
Abstract
A group of sulfonamides exhibiting antimicrobial activity was analyzed with the application of quantitative structure–activity/property relationships method. The purpose of this study was to show the common and differentiating characteristics of the analyzed chemical structures alike physicochemically as well as pharmacologically based on the quantum-chemical calculations both in vacuo and in the aquatic environment together with their microbiological activity and chromatographic retention data. The semiempirical and ab initio level of in silico molecular modeling was performed for calculations of statistically significant molecular descriptors to compare obtained results. The relationship between the structure and biological activity and physicochemical parameters data was able to class and describe analyzed molecules and the applied chemometric approaches (principal component analysis, factor analysis, and multiple regression analysis) revealed the influential features of the tested structures responsible for the antimicrobial activity.
Similar content being viewed by others

Introduction
Antibacterial sulfonamides are one of the oldest groups of chemotherapeutics and therefore the structure–property relationship, and especially the structure–biological activity of this group of compounds was the subject of a number of works. One of the first of these was the finding of the relationships between acid and alkaline dissociation constants and microbiological activity (Bell and Roblin 1942). In addition, one should mention the study of the relationship between the acid dissociation constant and the chemical shift of nuclear magnetic resonance spectra and microbiological activity (Seydel 1971; De Benedetti and Rastelli 1978), the relationship between microbiological activity and data on high-performance liquid chromatography retention (Rastelli et al. 1975; Henry et al. 1976; Brent et al. 1983), and the relationship between microbiological activity and physical parameters such as molar refraction or the sum of the electronegativity values of the atoms that make up the substituent in the sulfonamide group (Kaliszan et al. 1979). Previously, the chemometric analysis was presented covering the dependence of retention data from high-performance liquid chromatography and nonempirical parameters calculated by quantum-chemical methods and other structural parameters and data obtained by ultraviolet spectroscopy, and to a limited extent with the microbiological activity of sulfonamides (Bober et al. 1998). Since the significance of the parameters available from quantum-chemical calculations in the chromatographic structure-retention dependence (QSRR) as well as the structure–biological activity was discussed by Kaliszan (1987, 1997, 2007), it would be interesting to carry out such an analysis for data from quantum-chemical calculations using the ab initio method and data available from the literature on both microbial activity in vitro and other parameters conditioning the biological activity of sulfonamides for a slightly broader database, as well as previously determined chromatographic retention parameters and other physicochemical literature data.
In the literature, we can also find other studies with the use of structure–activity together with molecular docking with complexes and toxicity analysis of different antimicrobial sulfonamides (Agrawal et al. 2001; Krátký et al. 2012; Boufas et al. 2014; Singh 2015; Chen et al. 2017; Mondal et al. 2017; Swain et al. 2017; Mizdal et al. 2018; Naaz et al. 2018).
On the other hand, the significance of the parameters available from quantum-chemical calculations in relation to a structure–biological activity for different groups of compounds was as well already confirmed in the previous series of publications (Belka et al. 2012, 2013; Bober et al. 2012a, b; Stasiak et al. 2013; Kawczak et al. 2014, 2015, 2018a, b, c, 2019; Ciura et al. 2017). In view of the above, it was decided to check how the physicochemical descriptors obtained as a result of quantum-chemical methods are influencing the classification of sulfonamides, while all the performed calculations were possible on standard-class principal components. In addition, it would be advisable to determine quantitative relationships between structural parameters derived from quantum-chemical calculations and biological activity, all using standard statistical analysis methods. The subject of this work was to perform analysis for ab initio together with semiempirical data using the RM1 method of both isolated (in vacuo) molecules as well as hydration of molecules and hydrogen bonds for a number of antibacterial sulfonamides and to obtain chemometrically confirmed relationships between physicochemical parameters and biological parameters of antibacterial sulfonamides and nonempirical parameters characterizing their chemical structure.
Materials and methods
Molecules and analytes
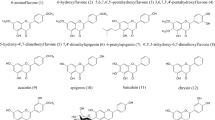
The following compounds were selected for the study from the group of antibacterial sulfonamides: 2-sulfapyridine, 3-sulfapyridine, 4-methylsulfathiazole, sulfacarbamide, sulfacetamide, sulfachloropyridazine, sulfadiazine, sulfadimethoxine, sulfaetidol, sulfafurazole, sulfaguanidine, sulfalene, sulfamerazine, sulfamethazine, sulfamethoxydiazine, sulfamethoxypyridazine, sulfamethizole, sulfamethoxazole, sulfamonomethoxine, sulfamoxole, sulfanilamide, sulfaquinoxaline, sulfasomidine, and sulfathiazole. All structural formulas of analyzed molecules are presented in Fig. 1.

Structural formulas of compounds studied
Biological activity and chromatographic retention data
The work uses literature data on microbial activity expressed as log 1/MIC, where MIC means the lowest concentration inhibiting the growth of Escherichia coli in vitro in μmol dm−3 (microbial inhibiting concentration) from the work of Krüger-Thiemer and Bünger (1965). In the presented work in the case of sulfonamides, the literature data relevant to the bioavailability and pharmacokinetics were taken from Rieder’s work (Von Rieder 1963). These are the binding of sulfonamide to plasma proteins expressed as the logarithm of the inverse (i.e., negative) constant α and logarithm of the constant β in the Langmuir adsorption equation (LG1_ALFA and LG_BETA, respectively) expressed as logarithms in accordance with the concept presented in Brent’s paper (Brent et al. 1983). Rieder’s constant α is the concentration of sulfonamide in the protein lacking proteins in μmol l−1 at half-saturation with protein adsorption (therefore the reciprocal of the constant is directly proportional to plasma protein binding (PB)) and the constant β is determined as the maximum specific adsorption of sulfonamide on plasma proteins at μmol g−1. The percentage of PB comes from the study of Othmer (2007).
The chromatographic data for some sulfonamides come from works of Koba et al. (2010) and Bober et al. (2011) for XTerra RP-18, XTerra RP-8, IAM PC C10/C3, AGP, Hypersil HSA, Nucleosil 100-5 OH, Discovery HS PEG, IC Pak Anion HR, IC Pak Cation M/D, Spheri Anion AX and Purospher STAR RP-18, Aluspher RP select B, Chromolith RP-18, and Supelcosil Plus ABZ.
Molecular descriptors
Nonempirical structural indicators (quantum-chemical indicators) were calculated during the study. The structure of compounds was studied by molecular modeling using the Gaussian 03W (Gaussian Inc., Wallingford, CT, USA). The geometry of the molecules was optimized using restricted Hartree-Fock 6–-1G (d, p) (6-31G**) (Official Gaussian Website 2019http://www.gaussian.com/). For compounds for which biological activity data were available, the optimal solutions would be optimization of the structures in aqueous medium, using the method of polarizable continuum model (Tomasi and Persico 1994; Tomasi et al. 2005; Caricato and Scalmani 2011); however, the size of most of the considered molecules on standard-class PC prevented this intention, and therefore we decided to use only ab initio results for the molecules in vacuo. On the other hand, semiempirical methods for the whole group of analyzed compounds in vacuo and in the aquatic environment were performed. The structures of the tested compounds were studied by molecular modeling using HyperChem v. 8.0 (Hypercube Inc., Gainesville, FL, USA) software. The geometry of the molecule was initially optimized by molecular mechanics MM+ and then using the semiempirical method RM1 (HyperChem® Computational Chemistry 1996). After completing the optimization, a single-point calculation was performed. The molecule was placed in a periodic box, in which dimensions were selected in such a way that the program has been placed within around 40 water molecules, and the optimization of the geometry was repeated in an environment of water molecules by RM1. Among the quantum-chemical indices were considered: total energy (TE), binding energy (BE), electron energy (EE), heat of formation (HF), energy of the highest-occupied molecular orbital, energy of the lowest vacant molecular orbital (ELUMO), and the difference of HOMO and LUMO energy defined as the energy gap (EG). Moreover, the following values were used: the spatial extent of the electron (ESE), the largest positive charge on the electron atoms (Q + MAX/MAX_POS), the largest negative charge on the electron atoms (Q-MIN/ MAX_NEG), the difference between the largest positive and negative charge (ΔQ/DELTA_Q), total dipole moment (TDM), the isotropic polarizability (IPOL) the mean polarizability (MPOL), and energy values for the most long-term transition of electron (EL) for which a power oscillator >0. The values of TE were expressed in atomic energy units (a.u.) = Hartree (1 Hartree = 2625.552 and 627.5095 kcal × mol−1 or 27.2116 eV), energy HOMO, LUMO, and EG expressed in eV (converted the above values into eV), and electron spatial extent of eBohr−3. The values of electron density and electron charges on the atoms were expressed in units of elementary charge (e−), dipole moment expressed in Debye (D), and the average polarizability in Bohr3 (Bohr = 0.5292 × 10−10 m = 0.5292 Å). For the structures optimized in aqueous medium, the following additional parameters were used: the surface of the molecule accessible to the solvent (SA), expressed in Å2, the volume of the molecule (V) in Å3, and the hydration energy (HE) in kcal mol−1 for both types of structures optimized by RM1 in vacuo and in the surrounding water molecules.
Furthermore, for the whole group of molecules, Dragon 7.0 (Kode Chemoinformatics, Pisa, Italy) software was used to calculate a huge set (>5200) of extra descriptors (Todeschini and Consonni 2010; Dragon 7 molecular descriptors 2019https://chm.kode-solutions.net/products_dragon.php).
Statistical analysis
The data that examined the biological activity of compounds were related to their structural indicators using multiparametric regression analysis/multiple regression analysis with a stepwise progressive method together with principal component analysis (PCA) and factor analysis (FA) implemented in Statistica 13 (StatSoft, Tulsa, OK, USA) on a personal computer. In PCA, a matrix of correlations has been diagonalized, and in FA, Varimax rotation has been performed.
Results and discussion
The numerical values of all 10 structural parameters derived from quantum-chemical calculations in vacuo from Gaussian software for the selected 24 antimicrobial sulfonamides are shown in Table 1S, the values of 16 parameters for the optimized structures in vacuo from HyperChem software are presented in Table 2S, and the structures in the aquatic environment in Table 3S are analyzed during the study. Table 1 presents the biological activity values for the sulfonamides and Table 2 presents the retention parameters obtained for the selected sulfonamides.
First of all, the analysis of PCA was performed for the geometry-optimized structures both for the isolated molecules or in a vacuum and in the aquatic environment to compare structural resemblance between the analyzed molecules.
In the case of the selected group of 24 sulfonamides, PCA in vacuo results from Gaussian are as follows: PC1 is around 32.98, PC2 30.72, and PC3 14.33% (Fig. 2). PC1 1 is mainly influenced by an EG, the highest negative charge of the atom of the molecule, TE, and the ELUMOs. PC2 is most influenced again by electron charges on atoms: the highest negative charge of the atom of the molecule, the ESE, and the IPOL. Analyzing the results of the hydrated form of sulfonamides from HyperChem, we get the following results: PC1 is around 45.15, PC2 17.40, and PC3 14.90%. PC1 1 is mainly influenced by TE, BE, and electron energy, and also EG, the difference between the largest positive and negative loads, the ELUMO, and HE. On the other side, PC2 is most influenced by energy values for the most long-term transition of the electron, the EG and TDM, but also electron energy and BE to a lesser extent. On the other hand, PCA in vacuo results from HyperChem are as follows: PC1 is around 49.66, PC2 16.46, and PC3 13.84%. PC1 1 is mainly influenced by EG, electron energy, TE, and BE,, and also the energy of the lowest vacant molecular orbitals, the difference between the largest positive and negative loads, the highest positive charge of the atom of the molecule, and energy values for the most long-term transition of the electron. PC2 is most influenced by TE and the highest negative charge of the atom of the molecule.

Three-dimensional scatter plots of the scores of the first three factors obtained by principal component analysis (PCA) of structural parameters derived from the quantum-chemical calculations in vacuo from the HyperChem software a and hydrated from the HyperChem software b for 23 antimicrobial sulfonamides (a PC1—49.66%, PC2—16.46%, and PC3—13.84%; b PC1—45.15%, PC2—17.40%, and PC3—14.90%)
In Fig. 2 presented with PCA, some of the sulfonamides sulfacarbamide, sulfacetamide, sulfamoxole, sulfanilamide, and sulfaguanidine in the case of structures in vacuo, and also sulfamoxole and sulfanilamide in the case of hydrated structures are out of the clusters formed by the rest of analyzed compounds.
Performed FA analysis for the molecules with the same calculation conditions coincides completely with the results obtained by PCA. Analyzing the molecules only with the use of HyperChem software, the following results were obtained: molecules in vacuo—FA1 49.66%, FA2 16.46%, and molecules in the aquatic environment—FA1 45.15%, FA2 17.40%.
Figure 3 with FA also shows some of the before-mentioned sulfonamides: sulfacarbamide, sulfacetamide, sulfamoxole, sulfanilamide, and sulfaguanidine in the case of both structures in vacuo and hydrated are out of the clusters formed by the rest of analyzed compounds.

Two-dimensional scatter plots of the scores first two factors obtained by FA structural parameters derived from the HyperChem quantum-chemical calculations from in vacuo a and hydrated b for selected antimicrobials (a FA1—49.66%, FA2—16.46%; b FA1—45.15%, FA2—17.40%)
In the next step, biological activity values were analyzed together with Gaussian and HyperChem descriptors (in vacuo from Gaussian, and in vacuo together within the aquatic environment from HyperChem). Since the values of biological activity were obtained for rather small collections of molecules (13–18 cases) for such small datasets, it was difficult to carry out external validation, so it was limited only to the internal validation leave-one-out together with progressive stepwise multiregression analysis; significant descriptors and equations were as follows:
Gaussian software in vacuo
HyperChem software in vacuo
HyperChem software aquatic environment
Analyzing the quantum-chemical parameters obtained for the biological and pharmacokinetic parameter values in the case of Gaussian in vacuo, the most influence for log1/MIC is IsoPol, for α IsoPol and MAX_POS, and for β TE and for PB again IsoPol and MAX_POS. On the other hand, in the case of HyperChem statistically important in vacuo quantum-chemical descriptors, we can distinguish MeanPol for log1/MIC and α, DELTA_Q for β and EE, and E_HOMO together with HF for PB, while in the aquatic environment, MeanPol for log1/MIC α and β and finally EE and E_HOMO for PB.
In the next step, so progressive stepwise multiregression analysis was performed, but with the use of a huge set of additional descriptors obtained by the professional software for the analyzed compounds with owned biological activity values and chromatographic retention data. Model equations for statistically significant descriptors are presented in the below set of derived equations:
The full list of molecular descriptors obtained from Dragon software characterizing biological activity with their designations is presented in Table 3.
Performed predictions with the use of professional software and a wide range of molecular descriptors provide a more detailed information about the studied molecules. The obtained statistically significant molecular descriptors belong to different classes, but we can distinguish some of the common one’s classes between them. In our opinion, it is difficult to indicate the most important equation between the proposed because the datasets are relatively small, and therefore we wanted to focus rather on the same block of descriptors appearing in the presented equations. Therefore, the most important group of descriptors characterizing biological activity of sulfonamides are the edge adjacency indices (SpMAD_EA(dm), Eig15_AEA, and Eig02_AEA(ed)) followed by 2D and 3D matrix-based descriptors (SpMAD_B(s), Wi_G/D) as appearing most often in proposed equations. Experimental equations confirmed the very important role of electronic properties of the molecules in antimicrobial activity of analyzed sulfonamides.
Finally, with possessed chromatographic retention parameters, also progressive stepwise multiregression analysis was performed. The chromatographic retention data parameters described with statistically significant descriptors from Gaussian, HyperChem, and Dragon software are presented in Table 4S. In the case of Gaussian, in vacuo statistically significant molecular descriptors characterizing chromatographic parameters the most often appear: IsoPol, ESE, and E_LUMO and finally MAX_POS and MAX_NEG. On the other hand, the most often appearing in vacuo descriptors from HyperChem are MeanPol, BE, and TE, while the most common descriptors from HyperChem in the aquatic environment also represent MeanPol, BE, and E_HOMO together with EL. By analyzing Dragon’s statistically significant descriptors described with the retention parameters on the different chromatographic columns, we can select not only the same descriptor groups, but also exactly the same in several cases, between them are also again edge adjacency indices (SM06_EA(bo), SM08_EA(bo), Eig08_AEA(ed), Eig03_EA(ri), and Eig09_EA(ri)), 2D atom pairs descriptors (F04[C–O], F06[C–S], B06[C–S], and B08[C–O]), 3D-MoRSE descriptors (Mor26p, Mor12i, and Mor12u), Burden eigenvalues (SpMin1_Bh(s), SpMax2_Bh(p)), and finally GETAWAY and WHIM descriptors. All additional information pay attention to the role of the lipophilicity of analyzed sulfonamides in connection with atom fragment elements and simultaneously shape the geometry and van der Waals surface area of the molecules.
Conclusion
Based on the above discussion of the results that may be put forward, the following conclusion is presented. The set of analyzed molecules, for which optimization of the structures in the aquatic environment and in a vacuum was performed, parameters characterizing the classification are mostly compatible in all cases. In a set containing molecules for which optimization of the structure of isolated molecules was carried out and molecular parameters were calculated by the Hartree–Fock method, 6–31G with polarization functions d, p and in the presence of water molecules, where molecular parameters were calculated by the semiempirical method RM1, shows a classification partially consistent with the chemical structure and biological activity. By analyzing quantum-chemical descriptors characterizing biological activity values obtained from HyperChem for the molecules in vacuo and in the aquatic environment, we can observe that they are comparable, but different from those for the Gaussian in vacuo molecules, while Dragon descriptors provide much more detailed information about the studied molecules of antimicrobial sulfonamides. Among the different parameters, the most important for the antimicrobial activity of sulfonamides confirmed repeated values of electronic property descriptors of analyzed molecules in proposed model equations characterizing both biological and retention parameters of sulfonamides. The above-presented method after extending the database with other amphoteric (zwitterionic) and monofunctional (acidic or alkaline) compounds could be used both for the initial classification of biologically relevant agents and could be included in the set of methods of QSAR analysis and design, as well as help in the verification of the hypothesis about functional dependencies between chromatographic or spectroscopic and nonempirical parameters. It seems advisable also to continue research in the extended database of molecules and their experimental values of pharmacological/physicochemical properties.
References
Agrawal VK, Srivastava R, Khadikar PV (2001) QSAR studies on some antimalarial sulfonamides. Bioorg Med Chem 9:3287–3293
Belka M, Konieczna L, Kawczak P, Ciesielski T, Slawinski J, Baczek T (2012) The chemometric evaluation of antitumor activity of novel benzensulfonamide derivatives based on their physiochemical properties. Lett Drug Des Discov 3:288–294
Belka M, Sławinski J, Konieczna L, Kawczak P, Ciesielski T, Baczek T (2013) Antitumor activity of novel benzensulfonamide derivatives in view of their physiochemical properties searched by principal component analysis. Med Chem 9:517–525
Bell PH, Roblin Jr RO (1942) Studies in chemotherapy. VII. A theory of the relation of structure to activity of sulfanilamide type compounds1. J Am. Chem Soc 64:2905–2917
Brent DA, Sabatka JJ, Minick DJ, Henry DW (1983) A simplified high-pressure liquid chromatography method for determining lipophilicity for structure-activity relationships. J. Med Chem 26:1014–1020
Bober L, Nasal A, Kuchta A, Kaliszan R (1998) Chemometric analysis of high-performance liquid chromatography retention data, structural parameters and biological activity of sulphonamides. Acta Chromatogr 8:48–69
Bober L, Koba M, Judycka-Proma U, Bączek T (2011) Pharmacological classification of drugs by principal component analysis applying molecular modeling descriptors and HPLC retention data. J Chromatogr Sci 49:758–763
Bober L, Kawczak P, Bączek T (2012a) QSAR analysis of compounds exhibiting general anesthetics’ properties. Lett Drug Des Discov 9:595–603
Bober L, Kawczak P, Bączek T (2012b) Pharmacological classification and activity evaluation of furan and thiophene amide derivatives applying semi-empirical ab initio molecular modeling methods. Int J Mol Sci 13:6665–6678
Boufas W, Dupont N, Berredjem M, Berrezag K, Becheker I, Berredjem H, Aouf NE (2014) Synthesis and antibacterial activity of sulfonamides. SAR and DFT studies. J Mol. Struct 1074:180–185
Ciura K, Belka M, Kawczak P, Bączek T, Markuszewski MJ, Nowakowska J (2017) Combined computational-experimental approach to predict blood-brain barrier (BBB) permeation based on “green” salting-out thin layer chromatography supported by simple molecular descriptors. J Pharm Biomed Anal 143:214–221
Caricato M, Scalmani G (2011) On the importance of the orbital relaxation in ground-state coupled cluster calculations in solution with the polarizable continuum model of solvation. J Chem Theory Comput 7:4012–4018
Chen C, Wang D, Wang H, Lin Z, Fang Z (2017) A SAR-based mechanistic study on the combined toxicities of sulfonamides and quorum sensing inhibitors on Escherichia coli. SAR QSAR Environ Res 28:595–608
De Benedetti PG, Rastelli A (1978) Electronic aspects of the antibacterial action of sulfanilamides. J Med Chem 21:1325–1327
Dragon 7 molecular descriptors. https://chm.kode-solutions.net/products_dragon.php. Accessed 2 Jan 2019
Henry D, Block JH, Anderson JL, Carlson GR (1976) Use of high-pressure liquid chromatography for quantitative structure-activity relationship studies of sulfonamides and barbiturates. J Med Chem 19:619–626
HyperChem® Computational Chemistry (1996) Part 1. Practical guide. Part 2 Theory and methods. Hypercube Inc., Waterloo
Kaliszan R, Ośmiałowski K, Kobyłczyk K (1979) In vitro activity of sulphonamides as a function of their molar refractivity. Chemotherapy 25:5–8
Kaliszan R (1987) Quantitative structure—chromatographic retention relationships. Wiley, New York
Kaliszan R (1997) Structure and retention in chromatography. A chemometric approach. Harwood Academic Publishers, Amsterdam
Kaliszan R (2007) QSRR: Quantitative Structure-(Chromatographic) Retention Relationships. Chem Rev 107:3212–3246
Kawczak P, Bober L, Bączek T (2014) Biological activity of compounds exhibiting local anesthetics’s properties evaluated by QSAR approach. Curr Pharm Anal 10:255–262
Kawczak P, Bober L, Bączek T (2015) QSPR analysis of some agonists and antagonists of α-adrenergic receptors. Med Chem Res 24:372–382
Kawczak P, Bober L, Bączek T (2018a) Activity evaluation of some psychoactive drugs with the application of QSAR/QSPR modeling methods. Med Chem Res 27:2279–2286
Kawczak P, Bober L, Bączek T (2018b) Application of QSAR analysis and different quantum chemical calculation methods in activity evaluation of selected fluoroquinolones. Comb Chem High Throughput Screen 21:468–475
Kawczak P, Bober L, Bączek T (2018c) QSAR analysis of selected antimicrobial structures belonging to nitro-derivatives of heterocyclic compounds. Lett Drug Des Discov. https://doi.org/10.2174/1570180815666181004112947
Kawczak P, Bober L, Bączek T (2019) Evaluation of chemotherapeutic activity of the selected bases analogues of nucleic acids supported by ab initio various quantum chemical calculations. Curr Comput Aided Drug Des. https://doi.org/10.2174/1573409915666190206212024
Koba M, Bober L, Judycka-Proma U, Bączek T (2010) Influence of HPLC retention data and molecular modeling descriptors on prediction of pharmacological classification of drugs using factor analysis method. Comb Chem High T Scr 13:765–776
Krátký M, Vinšová J, Volková M, Buchta V, Trejtnar F, Stolaříková J (2012) Antimicrobial activity of sulfonamides containing 5-chloro-2-hydroxybenzaldehyde and 5-chloro-2-hydroxybenzoic acid scaffold. Eur J Med Chem 50:433–440
Krüger-Thiemer E, Bünger P (1965) The role of the therapeutic regimen in dosage design. Part II Chemotherapy 10:129–144
Mizdal CR, Stefanello ST, Nogara PA, Antunes Soares FA, de Lourenço Marques L, de Campos MMA (2018) Molecular docking, and anti-biofilm activity of gold-complexed sulfonamides on Pseudomonas aeruginosa. Microb Pathog 125:393–400
Mondal S, Mandal SM, Mondal TK, Sinha C (2017) Spectroscopic characterization, antimicrobial activity, DFT computation and docking studies of sulfonamide Schiff bases. J Mol. Struct 1127:557–567
Naaz F, Srivastava R, Singh A, Singh N, Verma R, Singh VK, Singh RK (2018) Molecular modeling, synthesis, antibacterial and cytotoxicity evaluation of sulfonamide derivatives of benzimidazole, indazole, benzothiazole and thiazole. Bioorg Med Chem 26:3414–3428
Official Gaussian Website. http://www.gaussian.com/. Accessed 2 Jan 2019
Othmer K (2007) Kirk-Othmer Encyclopedia of Chemical Technology, 5th edn. Wiley, Hoboken
Rastelli A, de Benedetti PG, Battistuzzi GG (1975) Role of anionic, imidic, and amidic forms in structure-activity relations. Correlation of electronic indexes and bacteriostatic activity in sulfonamides. J Med Chem 18:963–967
Seydel JK (1971) Prediction of the in vitro activity of sulfonamides synthesized from simple amines by use of electronic data obtained from the simple amines. J. Med Chem 14:724–729
Singh S (2015) Computational design and chemometric QSAR modeling of Plasmodium falciparum carbonic anhydrase inhibitors. Bioorg Med Chem Lett 25:133–141
Stasiak J, Koba M, Bober L, Kawczak P, Baczek T (2013) The comparison between the calculated and HPLC-predicted lipophilicity parameters for selected groups of drugs. Comb Chem High Throughput Screen 16:603–617
Swain SS, Paidesetty SK, Padhy RN (2017) Antibacterial activity, computational analysis and host toxicity study of thymol-sulfonamide conjugates. Biomed Pharmacother 88:181–193
Todeschini R, Consonni V (2010) Molecular descriptors for chemoinformatics: volume I: alphabetical listing/volume ii: appendices, references, volume 41. Wiley, Weinheim
Tomasi J, Persico M (1994) Molecular interactions in solutions: an overview of methods based on continuous distributions of the solvent. Chem Rev 94:2027–2094
Tomasi J, Mennucci B, Cammi R (2005) Quantum mechanical continuum solvation models. Chem Rev 105:2999–3093
Von Rieder J (1963) Physicalisch-chemische und biologische untersuchungen an sulfonamiden. Arzneim Forsch 13:81–88
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kawczak, P., Bober, L. & Bączek, T. The comparison of semiempirical and ab initio molecular modeling methods in activity and property evaluation of selected antimicrobial sulfonamides. Med Chem Res 28, 778–787 (2019). https://doi.org/10.1007/s00044-019-02334-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-019-02334-4