactions and transcription in single cells [501, 504]. Hi-M was recently employed in the developing Drosophila embryo to show that cis-regulatory elements cooperate to form hubs early in development prior to transcription, facilitated by the pioneer factor Zelda [453]. Notably, in addition to multiple enhancers, hubs detected by these techniques sometimes contain multiple promoters, corroborating the evidence for promoter-promoter contacts from the conventional 3C-derived methods [453, 498].
Evidence from live-imaging studies at candidate loci suggests that physical contacts between enhancers and promoters are often highly dynamic [310, 311, 448]. For example, in the β-globin locus, interactions between the LCR and the γ-globin or the β-globin genes can switch very rapidly on the same DNA strand [505]. Nonetheless, the existence of bona fide hubs could be explained by the phenomenon of LLPS, rather than direct looping, whereby various interacting regulatory elements are integrated into phase condensates [453, 506, 507]. These LLPS-mediated hubs could be formed or dissolved by local changes in factor concentrations or other biochemical properties that still allow for a dynamic, adjustable system for gene expression control.
Pioneer and maintenance enhancers
So far we have been considering multiple active enhancers in a unit as generally serving the same role. However, there is a possibility that some enhancers may also help others to associate with promoters, possibly in addition to their direct activity on those same promoters. For example, in the vertebrate Hox cluster, a strong enhancer situated outside of the gene locus could prime the region, allowing for the emergence of novel enhancers [508], leading to the notion of “pioneer enhancers” [509] in an analogy to pioneer TFs. Consistent with this model, it was recently shown that deletion of a relatively weak distal enhancer in the mouse Sox9 sex determination locus leads to sex reversal, while the phenotypic effects of deleting stronger and more proximal Sox9 enhancers are much less pronounced [510]. Analysis of enhancer chains suggests that the promoter-connected enhancer is often more distal than downstream enhancers in the same chain, yet binds more tissue-appropriate TFs, suggesting that enhancer chains might involve pioneer enhancers [490]. One possibility is that pioneer enhancers are required for gene induction, while other enhancers are involved in the maintenance and fine-tuning of gene expression [511, 512].
Theoretically, cohesin/CTCF-associated enhancers are good candidates for pioneer enhancers for a number of reasons, most important of which is the selective requirement of cohesin for inducible gene control [513]. Cohesin-dependent enhancers also tend to be longer-range than their cohesin-independent counterparts [301, 349, 514]. One possibility therefore is that looping orchestrated by cohesin/CTCF-bound pioneer enhancers enables cohesin-independent contacts [349] (Fig. 3), which may be mediated by other factors (such as, for example, LDB1) and mechanisms, including LLPS. Further mechanistic analyses of multi-enhancer control of inducible genes will be needed to validate this model directly.

A model of pioneer and maintenance enhancers. A distal pioneer enhancer, potentially bound by CTCF and/or cohesin, initiates a chromatin loop with the gene promoter and increases accessibility of the locus. Subsequently, more proximal cohesin-independent maintenance enhancers become activated and initiate gene transcription. Blue and red circles denote inactive and active enhancers, respectively
Transcriptional effects of enhancer action
What are the consequences of enhancer action on promoters? Conceptually, the simplest model would suggest that when an enhancer comes into proximity with a compatible promoter, it transmits to it a “dose of activation” that results in a certain level of transcriptional output [107]. (In view of the evidence reviewed in the previous sections, we can assume that this proximity can be achieved by either direct looping or otherwise, and may be potentially facilitated by a pioneer enhancer). The “dose of activation” model, although necessarily crude, fits well with the current understanding of the generally transient contacts between regulatory elements, TFs and cofactors, as well as with the flexibility of enhancer organisation discussed above. This model is also supported by the fact that transcription itself is discontinuous, occurring in “bursts” during which RNAP complexes transcribe one by one from the TSS producing one or more transcripts, followed by a period of transcriptional silence (reviewed in [515]). Evidence from live imaging and single-cell transcriptomics indicates that enhancers increase the frequency of transcriptional bursts without significantly affecting their amplitude and duration [310, 505, 516].
One implication of the “dose of activation” model is that multiple enhancers controlling the same gene are expected to provide largely additive inputs to the promoter. In support of this notion, the number of enhancers correlates with the expression level of a gene across cell types [243, 278, 279, 349]. Likewise, the transcriptional effects of enhancer deletions within extended super-enhancer loci are also consistent with the generally additive effects of individual enhancers [517, 518]. Finally, considerations related to the “dose of activation” view also lie at the foundation of the Activity-by-Contact (ABC) model for estimating the contribution of a given enhancer to a gene’s transcriptional output. In ABC, each enhancer is considered separately and the “dose” of activation it contributes to a promoter depends on its individual activity in cis and the frequency of the respective enhancer–promoter contact [183, 184]. The ABC model showed a good performance in predicting the transcriptional effects of CRISPRi-mediated enhancer perturbations (one-by-one) in dozens of loci in human cell lines, particularly for the more proximal enhancers [183, 184].
However, it has long been known that additivity in enhancer action is not absolute, and some enhancers work together either synergistically or sub-additively. For example, a mixture of additive, synergistic and sub-additive effects was demonstrated in a live imaging analysis of candidate developmental enhancers in Drosophila embryos [519]. Sub-additivity may also underlie some cases of functional enhancer redundancy, such as for Drosophila “shadow enhancers” that can “back up” important developmental enhancers and enable them to accumulate deleterious mutations [520,519,522]. In turn, synergistic effects were observed in mammalian cell fate determination systems, for example, at Fgf5 enhancers during the differentiation of mouse embryonic stem cells to epiblast-like cells (EpiLCs) [523] and at up to 20% enhancers upon transdifferentiation of human leukemia B-cells into macrophages [524].
Some deviations from enhancer additivity could potentially be explained by steric constraints arising from enhancer competition [519] or involvement of “pioneer enhancers” [525], but these considerations may not readily explain other reported cases [523, 526]. Recent insights into the biophysics of enhancer–promoter relationships expand the scope for possible scenarios underpinning these effects. Most importantly, dynamic modelling studies have challenged the direct relationship between enhancer–promoter contacts and transcriptional bursting. Instead, they propose that an enhancer’s contact with a promoter may facilitate the transition of the promoter from a low-activity state to a highly active state [405, 406], which in turn may generate bursts at a higher frequency. Mechanistically, this could be achieved if rather than directly initiating transcriptional bursts, the “doses of activation” provided by enhancers are accumulated at promoters through some intermediate molecular tags (such as histone modifications, transcription factors or RNAP itself), with a certain level of such “tags” required for transition to an active state [406]. This way, promoters may have a “memory” (hysteresis) of enhancer action [405, 406], reviving some early debates on this topic [527]. It also follows from these models that the frequency of enhancer–promoter contacts may have non-linear effects on transcriptional output, consistent with experimental observations [405, 503].
In theory, the functional “decoupling” of enhancer–promoter contacts from transcriptional bursting may accommodate the existence of multi-enhancer hubs and non-looping mechanisms of enhancer–promoter communication. For example, such hubs formed through phase separation may help to cluster RNAP at gene promoters, prior to RNAP pause release that directly results in a transcriptional burst [506, 528, 529]. However, the exact interplay between these phenomena, and the extent to which they are jointly capable of explaining the complexity of gene control by multiple enhancers, remains to be fully understood.
Concluding remarks
Research over the last few decades has provided ample evidence for the key role of enhancers in metazoan gene regulation and the importance of their aberrations in disease. We now also have efficient tools for detection of enhancers and their target genes genome-wide in cell populations and, increasingly, in single cells. Jointly, these advances have improved our understanding of the regulatory logic and molecular mechanisms underpinning enhancer activity in cis and in trans. However, the bulk of evidence of enhancer–promoter communication to date is based on 3C-derived methods that presume physical looping between these elements and necessitate a largely pairwise view on their relationships. Modern technologies gradually make it possible to transcend these limitations, enabling more direct investigations of non-looping mechanisms and multi-enhancer regulatory logic. We are also beginning to expand our conceptual understanding of enhancer types and states beyond a simple “on” and “off”, with emerging notions such as poised and pioneer enhancers. Single cell methods, particularly those based on high-resolution live imaging, combined with powerful genetic and epigenetic perturbation tools and state-of-the-art computational analysis paradigms, have the potential to significantly advance our ability to probe these concepts, paving the way towards comprehensive quantitative models of cis-regulatory gene control.
References
Jacob F, Monod J (1961) Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol 3:318–356
Capecchi MR (1980) High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell 22:479–488
Grosschedl R, Birnstiel ML (1980) Spacer DNA sequences upstream of the T-A-T-A-A-A-T-A sequence are essential for promotion of H2A histone gene transcription in vivo. Proc Natl Acad Sci USA 77:7102–7106
Banerji J, Rusconi S, Schaffner W (1981) Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 27:299–308
Moreau P, Hen R, Wasylyk B et al (1981) The SV40 72 base repair repeat has a striking effect on gene expression both in SV40 and other chimeric recombinants. Nucleic Acids Res 9:6047–6068. https://doi.org/10.1093/nar/9.22.6047
Schaffner W (2015) Enhancers, enhancers—from their discovery to today’s universe of transcription enhancers. Biol Chem 396:311–327
Grosveld F, van Staalduinen J, Stadhouders R (2021) Transcriptional regulation by (super)enhancers: from discovery to mechanisms. Annu Rev Genomics Hum Genet. https://doi.org/10.1146/annurev-genom-122220-093818
Benoist C, Chambon P (1981) In vivo sequence requirements of the SV40 early promoter region. Nature 290:304–310
Gruss P, Dhar R, Khoury G (1981) Simian virus 40 tandem repeated sequences as an element of the early promoter. Proc Natl Acad Sci USA 78:943–947
de Villiers J, Schaffner W (1981) A small segment of polyoma virus DNA enhances the expression of a cloned beta-globin gene over a distance of 1400 base pairs. Nucleic Acids Res 9:6251–6264
Weber F, de Villiers J, Schaffner W (1984) An SV40 “enhancer trap” incorporates exogenous enhancers or generates enhancers from its own sequences. Cell 36:983–992
Boshart M, Weber F, Jahn G et al (1985) A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell 41:521–530
Struhl K (1984) Genetic properties and chromatin structure of the yeast gal regulatory element: an enhancer-like sequence. Proc Natl Acad Sci USA 81:7865–7869
Shepherd B, Garabedian MJ, Hung MC, Wensink PC (1985) Developmental control of Drosophila yolk protein 1 gene by cis-acting DNA elements. Cold Spring Harb Symp Quant Biol 50:521–526
Banerji J, Olson L, Schaffner W (1983) A lymphocyte-specific cellular enhancer is located downstream of the joining region in immunoglobulin heavy chain genes. Cell 33:729–740
Gillies SD, Morrison SL, Oi VT, Tonegawa S (1983) A tissue-specific transcription enhancer element is located in the major intron of a rearranged immunoglobulin heavy chain gene. Cell 33:717–728
Mercola M, Wang XF, Olsen J, Calame K (1983) Transcriptional enhancer elements in the mouse immunoglobulin heavy chain locus. Science 221:663–665
Hoover TR, Santero E, Porter S, Kustu S (1990) The integration host factor stimulates interaction of RNA polymerase with NIFA, the transcriptional activator for nitrogen fixation operons. Cell 63:11–22
Feng J, Goss TJ, Bender RA, Ninfa AJ (1995) Repression of the Klebsiella aerogenes nac promoter. J Bacteriol 177:5535–5538
Xu H, Hoover TR (2001) Transcriptional regulation at a distance in bacteria. Curr Opin Microbiol 4:138–144
Reitzer LJ, Magasanik B (1986) Transcription of glnA in E. coli is stimulated by activator bound to sites far from the promoter. Cell 45:785–792
Ninfa AJ, Reitzer LJ, Magasanik B (1987) Initiation of transcription at the bacterial glnAp2 promoter by purified E. coli components is facilitated by enhancers. Cell 50:1039–1046
Hiromi Y, Kuroiwa A, Gehring WJ (1985) Control elements of the Drosophila segmentation gene fushi tarazu. Cell 43:603–613
Hiromi Y, Gehring WJ (1987) Regulation and function of the Drosophila segmentation gene fushi tarazu. Cell 50:963–974
Kioussis D, Vanin E, deLange T et al (1983) Beta-globin gene inactivation by DNA translocation in gamma beta-thalassaemia. Nature 306:662–666
Driscoll MC, Dobkin CS, Alter BP (1989) Gamma delta beta-thalassemia due to a de novo mutation deleting the 5’ beta-globin gene activation-region hypersensitive sites. Proc Natl Acad Sci USA 86:7470–7474
Lettice LA, Heaney SJH, Purdie LA et al (2003) A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet 12:1725–1735
Sharpe J, Lettice L, Hecksher-Sørensen J et al (1999) Identification of Sonic hedgehog as a candidate gene responsible for the polydactylous mouse mutant Sasquatch. Curr Biol 9:97-S1
Long HK, Prescott SL, Wysocka J (2016) Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell 167:1170–1187
Spitz F, Furlong EEM (2012) Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13:613–626
Zaret KS, Carroll JS (2011) Pioneer transcription factors: establishing competence for gene expression. Genes Dev 25:2227–2241
Larson ED, Marsh AJ, Harrison MM (2021) Pioneering the developmental frontier. Mol Cell 81:1640–1650
Liang H-L, Nien C-Y, Liu H-Y et al (2008) The zinc-finger protein Zelda is a key activator of the early zygotic genome in Drosophila. Nature 456:400–403
Harrison MM, Li X-Y, Kaplan T et al (2011) Zelda binding in the early Drosophila melanogaster embryo marks regions subsequently activated at the maternal-to-zygotic transition. PLoS Genet 7:e1002266
Schulz KN, Bondra ER, Moshe A et al (2015) Zelda is differentially required for chromatin accessibility, transcription factor binding, and gene expression in the early Drosophila embryo. Genome Res 25:1715–1726
Boyer LA, Lee TI, Cole MF et al (2005) Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122:947–956
Iwafuchi-Doi M (2019) The mechanistic basis for chromatin regulation by pioneer transcription factors. Wiley Interdiscip Rev Syst Biol Med 11:e1427
Iwafuchi-Doi M, Donahue G, Kakumanu A et al (2016) The pioneer transcription factor FoxA maintains an accessible nucleosome configuration at enhancers for tissue-specific gene activation. Mol Cell 62:79–91
Echigoya K, Koyama M, Negishi L et al (2020) Nucleosome binding by the pioneer transcription factor OCT4. Sci Rep 10:11832
Dodonova SO, Zhu F, Dienemann C et al (2020) Nucleosome-bound SOX2 and SOX11 structures elucidate pioneer factor function. Nature 580:669–672
Jacobs J, Atkins M, Davie K et al (2018) The transcription factor Grainy head primes epithelial enhancers for spatiotemporal activation by displacing nucleosomes. Nat Genet 50:1011–1020
Ding J, Xu H, Faiola F et al (2012) Oct4 links multiple epigenetic pathways to the pluripotency network. Cell Res 22:155–167
Pardo M, Lang B, Yu L et al (2010) An expanded Oct4 interaction network: implications for stem cell biology, development, and disease. Cell Stem Cell 6:382–395
van den Berg DLC, Snoek T, Mullin NP et al (2010) An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell 6:369–381
King HW, Klose RJ (2017) The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. Elife 6:e22631
Bao X, Rubin AJ, Qu K et al (2015) A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome Biol 16:284
Engelen E, Akinci U, Bryne JC et al (2011) Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat Genet 43:607–611
Hu G, Schones DE, Cui K et al (2011) Regulation of nucleosome landscape and transcription factor targeting at tissue-specific enhancers by BRG1. Genome Res 21:1650–1658
Takaku M, Grimm SA, Shimbo T et al (2016) GATA3-dependent cellular reprogramming requires activation-domain dependent recruitment of a chromatin remodeler. Genome Biol 17:36
Vierbuchen T, Ling E, Cowley CJ et al (2017) AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol Cell 68:1067-1082.e12
Mir M, Reimer A, Haines JE et al (2017) Dense Bicoid hubs accentuate binding along the morphogen gradient. Genes Dev 31:1784–1794
Mir M, Stadler MR, Ortiz SA et al (2018) Dynamic multifactor hubs interact transiently with sites of active transcription in embryos. Elife 7:e40497
Dufourt J, Trullo A, Hunter J et al (2018) Temporal control of gene expression by the pioneer factor Zelda through transient interactions in hubs. Nat Commun 9:5194
Yamada S, Whitney PH, Huang S-K et al (2019) The drosophila pioneer factor zelda modulates the nuclear microenvironment of a dorsal target enhancer to potentiate transcriptional output. Curr Biol 29:1387-1393.e5
Smith RP, Taher L, Patwardhan RP et al (2013) Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat Genet 45:1021–1028
Zabidi MA, Stark A (2016) Regulatory enhancer-core-promoter communication via transcription factors and cofactors. Trends Genet 32:801–814
Winkler DD, Luger K (2011) The histone chaperone FACT: structural insights and mechanisms for nucleosome reorganization. J Biol Chem 286:18369–18374
Tolstorukov MY, Sansam CG, Lu P et al (2013) Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc Natl Acad Sci USA 110:10165–10170
Chan HM, Shikama N, La Thangue NB (2001) Control of gene expression and the cell cycle. Essays Biochem 37:87–96
Jin Q, Yu L-R, Wang L et al (2011) Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J 30:249–262
Raisner R, Kharbanda S, Jin L et al (2018) Enhancer activity requires CBP/P300 bromodomain-dependent histone H3K27 acetylation. Cell Rep 24:1722–1729
Alver BH, Kim KH, Lu P et al (2017) The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat Commun 8:14648
Narita T, Ito S, Higashijima Y et al (2021) Enhancers are activated by p300/CBP activity-dependent PIC assembly, RNAPII recruitment, and pause release. Mol Cell 81:2166-2182.e6
Chan HM, La Thangue NB (2001) p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci 114:2363–2373
Kim TK, Kim TH, Maniatis T (1998) Efficient recruitment of TFIIB and CBP-RNA polymerase II holoenzyme by an interferon-beta enhanceosome in vitro. Proc Natl Acad Sci USA 95:12191–12196
Allen BL, Taatjes DJ (2015) The mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol 16:155–166
Flanagan PM, Kelleher RJ, Sayre MH et al (1991) A mediator required for activation of RNA polymerase II transcription in vitro. Nature 350:436–438
Kelleher RJ, Flanagan PM, Kornberg RD (1990) A novel mediator between activator proteins and the RNA polymerase II transcription apparatus. Cell 61:1209–1215
Crump NT, Ballabio E, Godfrey L et al (2021) BET inhibition disrupts transcription but retains enhancer–promoter contact. Nat Commun 12:223
Chapuy B, McKeown MR, Lin CY et al (2013) Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24:777–790
Di Micco R, Fontanals-Cirera B, Low V et al (2014) Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep 9:234–247
Whyte WA, Orlando DA, Hnisz D et al (2013) Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153:307–319
Core LJ, Martins AL, Danko CG et al (2014) Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet 46:1311–1320
Andersson R, Gebhard C, Miguel-Escalada I et al (2014) An atlas of active enhancers across human cell types and tissues. Nature 507:455–461
Lam MTY, Cho H, Lesch HP et al (2013) Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 498:511–515
De Santa F, Barozzi I, Mietton F et al (2010) A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol 8:e1000384
Kim T-K, Hemberg M, Gray JM et al (2010) Widespread transcription at neuronal activity-regulated enhancers. Nature 465:182–187
Struhl K (2007) Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat Struct Mol Biol 14:103–105
Liang J, Zhou H, Gerdt C et al (2016) Epstein-Barr virus super-enhancer eRNAs are essential for MYC oncogene expression and lymphoblast proliferation. Proc Natl Acad Sci USA 113:14121–14126
Banerjee AR, Kim YJ, Kim TH (2014) A novel virus-inducible enhancer of the interferon-β gene with tightly linked promoter and enhancer activities. Nucleic Acids Res 42:12537–12554
Hsieh C-L, Fei T, Chen Y et al (2014) Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc Natl Acad Sci USA 111:7319–7324
Tsai P-F, Dell’Orso S, Rodriguez J et al (2018) A muscle-specific enhancer RNA mediates cohesin recruitment and regulates transcription in trans. Mol Cell 71:129-141.e8
Mousavi K, Zare H, Dell’orso S et al (2013) eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol Cell 51:606–617
Kanno T, Kanno Y, LeRoy G et al (2014) BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat Struct Mol Biol 21:1047–1057
Rahnamoun H, Lee J, Sun Z et al (2018) RNAs interact with BRD4 to promote enhanced chromatin engagement and transcription activation. Nat Struct Mol Biol 25:687–697
Arnold PR, Wells AD, Li XC (2019) Diversity and emerging roles of enhancer RNA in regulation of gene expression and cell fate. Front Cell Dev Biol 7:377
Métivier R, Penot G, Hübner MR et al (2003) Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115:751–763
Thanos D, Maniatis T (1995) Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 83:1091–1100
Panne D, Maniatis T, Harrison SC (2007) An atomic model of the interferon-beta enhanceosome. Cell 129:1111–1123
Kulkarni MM, Arnosti DN (2003) Information display by transcriptional enhancers. Development 130:6569–6575
Junion G, Spivakov M, Girardot C et al (2012) A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell 148:473–486
Arnosti DN, Kulkarni MM (2005) Transcriptional enhancers: Intelligent enhanceosomes or flexible billboards? J Cell Biochem 94:890–898
Rastegar S, Hess I, Dickmeis T et al (2008) The words of the regulatory code are arranged in a variable manner in highly conserved enhancers. Dev Biol 318:366–377
Uhl JD, Zandvakili A, Gebelein B (2016) A Hox transcription factor collective binds a highly conserved distal-less cis-regulatory module to generate robust transcriptional outcomes. PLoS Genet 12:e1005981
Lloret-Fernández C, Maicas M, Mora-Martínez C et al (2018) A transcription factor collective defines the HSN serotonergic neuron regulatory landscape. Elife 7:e32785
Crocker J, Ilsley GR, Stern DL (2016) Quantitatively predictable control of Drosophila transcriptional enhancers in vivo with engineered transcription factors. Nat Genet 48:292–298
Chen L, Capra JA (2020) Learning and interpreting the gene regulatory grammar in a deep learning framework. PLoS Comput Biol 16:e1008334
Avsec Ž, Weilert M, Shrikumar A et al (2021) Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat Genet 53:354–366
Villar D, Berthelot C, Aldridge S et al (2015) Enhancer evolution across 20 mammalian species. Cell 160:554–566
Carelli FN, Liechti A, Halbert J et al (2018) Repurposing of promoters and enhancers during mammalian evolution. Nat Commun 9:4066
Wong ES, Zheng D, Tan SZ et al (2020) Deep conservation of the enhancer regulatory code in animals. Science 370:eaax8137
Hare EE, Peterson BK, Iyer VN et al (2008) Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet 4:e1000106
Schmidt D, Wilson MD, Ballester B et al (2010) Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328:1036–1040
Dowell RD (2010) Transcription factor binding variation in the evolution of gene regulation. Trends Genet 26:468–475
Habib N, Wapinski I, Margalit H et al (2012) A functional selection model explains evolutionary robustness despite plasticity in regulatory networks. Mol Syst Biol 8:619
Arnold CD, Gerlach D, Spies D et al (2014) Quantitative genome-wide enhancer activity maps for five Drosophila species show functional enhancer conservation and turnover during cis-regulatory evolution. Nat Genet 46:685–692
Spivakov M (2014) Spurious transcription factor binding: non-functional or genetically redundant? BioEssays 36:798–806
Borok MJ, Tran DA, Ho MCW, Drewell RA (2010) Dissecting the regulatory switches of development: lessons from enhancer evolution in Drosophila. Development 137:5–13
Khoueiry P, Girardot C, Ciglar L et al (2017) Uncoupling evolutionary changes in DNA sequence, transcription factor occupancy and enhancer activity. Elife 6:e28440
Yang S, Oksenberg N, Takayama S et al (2015) Functionally conserved enhancers with divergent sequences in distant vertebrates. BMC Genomics 16:882
Hörnblad A, Bastide S, Langenfeld K et al (2021) Dissection of the Fgf8 regulatory landscape by in vivo CRISPR-editing reveals extensive intra- and inter-enhancer redundancy. Nat Commun 12:439
Snetkova V, Ypsilanti AR, Akiyama JA et al (2021) Ultraconserved enhancer function does not require perfect sequence conservation. Nat Genet 53:521–528
Wilczyński B, Furlong EEM (2010) Dynamic CRM occupancy reflects a temporal map of developmental progression. Mol Syst Biol 6:383
Voss TC, Hager GL (2014) Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet 15:69–81
Chen J, Zhang Z, Li L et al (2014) Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156:1274–1285
Krebs AR, Imanci D, Hoerner L et al (2017) Genome-wide single-molecule footprinting reveals high RNA polymerase II turnover at paused promoters. Mol Cell 67:411–422.e4
Stergachis AB, Debo BM, Haugen E et al (2020) Single-molecule regulatory architectures captured by chromatin fiber sequencing. Science 368:1449–1454
Rao S, Ahmad K, Ramachandran S (2021) Cooperative binding between distant transcription factors is a hallmark of active enhancers. Mol Cell 81:1651-1665.e4
Sönmezer C, Kleinendorst R, Imanci D et al (2021) Molecular co-occupancy identifies transcription factor binding cooperativity in vivo. Mol Cell 81:255-267.e6
Macdonald PM, Ingham P, Struhl G (1986) Isolation, structure, and expression of even-skipped: a second pair-rule gene of Drosophila containing a homeo box. Cell 47:721–734
Stanojevic D, Small S, Levine M (1991) Regulation of a segmentation stripe by overlapping activators and repressors in the Drosophila embryo. Science 254:1385–1387
Goto T, Macdonald P, Maniatis T (1989) Early and late periodic patterns of even skipped expression are controlled by distinct regulatory elements that respond to different spatial cues. Cell 57:413–422
Small S, Kraut R, Hoey T et al (1991) Transcriptional regulation of a pair-rule stripe in Drosophila. Genes Dev 5:827–839
Arnosti DN, Barolo S, Levine M, Small S (1996) The eve stripe 2 enhancer employs multiple modes of transcriptional synergy. Development 122:205–214
Farley EK, Olson KM, Zhang W et al (2015) Suboptimization of developmental enhancers. Science 350:325–328
Crocker J, Abe N, Rinaldi L et al (2015) Low affinity binding site clusters confer hox specificity and regulatory robustness. Cell 160:191–203
Datta RR, Ling J, Kurland J et al (2018) A feed-forward relay integrates the regulatory activities of Bicoid and Orthodenticle via sequential binding to suboptimal sites. Genes Dev 32:723–736
Keller SH, Jena SG, Yamazaki Y, Lim B (2020) Regulation of spatiotemporal limits of developmental gene expression via enhancer grammar. Proc Natl Acad Sci USA 117:15096–15103
Corces MR, Granja JM, Shams S et al (2018) The chromatin accessibility landscape of primary human cancers. Science 362:eaav1898
Meuleman W, Muratov A, Rynes E et al (2020) Index and biological spectrum of human DNase I hypersensitive sites. Nature 584:244–251
ENCODE Project Consortium, Moore JE, Purcaro MJ et al (2020) Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583:699–710
Parelho V, Hadjur S, Spivakov M et al (2008) Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 132:422–433
Creyghton MP, Cheng AW, Welstead GG et al (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA 107:21931–21936
Ernst J, Kheradpour P, Mikkelsen TS et al (2011) Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473:43–49
Rada-Iglesias A, Bajpai R, Swigut T et al (2011) A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470:279–283
Zentner GE, Tesar PJ, Scacheri PC (2011) Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res 21:1273–1283
Bonn S, Zinzen RP, Girardot C et al (2012) Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat Genet 44:148–156
Koenecke N, Johnston J, Gaertner B et al (2016) Genome-wide identification of Drosophila dorso-ventral enhancers by differential histone acetylation analysis. Genome Biol 17:196
Ngan CY, Wong CH, Tjong H et al (2020) Chromatin interaction analyses elucidate the roles of PRC2-bound silencers in mouse development. Nat Genet 52:264–272
Gisselbrecht SS, Palagi A, Kurland JV et al (2020) Transcriptional silencers in drosophila serve a dual role as transcriptional enhancers in alternate cellular contexts. Mol Cell 77:324-337.e8
Freire-Pritchett P, Schoenfelder S, Várnai C et al (2017) Global reorganisation of regulatory units upon lineage commitment of human embryonic stem cells. Elife 6:e21926
Crispatzu G, Rehimi R, Pachano T et al (2021) The chromatin, topological and regulatory properties of pluripotency-associated poised enhancers are conserved in vivo. Nat Commun 12(1):4344
Cruz-Molina S, Respuela P, Tebartz C et al (2017) PRC2 facilitates the regulatory topology required for poised enhancer function during pluripotent stem cell differentiation. Cell Stem Cell 20:689-705.e9
Spivakov M, Fisher AG (2007) Epigenetic signatures of stem-cell identity. Nat Rev Genet 8:263–271
Zeitlinger J, Stark A, Kellis M et al (2007) RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat Genet 39:1512–1516
Visel A, Blow MJ, Li Z et al (2009) ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457:854–858
Blow MJ, McCulley DJ, Li Z et al (2010) ChIP-Seq identification of weakly conserved heart enhancers. Nat Genet 42:806–810
May D, Blow MJ, Kaplan T et al (2011) Large-scale discovery of enhancers from human heart tissue. Nat Genet 44:89–93
ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74
Bernstein BE, Stamatoyannopoulos JA, Costello JF et al (2010) The NIH roadmap epigenomics mapping consortium. Nat Biotechnol 28:1045–1048
Martens JHA, Stunnenberg HG (2013) BLUEPRINT: mapping human blood cell epigenomes. Haematologica 98:1487–1489
Gerstein MB, Lu ZJ, Van Nostrand EL et al (2010) Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330:1775–1787
modENCODE Consortium, Roy S, Ernst J et al (2010) Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330:1787–1797
Boix CA, James BT, Park YP, Meuleman W, Kellis M (2021) Regulatory genomic circuitry of human disease loci by integrative epigenomics. Nature 590(7845):300–307
Skene PJ, Henikoff S (2017) An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6:e21856
Kaya-Okur HS, Wu SJ, Codomo CA et al (2019) CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10:1930
Bartosovic M, Kabbe M, Castelo-Branco G (2021) Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat Biotechnol. https://doi.org/10.1038/s41587-021-00869-9
Hainer SJ, Bošković A, McCannell KN et al (2019) Profiling of pluripotency factors in single cells and early embryos. Cell 177:1319-1329.e11
Ernst J, Kellis M (2012) ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 9:215–216
Hoffman MM, Buske OJ, Wang J et al (2012) Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat Methods 9:473–476
Marco E, Meuleman W, Huang J et al (2017) Multi-scale chromatin state annotation using a hierarchical hidden Markov model. Nat Commun 8:15011
Sethi A, Gu M, Gumusgoz E et al (2020) Supervised enhancer prediction with epigenetic pattern recognition and targeted validation. Nat Methods 17:807–814
Pradeepa MM, Grimes GR, Kumar Y et al (2016) Histone H3 globular domain acetylation identifies a new class of enhancers. Nat Genet 48:681–686
Zhang T, Zhang Z, Dong Q et al (2020) Histone H3K27 acetylation is dispensable for enhancer activity in mouse embryonic stem cells. Genome Biol 21:45
Shiraki T, Kondo S, Katayama S et al (2003) Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc Natl Acad Sci USA 100:15776–15781
Cvetesic N, Leitch HG, Borkowska M et al (2018) SLIC-CAGE: high-resolution transcription start site mapping using nanogram-levels of total RNA. Genome Res 28:1943–1956
Kouno T, Moody J, Kwon AT-J et al (2019) C1 CAGE detects transcription start sites and enhancer activity at single-cell resolution. Nat Commun 10:360
Patwardhan RP, Hiatt JB, Witten DM et al (2012) Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol 30:265–270
Arnold CD, Gerlach D, Stelzer C et al (2013) Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339:1074–1077
Kheradpour P, Ernst J, Melnikov A et al (2013) Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res 23:800–811
Gisselbrecht SS, Barrera LA, Porsch M et al (2013) Highly parallel assays of tissue-specific enhancers in whole Drosophila embryos. Nat Methods 10:774–780
Yáñez-Cuna JO, Arnold CD, Stampfel G et al (2014) Dissection of thousands of cell type-specific enhancers identifies dinucleotide repeat motifs as general enhancer features. Genome Res 24:1147–1156
Tewhey R, Kotliar D, Park DS et al (2018) Direct Identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell 172:1132–1134
Kwasnieski JC, Fiore C, Chaudhari HG, Cohen BA (2014) High-throughput functional testing of ENCODE segmentation predictions. Genome Res 24:1595–1602
Nguyen TA, Jones RD, Snavely AR et al (2016) High-throughput functional comparison of promoter and enhancer activities. Genome Res 26:1023–1033
Gilbert LA, Larson MH, Morsut L et al (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154:442–451
Hilton IB, D’Ippolito AM, Vockley CM et al (2015) Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33:510–517
Qi LS, Larson MH, Gilbert LA et al (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183
Li K, Liu Y, Cao H et al (2020) Interrogation of enhancer function by enhancer-targeting CRISPR epigenetic editing. Nat Commun 11:485
Adamson B, Norman TM, Jost M et al (2016) A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167:1867-1882.e21
Xie S, Duan J, Li B et al (2017) Multiplexed engineering and analysis of combinatorial enhancer activity in single cells. Mol Cell 66:285-299.e5
Pierce SE, Granja JM, Greenleaf WJ (2021) High-throughput single-cell chromatin accessibility CRISPR screens enable unbiased identification of regulatory networks in cancer. Nat Commun 12(1):2969
Fulco CP, Nasser J, Jones TR et al (2019) Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat Genet 51:1664–1669
Nasser J, Bergman DT, Fulco CP et al (2021) Genome-wide enhancer maps link risk variants to disease genes. Nature. https://doi.org/10.1038/s41586-021-03446-x
Dixit A, Parnas O, Li B et al (2016) Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167:1853-1866.e17
Jaitin DA, Weiner A, Yofe I et al (2016) Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-Seq. Cell 167:1883-1896.e15
Datlinger P, Rendeiro AF, Schmidl C et al (2017) Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods 14:297–301
Ting PY, Parker AE, Scott Lee J et al (2018) Guide Swap enables genome-scale pooled CRISPR–Cas9 screening in human primary cells. Nat Methods 15:941–946
Li L, Gao Y, Srivastava R et al (2020) Lentiviral delivery of combinatorial CAR/CRISPRi circuit into human primary T cells is enhanced by TBK1/IKKɛ complex inhibitor BX795. J Transl Med 18:1–12
Seki A, Rutz S (2018) Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Med 215:985–997
Spivakov M, Akhtar J, Kheradpour P et al (2012) Analysis of variation at transcription factor binding sites in Drosophila and humans. Genome Biol 13:R49
Heinz S, Romanoski CE, Benner C et al (2013) Effect of natural genetic variation on enhancer selection and function. Nature 503:487–492
Gotea V, Visel A, Westlund JM et al (2010) Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers. Genome Res 20:565–577
Shimell M, Simon J, Bender W, O’Connor M (1994) Enhancer point mutation results in a homeotic transformation in Drosophila. Science 264:968–971
Fu W, Duan H, Frei E, Noll M (1998) Shaven and sparkling are mutations in separate enhancers of the Drosophila Pax2 homolog. Development 125:2943–2950
Bhatia S, Bengani H, Fish M et al (2013) Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia. Am J Hum Genet 93:1126–1134
Wieczorek D, Pawlik B, Li Y et al (2010) A specific mutation in the distant sonic hedgehog (SHH) cis-regulator (ZRS) causes Werner mesomelic syndrome (WMS) while complete ZRS duplications underlie Haas type polysyndactyly and preaxial polydactyly (PPD) with or without triphalangeal thumb. Hum Mutat 31:81–89
Anderson C, Williams VC, Moyon B et al (2012) Sonic hedgehog acts cell-autonomously on muscle precursor cells to generate limb muscle diversity. Genes Dev 26:2103–2117
Lettice LA, Williamson I, Wiltshire JH et al (2012) Opposing functions of the ETS factor family define Shh spatial expression in limb buds and underlie polydactyly. Dev Cell 22:459–467
Lettice LA, Devenney P, De Angelis C, Hill RE (2017) The conserved sonic hedgehog limb enhancer consists of discrete functional elements that regulate precise spatial expression. Cell Rep 20:1396–1408
Smemo S, Campos LC, Moskowitz IP et al (2012) Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum Mol Genet 21:3255–3263
Gabbay M, Ellard S, De Franco E, Moisés RS (2017) Pancreatic agenesis due to compound heterozygosity for a novel enhancer and truncating mutation in the PTF1A gene. J Clin Res Pediatr Endocrinol 9:274–277
Fakhouri WD, Rahimov F, Attanasio C et al (2014) An etiologic regulatory mutation in IRF6 with loss- and gain-of-function effects. Hum Mol Genet 23:2711–2720
Wen X, Luca F, Pique-Regi R (2015) Cross-population joint analysis of eQTLs: fine mapping and functional annotation. PLoS Genet 11:e1005176
Kumar S, Ambrosini G, Bucher P (2017) SNP2TFBS—a database of regulatory SNPs affecting predicted transcription factor binding site affinity. Nucleic Acids Res 45:D139–D144
Shi W, Fornes O, Wasserman WW (2019) Gene expression models based on transcription factor binding events confer insight into functional cis-regulatory variants. Bioinformatics 35:2610–2617
Mitchelmore J, Grinberg NF, Wallace C, Spivakov M (2020) Functional effects of variation in transcription factor binding highlight long-range gene regulation by epromoters. Nucleic Acids Res 48:2866–2879
Corradin O, Scacheri PC (2014) Enhancer variants: evaluating functions in common disease. Genome Med 6:85
Maurano MT, Humbert R, Rynes E et al (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science 337:1190–1195
Smemo S, Tena JJ, Kim K-H et al (2014) Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507:371–375
Adrianto I, Wen F, Templeton A et al (2011) Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet 43:253–258
Wang S, Wen F, Wiley GB et al (2013) An enhancer element harboring variants associated with systemic lupus erythematosus engages the TNFAIP3 promoter to influence A20 expression. PLoS Genet 9:e1003750
Waszak SM, Delaneau O, Gschwind AR et al (2015) Population variation and genetic control of modular chromatin architecture in humans. Cell 162:1039–1050
Tehranchi AK, Myrthil M, Martin T et al (2016) Pooled ChIP-Seq links variation in transcription factor binding to complex disease risk. Cell 165:730–741
Watt S, Vasquez L, Walter K et al (2021) Genetic perturbation of PU.1 binding and chromatin looping at neutrophil enhancers associates with autoimmune disease. Nat Commun 12:2298
Degner JF, Pai AA, Pique-Regi R et al (2012) DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 482:390–394
Gate RE, Cheng CS, Aiden AP et al (2018) Genetic determinants of co-accessible chromatin regions in activated T cells across humans. Nat Genet 50:1140–1150
Kumasaka N, Knights AJ, Gaffney DJ (2019) High-resolution genetic mapping of putative causal interactions between regions of open chromatin. Nat Genet 51:128–137
Tehranchi A, Hie B, Dacre M, et al (2019) Fine-mapping-regulatory variants in diverse human populations. Elife 8:e39595
Grubert F, Zaugg JB, Kasowski M et al (2015) Genetic control of chromatin states in humans involves local and distal chromosomal interactions. Cell 162:1051–1065
Chen L, Ge B, Casale FP et al (2016) Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 167:1398-1414.e24
Banovich NE, Lan X, McVicker G et al (2014) Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet 10:e1004663
McVicker G, van de Geijn B, Degner JF et al (2013) Identification of genetic variants that affect histone modifications in human cells. Science 342:747–749
Greenwald WW, Li H, Benaglio P et al (2019) Subtle changes in chromatin loop contact propensity are associated with differential gene regulation and expression. Nat Commun 10:1054
Gorkin DU, Qiu Y, Hu M et al (2019) Common DNA sequence variation influences 3-dimensional conformation of the human genome. Genome Biol 20:255
Gibson G, Reed LK (2008) Cryptic genetic variation. Curr Biol 18:R989–R990
Paaby AB, Rockman MV (2014) Cryptic genetic variation: evolution’s hidden substrate. Nat Rev Genet 15:247–258
Fairfax BP, Humburg P, Makino S et al (2014) Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343:1246949
Burren OS, Rubio García A, Javierre B-M et al (2017) Chromosome contacts in activated T cells identify autoimmune disease candidate genes. Genome Biol 18:165
Knowles DA, Burrows CK, Blischak JD et al (2018) Determining the genetic basis of anthracycline-cardiotoxicity by molecular response QTL mapping in induced cardiomyocytes. Elife 7:e33480
Kang HM, Subramaniam M, Targ S et al (2018) Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol 36:89–94
Huang Y, McCarthy DJ, Stegle O (2019) Vireo: Bayesian demultiplexing of pooled single-cell RNA-seq data without genotype reference. Genome Biol 20:273
Andersson R, Sandelin A (2020) Determinants of enhancer and promoter activities of regulatory elements. Nat Rev Genet 21:71–87
Pekowska A, Benoukraf T, Zacarias-Cabeza J et al (2011) H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J 30:4198–4210
Henriques T, Scruggs BS, Inouye MO et al (2018) Widespread transcriptional pausing and elongation control at enhancers. Genes Dev 32:26–41
Andersson R, Refsing Andersen P, Valen E et al (2014) Nuclear stability and transcriptional directionality separate functionally distinct RNA species. Nat Commun 5:5336
Chen Y, Pai AA, Herudek J et al (2016) Principles for RNA metabolism and alternative transcription initiation within closely spaced promoters. Nat Genet 48:984–994
Kowalczyk MS, Hughes JR, Garrick D et al (2012) Intragenic enhancers act as alternative promoters. Mol Cell 45:447–458
Li G, Ruan X, Auerbach RK et al (2012) Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148:84–98
Dao LTM, Galindo-Albarrán AO, Castro-Mondragon JA et al (2017) Genome-wide characterization of mammalian promoters with distal enhancer functions. Nat Genet 49:1073–1081
Engreitz JM, Haines JE, Perez EM et al (2016) Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539:452–455
Diao Y, Fang R, Li B et al (2017) A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat Methods 14:629–635
Javierre BM, Burren OS, Wilder SP et al (2016) Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell 167:1369-1384.e19
Mumbach MR, Satpathy AT, Boyle EA et al (2017) Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat Genet 49:1602–1612
Chandra V, Bhattacharyya S, Schmiedel BJ et al (2021) Promoter-interacting expression quantitative trait loci are enriched for functional genetic variants. Nat Genet 53:110–119
Dao LTM, Spicuglia S (2018) Transcriptional regulation by promoters with enhancer function. Transcription 9:307–314
Zabidi MA, Arnold CD, Schernhuber K et al (2015) Enhancer–core-promoter specificity separates developmental and housekeeping gene regulation. Nature 518:556–559
Mikhaylichenko O, Bondarenko V, Harnett D et al (2018) The degree of enhancer or promoter activity is reflected by the levels and directionality of eRNA transcription. Genes Dev 32:42–57
Pachano T, Sánchez-Gaya V, Ealo T et al (2021) Orphan CpG islands amplify poised enhancer regulatory activity and determine target gene responsiveness. Nat Genet. https://doi.org/10.1038/s41588-021-00888-x
Scourzic L, Salataj E, Apostolou E (2021) Deciphering the complexity of 3D chromatin organization driving lymphopoiesis and lymphoid malignancies. Front Immunol 12:669881
Fernando N, Sciumè G, O’Shea JJ, Shih H-Y (2021) Multi-dimensional gene regulation in innate and adaptive lymphocytes: a view from regulomes. Front Immunol 12:655590
Yuan X, Scott IC, Wilson MD (2021) Heart enhancers: development and disease control at a distance. Front Genet 12:642975
Richart L, Bidard F-C, Margueron R (2021) Enhancer rewiring in tumors: an opportunity for therapeutic intervention. Oncogene 40:3475–3491
Tuan D, Solomon W, Li Q, London IM (1985) The “beta-like-globin” gene domain in human erythroid cells. Proc Natl Acad Sci USA 82:6384–6388
Reik A, Telling A, Zitnik G et al (1998) The locus control region is necessary for gene expression in the human beta-globin locus but not the maintenance of an open chromatin structure in erythroid cells. Mol Cell Biol 18:5992–6000
Bender MA, Bulger M, Close J, Groudine M (2000) Beta-globin gene switching and DNase I sensitivity of the endogenous beta-globin locus in mice do not require the locus control region. Mol Cell 5:387–393
Bungert J, Davé U, Lim KC et al (1995) Synergistic regulation of human beta-globin gene switching by locus control region elements HS3 and HS4. Genes Dev 9:3083–3096
Wijgerde M, Grosveld F, Fraser P (1995) Transcription complex stability and chromatin dynamics in vivo. Nature 377:209–213
Bulger M, Groudine M (1999) Looping versus linking: toward a model for long-distance gene activation. Genes Dev 13:2465–2477
Dorsett D (1999) Distant liaisons: long-range enhancer–promoter interactions in Drosophila. Curr Opin Genet Dev 9:505–514
Engel JD, Tanimoto K (2000) Looping, linking, and chromatin activity: new insights into beta-globin locus regulation. Cell 100:499–502
Ptashne M (1986) Gene regulation by proteins acting nearby and at a distance. Nature 322:697–701
West AG, Gaszner M, Felsenfeld G (2002) Insulators: many functions, many mechanisms. Genes Dev 16:271–288
Dillon N, Sabbattini P (2000) Functional gene expression domains: defining the functional unit of eukaryotic gene regulation. BioEssays 22:657–665
Carter D, Chakalova L, Osborne CS et al (2002) Long-range chromatin regulatory interactions in vivo. Nat Genet 32:623–626
Tolhuis B, Palstra R-J, Splinter E et al (2002) Looping and Interaction between hypersensitive sites in the active β-globin locus. Mol Cell 10:1453–1465
Dekker J, Rippe K, Dekker M, Kleckner N (2002) Capturing chromosome conformation. Science 295:1306–1311
Palstra R-J, Tolhuis B, Splinter E et al (2003) The beta-globin nuclear compartment in development and erythroid differentiation. Nat Genet 35:190–194
Simonis M, Klous P, Splinter E et al (2006) Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture–on-chip (4C). Nat Genet 38:1348–1354
Zhao Z, Tavoosidana G, Sjölinder M et al (2006) Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet 38:1341–1347
Dostie J, Richmond TA, Arnaout RA et al (2006) Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res 16:1299–1309
Lieberman-Aiden E, van Berkum NL, Williams L et al (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326:289–293
Hsieh T-HS, Weiner A, Lajoie B et al (2015) Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell 162:108–119
Hsieh T-HS, Fudenberg G, Goloborodko A, Rando OJ (2016) Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nat Methods 13:1009–1011
Krietenstein N, Abraham S, Venev SV et al (2020) Ultrastructural details of mammalian chromosome architecture. Mol Cell 78:554-565.e7
Hsieh T-HS, Cattoglio C, Slobodyanyuk E et al (2020) Resolving the 3D landscape of transcription-linked mammalian chromatin folding. Mol Cell 78:539-553.e8
Rao SSP, Huntley MH, Durand NC et al (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159:1665–1680
Mifsud B, Tavares-Cadete F, Young AN et al (2015) Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet 47:598–606
Schoenfelder S, Furlan-Magaril M, Mifsud B et al (2015) The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res 25:582–597
Schoenfelder S, Sugar R, Dimond A et al (2015) Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat Genet 47:1179–1186
Schoenfelder S, Javierre B-M, Furlan-Magaril M et al (2018) Promoter capture Hi-C: high-resolution, genome-wide profiling of promoter interactions. J Vis Exp 136:57320
Hughes JR, Roberts N, McGowan S et al (2014) Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat Genet 46:205–212
Davies JOJ, Telenius JM, McGowan SJ et al (2016) Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nat Methods 13:74–80
Golov AK, Abashkin DA, Kondratyev NV et al (2020) A modified protocol of Capture-C allows affordable and flexible high-resolution promoter interactome analysis. Sci Rep 10:15491
Hua P, Badat M, Hanssen LLP et al (2021) Defining genome architecture at base-pair resolution. Nature. https://doi.org/10.1038/s41586-021-03639-4
Montefiori LE, Sobreira DR, Sakabe NJ et al (2018) A promoter interaction map for cardiovascular disease genetics. Elife 7:e35788
Schoenfelder S, Mifsud B, Senner CE et al (2018) Divergent wiring of repressive and active chromatin interactions between mouse embryonic and trophoblast lineages. Nat Commun 9:4189
Novo CL, Javierre B-M, Cairns J et al (2018) Long-range enhancer interactions are prevalent in mouse embryonic stem cells and are reorganized upon pluripotent state transition. Cell Rep 22:2615–2627
Martin P, McGovern A, Orozco G et al (2015) Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci. Nat Commun 6:10069
Chesi A, Wagley Y, Johnson ME et al (2019) Genome-scale Capture C promoter interactions implicate effector genes at GWAS loci for bone mineral density. Nat Commun 10:1260
Ray-Jones H, Duffus K, McGovern A et al (2020) Mapping DNA interaction landscapes in psoriasis susceptibility loci highlights KLF4 as a target gene in 9q31. BMC Biol 18:47
Mumbach MR, Rubin AJ, Flynn RA et al (2016) HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods 13:919–922
Fang R, Yu M, Li G et al (2016) Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res 26:1345–1348
Fullwood MJ, Liu MH, Pan YF et al (2009) An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 462:58–64
Weintraub AS, Li CH, Zamudio AV et al (2017) YY1 Is a structural regulator of enhancer-promoter loops. Cell 171:1573-1588.e28
Pelikan RC, Kelly JA, Fu Y et al (2018) Enhancer histone-QTLs are enriched on autoimmune risk haplotypes and influence gene expression within chromatin networks. Nat Commun 9:2905
Nott A, Holtman IR, Coufal NG et al (2019) Brain cell type-specific enhancer–promoter interactome maps and diseaserisk association. Science 366:1134–1139
O’Mara TA, Spurdle AB, Glubb DM, Endometrial Cancer Association Consortium (2019) Analysis of promoter-associated chromatin interactions reveals biologically relevant candidate target genes at endometrial cancer risk loci. Cancers 11:1440
Shi C, Ray-Jones H, Ding J et al (2021) Chromatin looping links target genes with genetic risk loci for dermatological traits. J Invest Dermatol. https://doi.org/10.1016/j.jid.2021.01.015
Chen L, Cao W, Aita R et al (2021) Three-dimensional interactions between enhancers and promoters during intestinal differentiation depend upon HNF4. Cell Rep 34:108679
Kubo N, Ishii H, Xiong X et al (2021) Promoter-proximal CTCF binding promotes distal enhancer-dependent gene activation. Nat Struct Mol Biol 28:152–161
Bauman JGJ, Wiegant J, Borst P, van Duijn P (1980) A new method for fluorescence microscopical localization of specific DNA sequences by in situ hybridization of fluorochrome-labelled RNA. Exp Cell Res 128:485–490
Amano T, Sagai T, Tanabe H et al (2009) Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell 16:47–57
Williamson I, Eskeland R, Lettice LA et al (2012) Anterior-posterior differences in HoxD chromatin topology in limb development. Development 139:3157–3167
Giorgetti L, Heard E (2016) Closing the loop: 3C versus DNA FISH. Genome Biol 17:215
Ulianov SV, Tachibana-Konwalski K, Razin SV (2017) Single-cell Hi-C bridges microscopy and genome-wide sequencing approaches to study 3D chromatin organization. Bioessays 39(10):1700104
Fudenberg G, Imakaev M (2017) FISH-ing for captured contacts: towards reconciling FISH and 3C. Nat Methods 14:673–678
Beliveau BJ, Boettiger AN, Avendaño MS et al (2015) Single-molecule super-resolution imaging of chromosomes and in situ haplotype visualization using Oligopaint FISH probes. Nat Commun 6:7147
Ni Y, Cao B, Ma T et al (2017) Super-resolution imaging of a 2.5 kb non-repetitive DNA in the nuclear genome using molecular beacon probes. Elife 6:e21660
Fukaya T, Lim B, Levine M (2016) Enhancer control of transcriptional bursting. Cell 166:358–368
Chen H, Levo M, Barinov L et al (2018) Dynamic interplay between enhancer–promoter topology and gene activity. Nat Genet 50:1296–1303
Gu B, Swigut T, Spencley A et al (2018) Transcription-coupled changes in nuclear mobility of mammalian cis-regulatory elements. Science 359:1050–1055
Morgan SL, Chang EY, Mariano NC et al (2018) CRISPR-mediated reorganization of chromatin loop structure. J Vis Exp. https://doi.org/10.3791/57457
Kim JH, Rege M, Valeri J et al (2019) LADL: light-activated dynamic looping for endogenous gene expression control. Nat Methods 16:633–639
Vakoc CR, Letting DL, Gheldof N et al (2005) Proximity among distant regulatory elements at the beta-globin locus requires GATA-1 and FOG-1. Mol Cell 17:453–462
Drissen R, Palstra R-J, Gillemans N et al (2004) The active spatial organization of the beta-globin locus requires the transcription factor EKLF. Genes Dev 18:2485–2490
Song S-H, Hou C, Dean A (2007) A positive role for NLI/Ldb1 in long-range beta-globin locus control region function. Mol Cell 28:810–822
Ohlsson R, Renkawitz R, Lobanenkov V (2001) CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet 17:520–527
Baniahmad A, Steiner C, Köhne AC, Renkawitz R (1990) Modular structure of a chicken lysozyme silencer: involvement of an unusual thyroid hormone receptor binding site. Cell 61:505–514
Filippova GN, Fagerlie S, Klenova EM et al (1996) An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol Cell Biol 16:2802–2813
Köhne AC, Baniahmad A, Renkawitz R (1993) NeP1. A ubiquitous transcription factor synergizes with v-ERBA in transcriptional silencing. J Mol Biol 232:747–755
Vostrov AA, Quitschke WW (1997) The zinc finger protein CTCF binds to the APBbeta domain of the amyloid beta-protein precursor promoter. Evidence for a role in transcriptional activation. J Biol Chem 272:33353–33359
Bell AC, West AG, Felsenfeld G (1999) The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 98:387–396
Saitoh N, Bell AC, Recillas-Targa F et al (2000) Structural and functional conservation at the boundaries of the chicken beta-globin domain. EMBO J 19:2315–2322
Hark AT, Schoenherr CJ, Katz DJ et al (2000) CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405:486–489
Kanduri C, Pant V, Loukinov D et al (2000) Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr Biol 10:853–856
Xie X, Mikkelsen TS, Gnirke A et al (2007) Systematic discovery of regulatory motifs in conserved regions of the human genome, including thousands of CTCF insulator sites. Proc Natl Acad Sci USA 104:7145–7150
Barski A, Cuddapah S, Cui K et al (2007) High-resolution profiling of histone methylations in the human genome. Cell 129:823–837
Kim TH, Abdullaev ZK, Smith AD et al (2007) Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128:1231–1245
Shen Y, Yue F, McCleary DF et al (2012) A map of the cis-regulatory sequences in the mouse genome. Nature 488:116–120
Yusufzai TM, Tagami H, Nakatani Y, Felsenfeld G (2004) CTCF tethers an insulator to subnuclear sites, suggesting shared insulator mechanisms across species. Mol Cell 13:291–298
Hou C, Zhao H, Tanimoto K, Dean A (2008) CTCF-dependent enhancer-blocking by alternative chromatin loop formation. Proc Natl Acad Sci U S A 105:20398–20403
Splinter E, Heath H, Kooren J et al (2006) CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev 20:2349–2354
Rubio ED, Reiss DJ, Welcsh PL et al (2008) CTCF physically links cohesin to chromatin. Proc Natl Acad Sci USA 105:8309–8314
Wendt KS, Yoshida K, Itoh T et al (2008) Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451:796–801
Onn I, Heidinger-Pauli JM, Guacci V et al (2008) Sister chromatid cohesion: a simple concept with a complex reality. Annu Rev Cell Dev Biol 24:105–129
Michaelis C, Ciosk R, Nasmyth K (1997) Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 91:35–45
Guacci V, Koshland D, Strunnikov A (1997) A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell 91:47–57
Losada A, Hirano M, Hirano T (1998) Identification of Xenopus SMC protein complexes required for sister chromatid cohesion. Genes Dev 12:1986–1997
Nishiyama T (2019) Cohesion and cohesin-dependent chromatin organization. Curr Opin Cell Biol 58:8–14
de Wit E, Vos ESM, Holwerda SJB et al (2015) CTCF Binding Polarity Determines Chromatin Looping. Mol Cell 60:676–684
Sanborn AL, Rao SSP, Huang S-C et al (2015) Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci USA 112:E6456–E6465
Schmidt D, Schwalie PC, Ross-Innes CS et al (2010) A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res 20:578–588
Seitan VC, Faure AJ, Zhan Y et al (2013) Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res 23:2066–2077
Zuin J, Dixon JR, van der Reijden MIJA et al (2014) Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci USA 111:996–1001
Nora EP, Goloborodko A, Valton A-L et al (2017) Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169:930–944.e22
Rao SSP, Huang S-C, Glenn St Hilaire B et al (2017) Cohesin loss eliminates all loop domains. Cell 171:305-320.e24
Schwarzer W, Abdennur N, Goloborodko A et al (2017) Two independent modes of chromatin organization revealed by cohesin removal. Nature 551:51–56
Thiecke MJ, Wutz G, Muhar M et al (2020) Cohesin-dependent and -independent mechanisms mediate chromosomal contacts between promoters and enhancers. Cell Rep 32:107929
Hsieh TS, Cattoglio C, Slobodyanyuk E, Hansen AS, Darzacq X, Tjian R (2021) Enhancer-promoter interactions and transcription are maintained upon acute loss of CTCF, cohesin, WAPL, and YY1. BioRxiv. https://doi.org/10.1101/2021.07.14.452365
Croce LD, Di Croce L, Helin K (2013) Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol 20:1147–1155
Aranda S, Mas G, Di Croce L (2015) Regulation of gene transcription by polycomb proteins. Sci Adv 1:e1500737
Pirrotta V, Li H-B (2012) A view of nuclear polycomb bodies. Curr Opin Genet Dev 22:101–109
Bantignies F, Roure V, Comet I et al (2011) Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell 144:214–226
Eagen KP, Aiden EL, Kornberg RD (2017) Polycomb-mediated chromatin loops revealed by a subkilobase-resolution chromatin interaction map. Proc Natl Acad Sci USA 114:8764–8769
Ogiyama Y, Schuettengruber B, Papadopoulos GL et al (2018) Polycomb-dependent chromatin looping contributes to gene silencing during drosophila development. Mol Cell 71:73-88.e5
Rhodes JDP, Feldmann A, Hernández-Rodríguez B et al (2020) Cohesin disrupts polycomb-dependent chromosome interactions in embryonic stem cells. Cell Rep 30:820-835.e10
Chovanec P, Collier AJ, Krueger C et al (2021) Widespread reorganisation of pluripotent factor binding and gene regulatory interactions between human pluripotent states. Nat Commun 12:2098
Pachano T, Crispatzu G, Rada-Iglesias A (2019) Polycomb proteins as organizers of 3D genome architecture in embryonic stem cells. Brief Funct Genomics 18:358–366
Denholtz M, Bonora G, Chronis C et al (2013) Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell 13:602–616
Loubiere V, Papadopoulos GL, Szabo Q et al (2020) Widespread activation of developmental gene expression characterized by PRC1-dependent chromatin looping. Sci Adv 6:eaax4001
Heidari N, Phanstiel DH, He C et al (2014) Genome-wide map of regulatory interactions in the human genome. Genome Res 24:1905–1917
Bailey SD, Zhang X, Desai K et al (2015) ZNF143 provides sequence specificity to secure chromatin interactions at gene promoters. Nat Commun 2:6186
Ye B-Y, Shen W-L, Wang D et al (2016) ZNF143 is involved in CTCF-mediated chromatin interactions by cooperation with cohesin and other partners. Mol Biol 50:496–503
Beagan JA, Duong MT, Titus KR et al (2017) YY1 and CTCF orchestrate a 3D chromatin looping switch during early neural lineage commitment. Genome Res 27:1139–1152
Guo C, Gerasimova T, Hao H et al (2011) Two forms of loops generate the chromatin conformation of the immunoglobulin heavy-chain gene locus. Cell 147:332–343
Myslinski E, Gérard M-A, Krol A, Carbon P (2006) A genome scale location analysis of human Staf/ZNF143-binding sites suggests a widespread role for human Staf/ZNF143 in mammalian promoters. J Biol Chem 281:39953–39962
Zhou Q, Yu M, Tirado-Magallanes R et al (2021) ZNF143 mediates CTCF-bound promoter–enhancer loops required for murine hematopoietic stem and progenitor cell function. Nat Commun 12:43
Wen Z, Huang Z-T, Zhang R, Peng C (2018) ZNF143 is a regulator of chromatin loop. Cell Biol Toxicol 34:471–478
Jurata LW, Pfaff SL, Gill GN (1998) The nuclear LIM domain interactor NLI mediates homo- and heterodimerization of LIM domain transcription factors. J Biol Chem 273:3152–3157
Deng W, Lee J, Wang H et al (2012) Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 149:1233–1244
Magli A, Baik J, Pota P et al (2019) Pax3 cooperates with Ldb1 to direct local chromosome architecture during myogenic lineage specification. Nat Commun 10:2316
Monahan K, Horta A, Lomvardas S (2019) LHX2- and LDB1-mediated trans interactions regulate olfactory receptor choice. Nature 565:448–453
Deng W, Rupon JW, Krivega I et al (2014) Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell 158:849–860
Krivega I, Dale RK, Dean A (2014) Role of LDB1 in the transition from chromatin looping to transcription activation. Genes Dev 28:1278–1290
Soutourina J (2018) Transcription regulation by the mediator complex. Nat Rev Mol Cell Biol 19:262–274
Kagey MH, Newman JJ, Bilodeau S et al (2010) Mediator and cohesin connect gene expression and chromatin architecture. Nature 467:430–435
Lai F, Orom UA, Cesaroni M et al (2013) Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 494:497–501
Phillips-Cremins JE, Sauria MEG, Sanyal A et al (2013) Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153:1281–1295
El Khattabi L, Zhao H, Kalchschmidt J et al (2019) A pliable mediator acts as a functional rather than an architectural bridge between promoters and enhancers. Cell 178:1145-1158.e20
Bisgrove DA, Mahmoudi T, Henklein P, Verdin E (2007) Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci USA 104:13690–13695
Delmore JE, Issa GC, Lemieux ME et al (2011) BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146:904–917
Floyd SR, Pacold ME, Huang Q et al (2013) The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature 498:246–250
Wang R, Li Q, Helfer CM et al (2012) Bromodomain protein Brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J Biol Chem 287:10738–10752
Sabari BR, Dall’Agnese A, Boija A et al (2018) Coactivator condensation at super-enhancers links phase separation and gene control. Science 361:eaar3958
van Steensel B, Furlong EEM (2019) The role of transcription in shaping the spatial organization of the genome. Nat Rev Mol Cell Biol 20:327–337
Jubb AW, Boyle S, Hume DA, Bickmore WA (2017) Glucocorticoid receptor binding induces rapid and prolonged large-scale chromatin decompaction at multiple target loci. Cell Rep 21:3022–3031
Heinz S, Texari L, Hayes MGB et al (2018) Transcription elongation can affect genome 3D structure. Cell 174:1522-1536.e22
Brandão HB, Paul P, van den Berg AA et al (2019) RNA polymerases as moving barriers to condensin loop extrusion. Proc Natl Acad Sci USA 116:20489–20499
Li W, Notani D, Ma Q et al (2013) Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498:516–520
de Laat W, Duboule D (2013) Topology of mammalian developmental enhancers and their regulatory landscapes. Nature 502:499–506
Ghavi-Helm Y, Klein FA, Pakozdi T et al (2014) Enhancer loops appear stable during development and are associated with paused polymerase. Nature 512:96–100
Ing-Simmons E, Vaid R, Bing XY et al (2021) Independence of chromatin conformation and gene regulation during Drosophila dorsoventral patterning. Nat Genet 53:487–499
Rubin AJ, Barajas BC, Furlan-Magaril M et al (2017) Lineage-specific dynamic and pre-established enhancer–promoter contacts cooperate in terminal differentiation. Nat Genet 49:1522–1528
Siersbæk R, Madsen JGS, Javierre BM et al (2017) Dynamic rewiring of promoter-anchored chromatin loops during adipocyte differentiation. Mol Cell 66:420-435.e5
Oudelaar AM, Beagrie RA, Gosden M et al (2020) Dynamics of the 4D genome during in vivo lineage specification and differentiation. Nat Commun 11:2722
Dixon JR, Jung I, Selvaraj S et al (2015) Chromatin architecture reorganization during stem cell differentiation. Nature 518:331–336
Jin F, Li Y, Dixon JR et al (2013) A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 503:290–294
Yan J, Chen S-AA, Local A et al (2018) Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res 28:204–220
Sungalee S, Liu Y, Lambuta RA et al (2021) Histone acetylation dynamics modulates chromatin conformation and allele-specific interactions at oncogenic loci. Nat Genet 53:650–662
Bonev B, Mendelson Cohen N, Szabo Q et al (2017) Multiscale 3D genome rewiring during mouse neural development. Cell 171:557-572.e24
Alexander JM, Guan J, Li B et al (2019) Live-cell imaging reveals enhancer-dependent Sox2 transcription in the absence of enhancer proximity. eLife 8:e41769
Quinodoz SA, Ollikainen N, Tabak B et al (2018) Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174:744-757.e24
Imakaev M, Fudenberg G, McCord RP et al (2012) Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods 9:999–1003
Zuin J, Roth G, Zhan Y et al (2021) Nonlinear control of transcription through enhancer–promoter interactions. BioRxiv. https://doi.org/10.1101/2021.04.22.440891
Xiao J, Hafner A, Boettiger AN (2020) How subtle changes in 3D structure can create large changes in transcription. BioRxiv. https://doi.org/10.1101/2020.10.22.351395
Benabdallah NS, Williamson I, Illingworth RS et al (2019) Decreased enhancer-promoter proximity accompanying enhancer activation. Mol Cell 76:473-484.e7
Panigrahi A, O’Malley BW (2021) Mechanisms of enhancer action: the known and the unknown. Genome Biol 22:108
Heist T, Fukaya T, Levine M (2019) Large distances separate coregulated genes in living embryos. Proc Natl Acad Sci USA 116:15062–15067
Sabari BR, Dall’Agnese A, Young RA (2020) Biomolecular condensates in the nucleus. Trends Biochem Sci 45:961–977
Hnisz D, Shrinivas K, Young RA et al (2017) A phase separation model for transcriptional control. Cell 169:13–23
Chong S, Dugast-Darzacq C, Liu Z et al (2018) Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361:eaar2555
Mir M, Bickmore W, Furlong EEM, Narlikar G (2019) Chromatin topology, condensates and gene regulation: shifting paradigms or just a phase? Development 146:dev182766
Boija A, Klein IA, Sabari BR et al (2018) Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 175:1842-1855.e16
Gibson BA, Doolittle LK, Schneider MWG et al (2019) Organization of chromatin by intrinsic and regulated phase separation. Cell 179:470-484.e21
Sanulli S, Trnka MJ, Dharmarajan V et al (2019) HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 575:390–394
Zhang Y, Kutateladze TG (2019) Liquid-liquid phase separation is an intrinsic physicochemical property of chromatin. Nat Struct Mol Biol 26:1085–1086
Shrinivas K, Sabari BR, Coffey EL et al (2019) Enhancer features that drive formation of transcriptional condensates. Mol Cell 75:549-561.e7
Ryu J-K, Bouchoux C, Liu HW et al (2021) Bridging-induced phase separation induced by cohesin SMC protein complexes. Sci Adv 7:eabe5905
Chiariello AM, Corberi F, Salerno M (2020) The interplay between phase separation and gene-enhancer communication: a theoretical study. Biophys J 119:873–883
Nott TJ, Petsalaki E, Farber P et al (2015) Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell 57:936–947
Elbaum-Garfinkle S, Kim Y, Szczepaniak K et al (2015) The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc Natl Acad Sci USA 112:7189–7194
Alberti S, Gladfelter A, Mittag T (2019) Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 176:419–434
McSwiggen DT, Mir M, Darzacq X, Tjian R (2019) Evaluating phase separation in live cells: diagnosis, caveats, and functional consequences. Genes Dev 33:1619–1634
Cook PR (2010) A model for all genomes: the role of transcription factories. J Mol Biol 395:1–10
Cho W-K, Spille J-H, Hecht M et al (2018) Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361:412–415
Kent S, Brown K, Yang C-H et al (2020) Phase-separated transcriptional condensates accelerate target-search process revealed by live-cell single-molecule imaging. Cell Rep 33:108248
Guo YE, Manteiga JC, Henninger JE et al (2019) Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572:543–548
Hyman AA, Weber CA, Jülicher F (2014) Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol 30:39–58
Shlyueva D, Stampfel G, Stark A (2014) Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet 15:272–286
Schoenfelder S, Fraser P (2019) Long-range enhancer–promoter contacts in gene expression control. Nat Rev Genet 20:437–455
Dixon JR, Selvaraj S, Yue F et al (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485:376–380
Nora EP, Lajoie BR, Schulz EG et al (2012) Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485:381–385
Ji X, Dadon DB, Powell BE et al (2016) 3D chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell 18:262–275
Fudenberg G, Imakaev M, Lu C et al (2016) Formation of chromosomal domains by loop extrusion. Cell Rep 15:2038–2049
Davidson IF, Bauer B, Goetz D et al (2019) DNA loop extrusion by human cohesin. Science 366:1338–1345
Kim Y, Shi Z, Zhang H et al (2019) Human cohesin compacts DNA by loop extrusion. Science 366:1345–1349
Guo Y, Monahan K, Wu H et al (2012) CTCF/cohesin-mediated DNA looping is required for protocadherin α promoter choice. Proc Natl Acad Sci USA 109:21081–21086
Guo Y, Xu Q, Canzio D et al (2015) CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell 162:900–910
Nanni L, Ceri S, Logie C (2020) Spatial patterns of CTCF sites define the anatomy of TADs and their boundaries. Genome Biol 21:197
Dowen JM, Fan ZP, Hnisz D et al (2014) Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159:374–387
Dixon JR, Gorkin DU, Ren B (2016) Chromatin domains: the unit of chromosome organization. Mol Cell 62:668–680
Zhan Y, Mariani L, Barozzi I et al (2017) Reciprocal insulation analysis of Hi-C data shows that TADs represent a functionally but not structurally privileged scale in the hierarchical folding of chromosomes. Genome Res 27:479–490
Sexton T, Cavalli G (2015) The role of chromosome domains in shaping the functional genome. Cell 160:1049–1059
Lupiáñez DG, Spielmann M, Mundlos S (2016) Breaking TADs: how alterations of chromatin domains result in disease. Trends Genet 32:225–237
Lupiáñez DG, Kraft K, Heinrich V et al (2015) Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161:1012–1025
Melo US, Schöpflin R, Acuna-Hidalgo R et al (2020) Hi-C identifies complex genomic rearrangements and TAD-shuffling in developmental diseases. Am J Hum Genet 106:872–884
Bintu B, Mateo LJ, Su J-H et al (2018) Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362:eaau1783
Ghavi-Helm Y, Jankowski A, Meiers S et al (2019) Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression. Nat Genet 51:1272–1282
Williamson I, Kane L, Devenney PS et al (2019) Developmentally regulated expression is robust to TAD perturbations. Development 146:dev179523
Paliou C, Guckelberger P, Schöpflin R et al (2019) Preformed chromatin topology assists transcriptional robustness of during limb development. Proc Natl Acad Sci USA 116:12390–12399
Finn EH, Pegoraro G, Brandão HB et al (2019) Extensive heterogeneity and intrinsic variation in spatial genome organization. Cell 176:1502-1515.e10
Espinola SM, Götz M, Bellec M et al (2021) Cis-regulatory chromatin loops arise before TADs and gene activation, and are independent of cell fate during early Drosophila development. Nat Genet 53:477–486
Hug CB, Grimaldi AG, Kruse K, Vaquerizas JM (2017) Chromatin architecture emerges during zygotic genome activation independent of transcription. Cell 169:216-228.e19
Zhang H, Emerson DJ, Gilgenast TG et al (2019) Chromatin structure dynamics during the mitosis-to-G1 phase transition. Nature 576:158–162
Vian L, Pękowska A, Rao SSP et al (2018) The energetics and physiological impact of cohesin extrusion. Cell 173:1165-1178.e20
Barrington C, Georgopoulou D, Pezic D et al (2019) Enhancer accessibility and CTCF occupancy underlie asymmetric TAD architecture and cell type specific genome topology. Nat Commun 10:2908
Furlan-Magaril M, Ando-Kuri M, Arzate-Mejía RG et al (2021) The global and promoter-centric 3D genome organization temporally resolved during a circadian cycle. Genome Biol 22:162
Naumova N, Imakaev M, Fudenberg G et al (2013) Organization of the mitotic chromosome. Science 342:948–953
Nuebler J, Fudenberg G, Imakaev M et al (2018) Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc Natl Acad Sci USA 115:E6697–E6706
Falk M, Feodorova Y, Naumova N et al (2019) Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 570:395–399
van Steensel B, Belmont AS (2017) Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169:780–791
Robson MI, de Las Heras JI, Czapiewski R et al (2017) Constrained release of lamina-associated enhancers and genes from the nuclear envelope during T-cell activation facilitates their association in chromosome compartments. Genome Res 27:1126–1138
Smith CL, Poleshko A, Epstein JA (2021) The nuclear periphery is a scaffold for tissue-specific enhancers. Nucleic Acids Res 49:6181–6195
Woodcock CL, Ghosh RP (2010) Chromatin higher-order structure and dynamics. Cold Spring Harb Perspect Biol 2:a000596
Barbieri M, Chotalia M, Fraser J et al (2012) Complexity of chromatin folding is captured by the strings and binders switch model. Proc Natl Acad Sci USA 109:16173–16178
Symmons O, Pan L, Remeseiro S et al (2016) The Shh topological domain facilitates the action of remote enhancers by reducing the effects of genomic distances. Dev Cell 39:529–543
Kaul A, Bhattacharyya S, Ay F (2020) Identifying statistically significant chromatin contacts from Hi-C data with FitHiC2. Nat Protoc 15:991–1012
Bhattacharyya S, Chandra V, Vijayanand P, Ay F (2019) Identification of significant chromatin contacts from HiChIP data by FitHiChIP. Nat Commun 10:4221
Holgersen EM, Gillespie A, Leavy OC et al (2021) Identifying high-confidence capture Hi-C interactions using CHiCANE. Nat Protoc 16:2257–2285
Cairns J, Freire-Pritchett P, Wingett SW et al (2016) CHiCAGO: robust detection of DNA looping interactions in capture Hi-C data. Genome Biol 17:127
Sanyal A, Lajoie BR, Jain G, Dekker J (2012) The long-range interaction landscape of gene promoters. Nature 489:109–113
Furlong EEM, Levine M (2018) Developmental enhancers and chromosome topology. Science 361:1341–1345
Li X, Noll M (1994) Compatibility between enhancers and promoters determines the transcriptional specificity of gooseberry and gooseberry neuro in the Drosophila embryo. EMBO J 13:400–406
Butler JE, Kadonaga JT (2001) Enhancer–promoter specificity mediated by DPE or TATA core promoter motifs. Genes Dev 15:2515–2519
Fudenberg G, Kelley DR, Pollard KS (2020) Predicting 3D genome folding from DNA sequence with Akita. Nat Methods 17:1111–1117
Avsec Ž, Agarwal V, Visentin D et al (2021) Effective gene expression prediction from sequence by integrating long-range interactions. BioRxiv. https://doi.org/10.1101/2021.04.07.438649
Oh S, Shao J, Mitra J et al (2021) Enhancer release and retargeting activates disease-susceptibility genes. Nature. https://doi.org/10.1038/s41586-021-03577-1
Fulco CP, Munschauer M, Anyoha R et al (2016) Systematic mapping of functional enhancer–promoter connections with CRISPR interference. Science 354:769–773
Osterwalder M, Barozzi I, Tissières V et al (2018) Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554:239–243
Montavon T, Soshnikova N, Mascrez B et al (2011) A regulatory archipelago controls Hox genes transcription in digits. Cell 147:1132–1145
Lonfat N, Montavon T, Darbellay F et al (2014) Convergent evolution of complex regulatory landscapes and pleiotropy at Hox loci. Science 346:1004–1006
Perry MW, Boettiger AN, Levine M (2011) Multiple enhancers ensure precision of gap gene-expression patterns in the Drosophila embryo. Proc Natl Acad Sci USA 108:13570–13575
Miller SW, Posakony JW (2020) Disparate expression specificities coded by a shared Hox-C enhancer. Elife 9:e39876
Allahyar A, Vermeulen C, Bouwman BAM et al (2018) Enhancer hubs and loop collisions identified from single-allele topologies. Nat Genet 50:1151–1160
Patrinos GP, de Krom M, de Boer E et al (2004) Multiple interactions between regulatory regions are required to stabilize an active chromatin hub. Genes Dev 18:1495–1509
Gavrilov AA, Razin SV (2008) Spatial configuration of the chicken alpha-globin gene domain: immature and active chromatin hubs. Nucleic Acids Res 36:4629–4640
Markenscoff-Papadimitriou E, Allen WE, Colquitt BM et al (2014) Enhancer interaction networks as a means for singular olfactory receptor expression. Cell 159:543–557
Ing-Simmons E, Seitan VC, Faure AJ et al (2015) Spatial enhancer clustering and regulation of enhancer-proximal genes by cohesin. Genome Res 25:504–513
Song W, Sharan R, Ovcharenko I (2019) The first enhancer in an enhancer chain safeguards subsequent enhancer–promoter contacts from a distance. Genome Biol 20:197
Farooq U, Saravanan B, Islam Z et al (2021) An interdependent network of functional enhancers regulates transcription and EZH2 loading at the INK4a/ARF locus. Cell Rep 34:108898
Wang Q, Sawyer IA, Sung M-H et al (2016) Cajal bodies are linked to genome conformation. Nat Commun 7:10966
Zhu I, Song W, Ovcharenko I, Landsman D (2021) A model of active transcription hubs that unifies the roles of active promoters and enhancers. Nucleic Acids Res 49:4493–4505
Apostolou E, Thanos D (2008) Virus Infection Induces NF-kappaB-dependent interchromosomal associations mediating monoallelic IFN-beta gene expression. Cell 134:85–96
Schoenfelder S, Sexton T, Chakalova L et al (2010) Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet 42:53–61
Noordermeer D, Leleu M, Splinter E et al (2011) The dynamic architecture of Hox gene clusters. Science 334:222–225
Beagrie RA, Scialdone A, Schueler M et al (2017) Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543:519–524
Oudelaar AM, Davies JOJ, Hanssen LLP et al (2018) Single-allele chromatin interactions identify regulatory hubs in dynamic compartmentalized domains. Nat Genet 50:1744–1751
Oudelaar AM, Harrold CL, Hanssen LLP et al (2019) A revised model for promoter competition based on multi-way chromatin interactions at the α-globin locus. Nat Commun 10:5412
Boettiger A, Murphy S (2020) Advances in chromatin imaging at kilobase-scale resolution. Trends Genet 36:273–287
Cardozo Gizzi AM, Cattoni DI, Fiche J-B et al (2019) Microscopy-based chromosome conformation capture enables simultaneous visualization of genome organization and transcription in intact organisms. Mol Cell 74:212-222.e5
Nir G, Farabella I, Pérez Estrada C et al (2018) Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLoS Genet 14:e1007872
Mateo LJ, Murphy SE, Hafner A et al (2019) Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 568:49–54
Cardozo Gizzi AM, Espinola SM, Gurgo J et al (2020) Direct and simultaneous observation of transcription and chromosome architecture in single cells with Hi-M. Nat Protoc 15:840–876
Bartman CR, Hsu SC, Hsiung CC-S et al (2016) Enhancer regulation of transcriptional bursting parameters revealed by forced chromatin looping. Mol Cell 62:237–247
Lim B, Levine MS (2021) Enhancer–promoter communication: hubs or loops? Curr Opin Genet Dev 67:5–9
Lim B, Heist T, Levine M, Fukaya T (2018) Visualization of transvection in living drosophila embryos. Mol Cell 70:287-296.e6
Gonzalez F, Duboule D, Spitz F (2007) Transgenic analysis of Hoxd gene regulation during digit development. Dev Biol 306:847–859
Darbellay F, Duboule D (2016) Topological domains, metagenes, and the emergence of pleiotropic regulations at Hox loci. Curr Top Dev Biol 116:299–314
Gonen N, Futtner CR, Wood S et al (2018) Sex reversal following deletion of a single distal enhancer of. Science 360:1469–1473
Sen R, Grosschedl R (2010) Memories of lost enhancers. Genes Dev 24:973–979
Song W, Ovcharenko I (2018) Dichotomy in redundant enhancers points to presence of initiators of gene regulation. BMC Genomics 19:947
Cuartero S, Weiss FD, Dharmalingam G et al (2018) Control of inducible gene expression links cohesin to hematopoietic progenitor self-renewal and differentiation. Nat Immunol 19:932–941
Kane L, Williamson I, Flyamer IM et al (2021) Cohesin is required for long-range enhancer action. Biorxiv. https://doi.org/10.1101/2021.06.24.449812
Tunnacliffe E, Chubb JR (2020) What is a transcriptional burst? Trends Genet 36:288–297
Larsson AJM, Johnsson P, Hagemann-Jensen M et al (2019) Genomic encoding of transcriptional burst kinetics. Nature 565:251–254
Hay D, Hughes JR, Babbs C et al (2016) Genetic dissection of the α-globin super-enhancer in vivo. Nat Genet 48:895–903
Dukler N, Gulko B, Huang Y-F, Siepel A (2016) Is a super-enhancer greater than the sum of its parts? Nat Genet 49:2–3
Bothma JP, Garcia HG, Ng S et al (2015) Enhancer additivity and non-additivity are determined by enhancer strength in the Drosophila embryo. eLife 4:e07956
Hong J-W, Hendrix DA, Levine MS (2008) Shadow enhancers as a source of evolutionary novelty. Science 321:1314
Barolo S (2012) Shadow enhancers: frequently asked questions about distributed cis-regulatory information and enhancer redundancy. BioEssays 34:135–141
Cannavò E, Khoueiry P, Garfield DA et al (2016) Shadow enhancers are pervasive features of developmental regulatory networks. Curr Biol 26:38–51
Thomas HF, Kotova E, Jayaram S et al (2021) Temporal dissection of an enhancer cluster reveals distinct temporal and functional contributions of individual elements. Mol Cell 81:969-982.e13
Choi J, Lysakovskaia K, Stik G et al (2021) Evidence for additive and synergistic action of mammalian enhancers during cell fate determination. Elife 10:e65381
Shin HY, Willi M, HyunYoo K et al (2016) Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat Genet 48:904–911
Scholes C, Biette KM, Harden TT, DePace AH (2019) Signal integration by shadow enhancers and enhancer duplications varies across the drosophila embryo. Cell Rep 26:2407-2418.e5
Schaffner W (1988) Gene regulation. A hit-and-run mechanism for transcriptional activation? Nature 336:427–428
Basu S, Mackowiak SD, Niskanen H et al (2020) Unblending of transcriptional condensates in human repeat expansion disease. Cell 181:1062-1079.e30
Cho W-K, Jayanth N, English BP et al (2016) RNA Polymerase II cluster dynamics predict mRNA output in living cells. Elife 5:e13617
Acknowledgements
The authors thank all members of the Functional Gene Control Group, Juanma Vaquerizas and Elizabeth Ing-Simmons for helpful discussions, and Monica Della Rosa and the anonymous reviewers for their valuable comments to the manuscript. Research in the group is supported by the Medical Research Council of the UK (MRC), through MRC Investigator funding to M.S. (MC-A652-5QA20).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ray-Jones, H., Spivakov, M. Transcriptional enhancers and their communication with gene promoters. Cell. Mol. Life Sci. 78, 6453–6485 (2021). https://doi.org/10.1007/s00018-021-03903-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-021-03903-w