
Abstract
Key message
This first pan-Mediterranean analysis of genetic diversity in wild narrow-leafed lupin revealed strong East–West genetic differentiation of populations, an historic eastward migration, and signatures of genetic adaptation to climatic variables.
Abstract
Most grain crops suffer from a narrow genetic base, which limits their potential for adapting to new challenges such as increased stresses associated with climate change. Plant breeders are returning to the wild ancestors of crops and their close relatives to broaden the genetic base of their crops. Understanding the genetic adaptation of these wild relatives will help plant breeders most effectively use available wild diversity. Here, we took narrow-leafed lupin (Lupinus angustifolius L.) as a model to understand adaptation in a wild crop ancestor. A set of 142 wild accessions of narrow-leafed lupin from across the Mediterranean basin were subjected to genotyping-by-sequencing using Diversity Arrays Technology. Phylogenetic, linkage disequilibrium and demographic analyses were employed to explore the history of narrow-leafed lupin within the Mediterranean region. We found strong genetic differentiation between accessions from the western and eastern Mediterranean, evidence of an historic West to East migration, and that eastern Mediterranean narrow-leafed lupin experienced a severe and recent genetic bottleneck. We showed that these two populations differ for flowering time as a result of local adaptation, with the West flowering late while the East flowers early. A genome-wide association study identified single nucleotide polymorphism markers associated with climatic adaptation. Resolving the origin of wild narrow-leafed lupin and how its migration has induced adaptation to specific regions of the Mediterranean serves as a useful resource not only for developing narrow-leafed lupin cultivars with greater resilience to a changing climate, but also as a model which can be applied to other legumes.
Similar content being viewed by others


Introduction
Agriculture is facing the ‘perfect storm’ of factors that threaten global food security including continued population growth, a rapidly changing climate, reduced water available to agriculture, soil erosion and increasing costs of fertilisers (Abberton et al. 2015; Gomiero 2016). A major concern is the lack of diversity in modern cropping systems to adapt to these challenges. For example, the human population derives more than 50% of its calorific intake from just three crop species (Awika 2011). This is largely reflected in the concentration of research and breeding investments into major crops, which serves to widen the gap between well-resourced major crop species and minor crops (also known as ‘neglected under-utilised species’). A wider range of crop species is required to efficiently exploit all available agricultural environmental niches in a sustainable manner (Abberton et al. 2015). Legumes have a special role in this given their ability to raise soil fertility from biological nitrogen fixation, system sustainability and improve human health (Foyer et al. 2016). Depleted diversity is also apparent within most modern crops relative to their wild progenitors due to historic population bottlenecks associated with domestication, breeding and geographic translocation around the world (Gepts 2010). Therefore, there is a strong need to understand and harness adaptive diversity from the wild ancestors and relatives of crop species (Berger et al. 2013; Maxted et al. 2012).
Evolutionary adaptation is one way that wild species may overcome the changes in their environment. For example, climate change has proven to act as a strong selective agent, driving new directions for natural selection in evolutionary adaptation (Hoffmann and Sgro 2011). Rapid adaptive evolutionary changes in response to climatic fluctuations have been reported in both plants and animals, such as those altering flowering phenology of Brassica rapa (Franks et al. 2007) and changes in migratory behaviour of birds (Pulido and Berthold 2010). However, the success of evolutionary adaptation is contingent on the availability of genetic diversity within species and genetic control (heritability) of adaptive traits. A recent study in New World lupin species showed there is more frequent genome-wide adaptation in rapidly diversifying species, as opposed to slowly diversifying species and plant species more generally (Nevado et al. 2016). The genetic basis of local adaptation can be explored using a genome-wide scan approach, as demonstrated for soil salinity adaptation of Medicago truncatula populations (Friesen et al. 2010). Migration by natural means or by human intervention (in the case of agricultural species) is another driver of adaptive change, particularly for sessile species such as plants. For example, a genetic differentiation analysis of wild barley using diversity array technology (DArT) and single nucleotide polymorphism (SNP) markers showed that wild barley migrated from Near East Fertile Crescent to Tibetan Plateau, where they have been adapted to the Tibet high altitude and its severe climate (Dai et al. 2012).
Maximising our exploitation of the diversity in wild populations, including materials from diverse climates, will significantly assist breeding and conservation planning (Franks et al. 2007; Hoffmann and Sgro 2011). Despite its importance, relatively little research has focused on how crop wild relatives adapt to their native environments, with much research focusing instead on the model plants Arabidopsis thaliana (Weigel and Nordborg 2015) and Medicago truncatula (Yoder et al. 2014). The capability of next generation sequencing technologies to generate data for a range of non-model species has transformed our ability to identify the genes underpinning adaptation (Stapley et al. 2010). For example, the adaptation of Arabidopsis lyrata to serpentine soils was found at least partly due to gene copy number variants (Turner et al. 2010). One productive avenue of research is to identify genetic variation for climatic adaptation by sampling accessions across environmental gradients such as aridity or temperature (Berger and Ludwig 2014). The analysis may be considered a “reverse ecology” approach for identifying loci that may underlie adaptation to clinal climate variation by genome-wide association with bioclimatic variables as proxy traits for environmental adaptation—without prior knowledge of specific traits mediating that adaptation (Yoder et al. 2014). The rationale of the approach is that if there is genetic adaptation along climatic gradients, then regions of the genome under strong genetic selection for adaptation may be identified. One aim of this study is to test this hypothesis.
The critical need for understanding and conserving crop wild relative diversity motivated Vincent et al. (2013) to develop a prioritised crop wild relative inventory for 1667 taxa. Here, we focus on one of those taxa—narrow-leafed lupin (Lupinus angustifolius L.)—as a model to study migration and climatic adaptation in the ancestral wild populations of this species. Narrow-leafed lupin (Lupinus angustifolius L.) is an important minor grain legume crop, being grown over 750,000 hectares mainly in Australia, Poland, Russia and Germany (FAO 2014). It has a unique adaptation to infertile, light-textured and acidic soils, where many other crops fail to thrive (Gladstones 1998). As with other members of the Lupinus genus, narrow-leafed lupin has the ability for co-adaptation with nitrogen-fixing root symbiont Bradyrhizobium sp. and to effectively mobilise soil phosphorus, which makes them of great importance in sustainable farming in rotation with cereals (Lambers et al. 2013; Seymour et al. 2012). Currently, narrow-leafed lupin grain is used mainly for animal feed and in aquaculture but interest is growing in its use in the human diet (e.g. as components in bread, pasta and other products), due to the excellent nutritional properties of the seeds, being gluten-free, high in protein and dietary fibre, and low in starch and fat (Caballero et al. 2015).
Wild narrow-leafed lupin exhibits the winter-annual life-cycle typical of many Mediterranean species. Seeds germinate in the autumn with the onset of rains, establish in rosette form during the winter and then in spring when temperature rises and photoperiod increases, growth increases rapidly and the plants transition to flowering (Kurlovich and Heinanen 2002). Time of flowering of wild lupins is mediated by their responsiveness to vernalisation (cold conditions), day length and ambient temperature (Berger et al. 2012b). We are only now beginning to understand the genetic control of phenology in narrow-leafed lupin. While wild narrow-leafed lupin has a Mediterranean distribution, its adaptation to summer cultivation in northern Europe and winter cultivation in southern Australia was achieved through the removal of the vernalisation response, which was recently shown to be controlled by a mutation in a Flowering Locus T (FT) gene (Nelson et al. 2017). A recent study in three Old World lupin species, including L. angustifolius, L. albus and L. luteus, demonstrated that phenology in all three has been under strong selection along aridity gradients (Jens Berger, unpublished data). While narrow-leafed lupin domestication was only completed in the twentieth century, there is extensive evidence of its use by Mediterranean cultures for centuries (Gladstones 1970). This is reflected in enlarged seeds on plants growing near agricultural sites (Gladstones and Crosbie 1979) and a wealth of morphology diversity in the Aegean region consistent with extensive movement of seed around that region (Clements and Cowling 1994). The centre of origin of narrow-leafed lupin itself is unknown.
Narrow-leafed lupin cultivars possess a very limited adaptive range relative to their wild ancestors (Berger et al. 2012a). Fortunately, extensive and well-annotated collections of genetic resources of wild narrow-leafed lupin are available (Berger et al. 2013; Wolko et al. 2011). This, together with the recent release of a comprehensive reference genome for narrow-leafed lupin (Hane et al. 2017), provides an excellent opportunity to study regional adaptation in wild narrow-leafed lupin within their full native range. To this end, using different genomics and bioinformatics approaches, we addressed the following questions: (1) How does genetic diversity in narrow-leafed lupin vary across its native range? (2) What can we deduce about the demographic history of this species? and (3) Can we detect regions of the genome associated with climatic adaptation? The information will be valuable to understand the origins of a wild crop ancestor such as narrow-leafed lupin, which serves as a model to identify sources of genetic and adaptive diversity to adapt crops to changing environments.
Materials and methods
Plant materials and climatic variables
A total of 142 wild accessions of narrow-leafed lupin from 11 countries across the Mediterranean basin were obtained from the Australian Lupin Collection, Department of Agriculture and Food Western Australia (DAFWA; Online Resource 1). This subset of 142 accessions were selected from 1248 wild accessions characterised by Berger et al. (2012a) using 137 array-based DArT markers, and encompassed the maximum genetic and phenotypic diversity from across the geographic range for this species (Gladstones 1998). Phenotypic data including alkaloid status, pod dehiscence, physical dormancy (hard versus soft seededness), rain, soil pH at collection site, flower colour, time to flowering from sowing date (flowering time), height at maturity and 100 seed weight were from a study of Gladstones and Crosbie (1979). Additional passport information was kindly provided by the Australian Lupin Collection, including the geographic coordinates of accessions from geo-referencing of collection site data. Geographic coordinates for each accession were used to extract 19 climatic variables drawn from the years 1960–1990 from WorldClim (http://www.worldclim.org/bioclim) (Hijmans et al. 2005; WorldClim 2006), using ArcMap GIS (Geographic Information System) software 10.3.1 version (Esri, CA, USA). Information on geographic, phenotypic and WorldClim variables are provided in Online Resource 1.
DArTseq genotyping
DNA was extracted from leaves of single plants from each accession using a standard CTAB method (Doyle and Doyle 1990). The quality and quantity of extracted DNA were assessed using standard agarose electrophoresis and Qubit™ assays (http://www.Invitrogen.com/qubit).
The DNA concentration of each sample was adjusted to 20 ng/μL and subjected to DArTSeq™ (hereafter, DArTseq) genotyping at Diversity Arrays Technology Pty Ltd, Canberra, Australia (Sansaloni et al. 2011). DNA samples are processed in digestion/ligation reactions principally as per Kilian et al. (2012), but replacing a single PstI-compatible adaptor with two different adaptors corresponding to two different Restriction Enzyme overhangs. The PstI-compatible adapter was designed to include Illumina flowcell attachment sequence, sequencing primer sequence and staggered, varying length barcode region, similar to the sequence reported by Elshire et al. (2011). Reverse adapter contained flowcell attachment region and MseI-compatible overhang sequence.
Only “mixed fragments” (PstI-HpaII) were effectively amplified in 30 rounds of PCR using the following reaction conditions: 94 °C for 1 min, then 30 cycles of 94 °C for 20 s, 58 °C for 30 s and 72 °C for 45 s, finishing with 72 °C for 7 min. After, PCR equimolar amounts of amplification products from each sample of the 96-well microtiter plate were bulked and applied to c-Bot (Illumina) bridge PCR followed by sequencing on Illumina Hiseq2500. The sequencing (single read) was run for 77 cycles.
Sequences generated from each lane were processed using proprietary DArT analytical pipelines. In the primary pipeline, the fastq files were first processed to filtre away poor quality sequences, applying more stringent selection criteria to the barcode region compared to the rest of the sequence. Approximately 2,500,000 (± 7%) sequences per barcode/sample were used in marker calling. Finally, identical sequences were collapsed into “fastqcall” files. These files were used in the secondary pipeline for DArT PL’s proprietary SNP and SilicoDArT (presence/absence of restriction fragments in representation) calling algorithms (DArTsoft14). Co-dominant SNP markers were the focus of the study, as they are able to distinguish heterozygous and homozygous loci, which was necessary for downstream analyses, and were filtered using different thresholds for various analyses. For demography analyses, SNP markers which had positions mapped to pseudo-chromosomes were used. For phylogenetic, population structure, linkage disequilibrium and association studies, SNPs with more than ≥ 25% missing data or ≥ 12.8% heterozygosity were eliminated. DArTseq reads were aligned to the L. angustifolius cv. Tanjil reference genome (Hane et al. 2017) using NUCmer aligner distributed with MUMmer (Delcher et al. 1999), with setting the minimum cluster length parameter (-c flag) to 25 bp, and allowing ≤ 3 matches. Then, the position of SNPs relative to Tanjil reference genome was determined from SNP positions on the DArTseq reads and the reads’ match position on the reference, summarised in Variant Call Format (VCF) relative to the lupin reference genome (Hane et al. 2017). The VCF file was validated using the Genome Analysis Toolkit (GATK) (McKenna et al. 2010).
Assessing population structure and climate adaptation across the Mediterranean basin
Two different methods were employed to identify population structure: Principal Component Analysis (PCA) was performed using EIGENSTRAT to assess genetic diversity and to correct for population stratification (Price et al. 2006). We also used fastSTRUCTURE at K = 2 to K = 12, using default parameters (Raj et al. 2014). The estimation of optimum K was obtained using the algorithm implemented in fastSTRUCTURE to choose model complexity (Raj et al. 2014). To understand the relationship between phylogeny and geographical distribution, we used GenGIS 2.4.0 (Parks et al. 2013). The unrooted distance-based phylogeny tree, which was implemented as Newick format in GenGIS, was obtained using Neighbor Joining method within the NEIGHBOR package in PHYLIP (Felsenstein 1989).
A Pearson correlation coefficient heat-map was calculated using the heat-map function in R version 3.3.0 (R Core Team 2016) for all phenotypic characteristics and climate data extracted from WorldClim.
To test the association between the SNP markers and the WorldClim variables which were significantly correlated with important agronomic traits such as flowering time—referred to hereafter as climatic variables—we carried out genome-wide association studies (GWAS) using GAPIT (Lipka et al. 2012) with the first two principal components as covariates, for a dominant model and a minor allele frequency (MAF) cutoff of 0.01.
Next, SNPs that were highly associated with our traits of interest were analysed using SnpEff version 4.3 (Cingolani et al. 2012), which annotates genetic variants and predicts their effects on genes. SnpEff annotations were compared with the predicted proteins reported by Hane et al. (2017).
GenStat version 16.2 (VSN International 2011) was used to perform analysis of variance (ANOVA) among groups of our accessions (East/West Mediterranean) which were inferred from GenGIS phylogeny result. This was undertaken to estimate the significance of variation between phenotypic traits and climatic variables of different samples belonging to separate geographic groups.
Linkage disequilibrium (LD) as measured by r2 was calculated for all values (–ld-window-r2 0) for every SNP within a window of 1 Mb using Plink (Purcell et al. 2007). The mean r2 values pooled over all 20 chromosomes for western and eastern wild germplasm were calculated and plotted using R v3.3.0 (R Core Team 2016). Analysis of haplotype blocks was performed using Haploview (Barrett et al. 2005).
Demographic analyses
To infer the demographic history of the wild populations of L. angustifolius, we used the models implemented in the software dadi (Gutenkunst et al. 2009). This method uses the site frequency spectrum (SFS) of populations, i.e. the distribution of allele frequencies across SNPs, to infer the demographic history of populations. The observed SFS was compared to the expected SFS under different demographic models, and using Maximum Likelihood, estimated both the likelihood of the different models and their parameters values.
For SFS analysis, we included all 38,948 SNPs with known position in the pseudo-chromosomes of the L. angustifolius cv. Tanjil genome assembly (Hane et al. 2017). Based on the fastSTRUCTURE and phylogeny results, we defined three wild populations for this analysis: the western population consisting of samples from Morocco, Portugal and Spain (n = 74); the central population containing samples from France and Italy (n = 21); and the eastern population containing the samples from Greece, Cyprus and Turkey (n = 45). As the ancestral state of each SNP is uncertain, we folded the SFS in dadi before fitting the Isolation with Migration (IM) model. Preliminary analysis suggested that singletons, i.e. alleles found only once in the population, were under-represented in all populations, suggesting a bias in the SNP calling protocol. To avoid biasing the demographic inferences due to this, we excluded all singletons from the calculations of the SFS.
We performed two types of analyses. First, we investigated the demographic history of each population separately, by fitting the default single population models available in dadi (Table 1). We then investigated the Isolation with Migration (IM) model, which compares pairs of populations and estimates their relative size, time of divergence, the population size changes for each population, and the amount of gene flow in each direction (Table 1). To ensure convergence of the parameter estimates, for each model investigated we performed 20 runs with random starting values for each parameter and large search regions. After analysis of these results, we defined smaller regions of parameter space with high likelihood and performed a second round of 20 searches from random starting points within this region.
Results
DArTseq analysis generated 45,230 co-dominant SNPs markers in 142 wild narrow-leafed lupin accessions. For the demography analyses, we used a subset of SNPs that mapped to pseudo-chromosomes which included 38,948 SNPs. For other analyses, loci with excessive missing and heterozygote values were removed (16,714 SNPs remained). Due to the evidence of whole-genome triplication in the genome of narrow-leafed lupin (Hane et al. 2017; Kroc et al. 2014), we kept those SNPs that had a maximum of three matches in the reference genome (11,690 SNPs remained). Consequently, these 11,690 SNPs were utilised for phylogenetic, population structure, linkage disequilibrium and association studies. The allelic profile per accession for these 45,230 and 11,690 SNP sets are reported in the Online Resource 2 and 3, respectively.
Population structure
Principal component analysis (PCA) depicted a vivid division between East and West Mediterranean accessions (Fig. 1). The first and second principal components (PCs), together accounted for 29% of the genetic variation, and despite a few exceptions (mainly from Italy and France), it differentiated the genotypes into two groups, western and eastern Mediterranean comprising 97 and 45 accessions, respectively (Online Resource 4).

Principal component analysis (PCA) for 142 accessions of wild narrow-leafed lupin labelled by country of origin
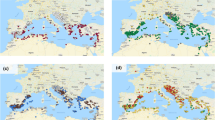
The second method we used to probe the relationships among accessions was the Bayesian clustering algorithm applied in fastSTRUCTURE programme (Raj et al. 2014). To reveal population structure, we investigated different numbers of populations from 2 to 12 (K2–12) (Online Resource 5). The internal algorithm in fastSTRUCTURE for multiple choices of K determined that population numbers K = 7 or K = 8 best explained the variation in the dataset. To determine which one of these two models (K = 7 or K = 8) better fitted the data, we subjected both models to phylogenetic analysis. Again, as both models appeared plausible, the simpler model (K = 7) was selected (Fig. 2a, b). We used a frequency threshold of > 0.7 to assign accessions to their corresponding populations. Where accessions had population affinity values below 0.7, they were categorised as Admixed. Structure grouping depicted seven populations: one population for the entire eastern Mediterranean region (Population 5) and the remaining six populations for the western Mediterranean region indicating greater stratification centred on the Iberian Peninsula. Admixed populations were distributed across the Mediterranean but were particularly frequent at the intersection of East and West (centred on Italy).

Population stratification among 142 accessions of wild narrow-leafed lupin (K = 7) using fastSTRUCTURE. Each colour denotes a population affiliation. a Seven population groups identified among 142 wild narrow-leafed lupin accessions. b GenGIS plot showing geographic distribution of seven fastSTRUCTURE groups. Dots represent collection sites of accessions colour-coded by fastSTRUCTURE group (K = 7), with grey shading denoting admixed accessions. Pie charts represent the frequencies of populations within each country using the same colour coding (colour figure online)
Phylogenetic analyses
Phylogenetic relationships were captured within an unrooted tree based on distances calculated from 11,690 DArTseq SNP markers. This shows a distinct East/West Mediterranean division, with most accessions from Morocco, Algeria, Spain, Portugal and France clustering together as a western group, while the remaining accessions mainly clustered together as an eastern group (Fig. 3). This delineation into East/West groups was generally congruent with the PCA and fastSTRUCTURE-defined groupings (Online Resource 4).

Phylogenetic analysis of 142 accessions of wild narrow-leafed lupin using GenGIS. Accessions are colour coded based on their country of origin, as following: orange Israel, dark green Syria, turquoise blue Cyprus, sky blue Turkey, purple Greece, dark blue Italy, pink France, red Spain, yellow Portugal, light green Morocco, black Algeria (colour figure online)
The phylogenetic grouping of most accessions corresponded to their geographical locations with the exception of three accessions from Spain and Portugal which were clustered within the eastern group (accessions P22666, P28221 and P26423), and seven accessions from Italy and one accession from Greece that fell within the western clusters (Accessions P20720, P20724, P25040, P25051, P25052, P26107, P26109 and P26991).
Investigating historic population bottlenecks
To explore further how genomic diversity differs between the distinct eastern and western Mediterranean population groups and to infer the historic levels of genetic diversity in each population group, we investigated linkage disequilibrium (LD) in these two population groups and conducted demographic analyses. The western population group showed far lower average maximum LD (0.2) than the eastern population group (0.4), and the LD decayed more rapidly in the western populations (Fig. 4). We investigated population bottleneck further through demographic analyses.

Decay of genome-wide linkage disequilibrium in 142 wild narrow-leafed lupin accessions for western and eastern populations
Demography analyses
The different single population models investigated returned very consistent results, with the western population group showing relatively small population size changes and an older age, while the eastern population group appearing much younger, having experienced a recent and severe bottleneck, and to have subsequently increased its population size considerably (Table 2). There was also evidence that a small central group (comprising accessions identified as admixed in the fastSTRUCTURE analysis) was distinguishable from western and eastern groups. Like the eastern population group, the central population group showed evidence of a recent and severe bottleneck followed by population increase.
We then considered the Isolation with Migration (IM) model with each pair of populations. The IM model had six free parameters: the relative size of the two populations after they split, time of the split, modern population sizes of each population and effective migration rates in two directions. We found that population split sizes were highly unequal, with each pairwise comparison suggesting that the eastern population emerged from a very small proportion of the ancestral population (s = 0.95–0.99, Table 3). The extent of migration from West to East was much higher than in the reverse direction (m12 = 0.06–1.13 while m21 = 9.9–15.11, Table 3).
Investigating adaptation of wild narrow-leafed lupin in West/East Mediterranean
We investigated adaptation parameters distinguishing West and East populations. Climatic variables from the sites of origin of each accession (extracted from the WorldClim database) were correlated with phenotypic traits (number of days to flowering, hard/soft seededness, pod dehiscence status, height at maturity and 100-seed weight). As separate correlation analyses for Western and Eastern populations showed similar trends, we only present one correlation matrix for both West and East together (Fig. 5). Variation in flowering time was significantly (at p < 0.01 and degree of freedom of 140) associated with some climatic/eco-geographic variables, including precipitation in different months/quarters (BIO14 and BIO17, respectively), latitude and annual mean temperature (BIO1). Positive correlations were observed between flowering time and latitude, and between flowering time and precipitation in the driest month and driest quarter, while the correlation between flowering time and annual mean temperature was negative.

Pearson correlation heat-map of climatic and geographic variables with phenotypic traits for 142 narrow-leafed lupin accessions from across the Mediterranean Basin. WorldClim data abbreviations are as follows: BIO1 annual mean temperature, BIO2 mean diurnal range [mean of monthly (max temp − min temp)]; BIO3 isothermality (BIO2/BIO7) (× 100), BIO4 temperature seasonality (standard deviation × 100), BIO5 max temperature of warmest month, BIO6 min temperature of coldest month, BIO7 temperature annual range (BIO5–BIO6), BIO8 mean temperature of wettest quarter, BIO9 mean temperature of driest quarter, BIO10 mean temperature of warmest quarter, BIO11 mean temperature of coldest quarter, BIO12 annual precipitation, BIO13 precipitation of wettest month, BIO14 precipitation of driest month, BIO15 precipitation seasonality (coefficient of variation), BIO16 precipitation of wettest quarter, BIO17 precipitation of driest quarter, BIO18 precipitation of warmest quarter, BIO19 precipitation of coldest quarter. Temperatures are in degrees Celsius × 10 and precipitation in millimetres
Exploring phenotypic variation corresponding to geographical location
We next sought to determine how geographical location (East or West Mediterranean, inferred from phylogenetic tree) could explain observed phenotypic variation. In total, 53 and 89 accessions were labelled as eastern and western groups, respectively (Online Resource 1). A one-way analysis of variance (ANOVA) indicated there was significant (p < 0.05) variation in phenotypic traits (such as flowering time) and climatic variables (e.g. Precipitation of driest month/quarter, etc.) between samples from West/East Mediterranean. Details of significant variation traits are provided in Table 4. It was clear that flowering time differed between West (late flowering) and East (early flowering) populations (p < 0.05 Table 4) and that flowering time was significantly correlated with some climatic variables such as precipitation of driest month/quarter, etc. Based on this, we next looked for regions of the narrow-leafed lupin genome that appear to be associated with adaptation.
Identifying regions of the genome associated with adaptation
First, we focused in more detail on the distribution of LD across the genome. Vernalisation is a key driver of flowering time in narrow-leafed lupin, which is controlled by the FT homologue LanFTc1 at the Ku locus in domesticated varieties (Nelson et al. 2017). We, therefore, compared the haplotype block of East (Fig. 6a) and West (Fig. 6b) populations in the LanFTc1 gene region on chromosome NLL-10. Although the overall pattern of haplotype blocks in the LanFTc1 region was similar between West/East populations, the comparison revealed evidence of greater historical recombination within western Mediterranean population compared to East (Fig. 6a, b).

Comparison of haplotype blocks between East (a) and West (b) Mediterranean accessions of wild narrow-leafed lupin on chromosome NLL-10 in the LanFTc1 gene region (flanked by markers indicated with yellow arrows). Each colourful diamond shows correlation between two markers. The extent of correlation between markers is shown by the shade of colours, with white denoting recombination and red denoting strong linkage disequilibrium. Purple and grey diamonds indicate uninformative and monomorphic marker comparisons, respectively. Black triangles show the haplotype blocks (colour figure online)
Second, we investigated association between markers and phenotypic traits (i.e. flowering time, flower colour, hard/soft seededness, alkaloid status, pod dehiscence, height at maturity and 100 seed weight), as well as association between markers and climatic variables. Although GWAS analyses did not highlight any significant association between markers and phenotypic traits, it pointed out a strong association with two of the climatic variables. A SNP on position 4,582,844 bp of chromosome NLL-07 (LaDArT_SNP18940) was found to be strongly associated (p = 1.143e−08) with precipitation of driest month/quarter and LaDArT_SNP09086 locus on chromosome NLL-05 (position: 2,350,287 bp) was significantly associated (p = 1.089e−08) with annual mean temperature (Fig. 7). The genic composition of these genomic regions was then investigated. Taking a conservative approach, we considered the entire regions flanked by non-associated SNP markers as potentially containing gene candidates. This revealed 31 genes in an interval spanning 240 Kb on chromosome NLL-07 around the SNP associated with precipitation of driest month/quarter and 5 genes in an interval spanning 47 Kb on NLL-05 around the SNP associated with annual mean temperature. These genes and their functional annotations are listed in Online Resource 6. No standout candidates (such as flowering time genes) based on functional annotations were identified.

Manhattan plot of genome-wide association study (GWAS) using 11,690 SNPs markers for precipitation of driest month/quarter (mm) (a) and annual mean temperature (°C × 10) (b) on 142 wild narrow-leafed lupin accessions. The X-axis represents physical distance along the 20 narrow-leafed lupin chromosomes, NLL-01 to NLL-20. SNPs above the threshold line [green line; − log10(p) = 6] are highly significantly associated with precipitation of driest month and annual mean temperature, respectively (colour figure online)
Discussion
Distribution pattern and migration of narrow-leafed lupin
This is the first pan-Mediterranean analysis of genetic diversity in wild narrow-leafed lupin. We observed a strong East–West division in wild narrow-leafed lupin, an eastward migration, and greater population structure in the western Mediterranean.
Migration of plant populations is influenced by a large number of factors. The complex geological history of the Mediterranean region along with climate fluctuations enforce plant migration, strong selection pressure on plant traits and shifts in plants species’ distribution. These two natural phenomena accompanied by the impact of human activities have shaped plant variation and evolution in the Mediterranean region (Thompson 2005). Thompson (2005) described three main poles of plant diversity in the Mediterranean region, which include (1) the Iberian Peninsula, (2) the Balkans and Aegean, and (3) Anatolia and Cyprus. At a wider genus level, Gladstones (1998) postulated western Asia (Turkey, Syria and nearby area) as the probable origin of smooth-seeded Mediterranean lupins, including narrow-leafed lupin, and that they have spread westwards along Mediterranean. Early phenotypic studies suggested the Aegean region as the centre of diversity of narrow-leafed lupin (Clements and Cowling 1994), with significant distribution of wild types in North Africa and Iberia (Gladstones 1998). The results of our demographic analysis strongly suggest that narrow-leafed lupin originated in the western part of its current distribution range, and that range expansion proceeded eastwards and was accompanied by strong founder effects (Table 2). The current populations seem to be still exchanging large amount of migrants, particularly from western towards eastern populations (Table 3). There is substantial admixture in the central region between West and East, centred on Italy and France (Fig. 2).
At present, it is not possible to estimate absolute ages for separation between western and eastern Mediterranean narrow-leafed lupin. Our results are consistent with east–west vicariance, one of the two common distribution patterns of higher plants in Mediterranean basin, which is attributed to population isolation in association with climatic change and human immigration as described by Thompson (2005). One well-known example of east–west vicariance is Mediterranean oak, in which extremes of morphological diversity were observed in a single variable complex (Barbero et al. 1992). It is assumed that the two taxa belonging to western Mediterranean oak split up due to the effect of glaciation during the refuge period. Owing to its particular geography, the Iberian Peninsula hosted multiple glacial refugia, which were “isolated from one another by the harsh climate of the high central Iberian plateau” (Gómez and Lunt 2007 p. 156). A similar population history could explain the greater population structure observed in western Mediterranean wild narrow-leafed lupins. Interestingly, great morphological diversity is also reported in lupins of eastern Mediterranean (Clements and Cowling 1994) despite the lower genetic diversity observed in that group in this present study (Figs. 1, 2, 4). This could be due to the fact that centre of earlier civilization was in Eastern Mediterranean, and as a result the semi-wild populations of narrow-leafed lupin in that region may have undergone more human selection and physical relocation.
Consistent with demography (Table 2) and population structure results (Fig. 2), the observed higher extent of LD in the eastern population compared to the western population (Fig. 4) could be an indicator of severe founder effect that gave rise to this population, whereby a small number of ancestral haplotypes from the western Mediterranean contributed to the emergence of today’s eastern population haplotypes. The lower extent of LD in the western population is an indicator of more recombination events which broke down the ancestral haplotypes of this population. This higher rate of recombination could be due to the older age, larger population size and greater diversity.
Recognising Iberian Peninsula as a variability hotspot for the wild ancestral populations of narrow-leafed lupin will help in planning to collect future genetic resources efficiently. The similar approach could be applied to other crop ancestral populations to assist their conservation plans and ensure the genetic resources are exploited effectively.
Selection for climatic adaptation
Adaptation is a continuous dynamic process which helps species to keep up with constantly changing environments. The strong seasonal climate of the Mediterranean basin, in particular the association of warm seasons with an effective drought which restrict plant growth, has had a fundamental impact on plant evolution in the region (Thompson 2005), including lupin species as studied by Berger et al. (2017).
Clements and Cowling (1994) study of wild narrow-leafed lupin from the Aegean region showed a negative correlation between flowering times with winter temperature, but positive correlation with the rainfall of the site. As revealed by ANOVA analysis in our study, the western Mediterranean has higher precipitation over the driest month/quarter and a lower annual mean temperature and is late flowering, in comparison with the eastern Mediterranean (Table 4). Shifts in phenology in response to climate change has been reported in many species, such as shifts in flowering time of Brassica rapa in response to drought (Franks et al. 2007; Parmesan and Yohe 2003; Pulido and Berthold 2010).
Flowering time is a key adaptation trait, which is affected by the interaction of genetic factors with environmental cues. Following up with the significant variation between flowering time of western and eastern population, we compared the haplotype block surrounding the main flowering time gene LanFTc1 between these two populations. This revealed evidence of recombination in the 571 Kbp region flanking LanFTc1 within the western population, while that region is monomorphic in the eastern population (Fig. 6a, b). This differentiation in flowering phenology could be explained in light of the migration story of narrow-leafed lupin from West to East. Although irregular in pattern, a West/East bipolar climate has ruled over the Mediterranean basin for at least the last 1000 years (Roberts et al. 2012). To effectively deal with climatic changes resulted from this migration, it appears that selection opted for opportunistic adaptation to the warmer and drier climate of the eastern Mediterranean. Hence, earlier flowering in the eastern Mediterranean population help plants escape drought and heat. This selection may have predated the first use of narrow-leafed lupin by humans and certainly before its domestication was completed in the twentieth Century. So, while early flowering is one of the main selection targets of current lupin breeding, it seems that this trait may have already been under natural selection prior to any domestication event.
SNP markers associated with climatic variables
Applying trait association analysis to the climatic variables inferred from WorldClim climatic factors highlighted two SNP markers associated with precipitation over the driest month/quarter and annual mean temperature (Fig. 7a, b). There were no clear gene candidates in the respective genomic regions of chromosomes NLL-07 and NLL-05, respectively, based on their functional annotations (Online Resource 6). However, it should be emphasised that the resolution afforded by DArTseq genotyping (approximately one SNP per 13 Kbp of assembled genome) in a sample size of 142 accessions is rather low. Nevertheless, these genomic regions provide starting points for follow-on association studies where we are phenotyping an enlarged set of accessions for flowering time and vernalisation response to identify genes associated with environmental adaptation of narrow-leafed lupin. We are also increasingly using whole-genome resequencing to increase the density of marker genotyping in such studies. A genome–environment association study in 1943 georeferenced sorghum landraces using 404,627 SNPs found signatures of local adaptation at two genes. Maturity1, controlling photoperiod sensitivity, was strongly associated with the minimum temperature of the coldest month and Tannin1, controlling grain tannins, with mean temperature of the warmest quarter (Lasky et al. 2015).
Summary conclusion and future directions
For the first time, this study unravelled the diversity and migration history of wild narrow-leafed lupin in the Mediterranean region. This information can be used to help genetic resource managers target regions for sampling wild diversity and to help plant breeders harness genetic diversity in their breeding programmes. Our genome-wide association study between SNP markers and climatic variables could serve as an initial foray into a potential application of combining genetic and spatial datasets. Finally, our comprehensive analytical approach could be applied to understand better other ancestral populations to help build adaptability and reliance into major and minor crops alike.
Author contribution statement
MMD, MNN and WE conceived the design of this study with input on analytical approaches by DE, PEB and JKH. AK, LGK, KBS and JDB contributed to the DArTseq genotyping. BN and DF conducted the demography analyses. MMD, JKH and PEB identified SNP positions related to narrow-leafed reference genome. BH and PEB provided MMD guidance on coding. MMD and WE performed ANOVA analyses. MMD conducted all other analyses. MMD and MNN led the manuscript preparation. All authors have read and approved the final manuscript.
References
Abberton M, Batley J, Bentley A, Bryant J, Cai H, Cockram J, Costa de Oliveira A, Cseke LJ, Dempewolf H, De Pace C, Edwards D, Gepts P, Greenland A, Hall AE, Henry R, Hori K, Howe GT, Hughes S, Humphreys M, Lightfoot D, Marshall A, Mayes S, Nguyen HT, Ogbonnaya FC, Ortiz R, Paterson AH, Tuberosa R, Valliyodan B, Varshney RK, Yano M (2015) Global agricultural intensification during climate change: a role for genomics. Plant Biotechnol J 14:1095–1098
Awika J (2011) Major cereal grains production and use around the world. In: Advances in cereal science: implications to food processing and health promotion. ACS symposium series. American Chemical Society, Washington DC, pp 1–13
Barbero M, Loisel R, Quézel P (1992) Biogeography, ecology and history of Mediterranean Quercus ilex ecosystems. In: Romane F, Terradas J (eds) Quercus ilex L. ecosystems: function, dynamics and management. Springer Netherlands, Dordrecht, pp 19–34
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21(2):263–265
Berger J, Ludwig C (2014) Contrasting adaptive strategies to terminal drought-stress gradients in Mediterranean legumes: phenology, productivity, and water relations in wild and domesticated Lupinus luteus L. J Exp Bot 65(21):6219–6229
Berger J, Buirchell B, Luckett D, Nelson M (2012a) Domestication bottlenecks limit genetic diversity and constrain adaptation in narrow-leafed lupin (Lupinus angustifolius L.). Theor Appl Genet 124(4):637–652
Berger J, Buirchell B, Luckett D, Palta J, Ludwig C, Liu DL (2012b) How has narrow-leafed lupin changed in its 1st 40 years as an industrial, broad-acre crop? AG × E-based characterization of yield-related traits in Australian cultivars. Field Crops Res 126:152–164
Berger JD, Clements JC, Nelson MN, Kamphuis LG, Singh KB, Buirchell B (2013) The essential role of genetic resources in narrow-leafed lupin improvement. Crop Pasture Sci 64(4):361–373
Berger JD, Shrestha D, Ludwig C (2017) Reproductive strategies in Mediterranean legumes: trade- offs between phenology, seed Size and vigor within and between wild and domesticated Lupinus species collected along aridity gradients. Front Plant Sci 8:548
Caballero B, Finglas P, Toldrá F (2015) Encyclopedia of food and health, 1st edn. Academic Press, Oxford, UK
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6(2):80–92
Clements J, Cowling W (1994) Patterns of morphological diversity in relation to geographical origins of wild Lupinus angustifolius from the Aegean region. Genet Resour Crop Evol 41(2):109–122
Dai F, Nevo E, Wu D, Comadran J, Zhou M, Qiu L, Chen Z, Beiles A, Chen G, Zhang G (2012) Tibet is one of the centers of domestication of cultivated barley. Proc Natl Acad Sci 109(42):16969–16973
Delcher AL, Kasif S, Fleischmann RD, Peterson J, White O, Salzberg SL (1999) Alignment of whole genomes. Nucleic Acids Res 27(11):2369–2376
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12(1):13–15
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6(5):e19379
FAO (2014) FAOSTAT database collections. Food and agriculture organization of the United Nations. http://faostat.fao.org. Accessed 03 Nov 2016
Felsenstein J (1989) PHYLIP-phylogeny inference package (version 3.2). Cladistics 5:164–166
Foyer CH, Lam H-M, Nguyen HT, Siddique KH, Varshney RK, Colmer TD, Cowling W, Bramley H, Mori TA, Hodgson JM, Cooper JW, Miller AJ, Kunert K, Vorster J, Cullis C, Ozga JA, Wahlqvist ML, Liang Y, Shou H, Shi K, Yu J, Fodor N, Kaiser BN, Wong F-L, Valliyodan B, Considine MJ (2016) Neglecting legumes has compromised human health and sustainable food production. Nat Plants 2:16112
Franks SJ, Sim S, Weis AE (2007) Rapid evolution of flowering time by an annual plant in response to a climate fluctuation. Proc Natl Acad Sci 104(4):1278–1282
Friesen ML, Cordeiro MA, Penmetsa RV, Badri M, Huguet T, Aouani ME, Cook DR, Nuzhdin SV (2010) Population genomic analysis of Tunisian Medicago truncatula reveals candidates for local adaptation. Plant J 63(4):623–635
Gepts P (2010) Crop domestication as a long-term selection experiment. Plant Breed Rev 24(Part 2):1–44
Gladstones J (1970) Lupins as crop plants. Field Crop Abstr 23(2):123–148
Gladstones J (1998) Distribution, origin, taxonomy, history and importance. In: Gladstones JS, Atkins C, Hamblin J (eds) Lupins as crop plants: biology, production and utilization. Cambridge University Press, Cambridge, UK, pp 1–40
Gladstones JS, Crosbie G (1979) Lupin wild types introduced into Western Australia to 1973. Collection site data, preliminary ratings of field characteristics and disease reactions, and measurements of seed protein and oil contents. Technical Bulletin-Western Australian Department of Agriculture (Australia)
Gómez A, Lunt DH (2007) Refugia within refugia: patterns of phylogeographic concordance in the Iberian Peninsula. In: Weiss S, Ferrand N (eds) Phylogeography of southern european refugia: evolutionary perspectives on the origins and conservation of European biodiversity. Springer Netherlands, Dordrecht, pp 155–188
Gomiero T (2016) Soil degradation, land scarcity and food security: reviewing a complex challenge. Sustainability 8(3):281
Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD (2009) Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet 5(10):e1000695
Hane JK, Ming Y, Kamphuis LG, Nelson MN, Garg G, Atkins CA, Bayer PE, Bravo A, Bringans S, Cannon S (2017) A comprehensive draft genome sequence for lupin (Lupinus angustifolius), an emerging health food: insights into plant-microbe interactions and legume evolution. Plant Biotechnol J 15(3):318–330
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25(15):1965–1978
Hoffmann AA, Sgro CM (2011) Climate change and evolutionary adaptation. Nature 470(7335):479–485
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H, Caig V, Heller-Uszynska K, Jaccoud D, Hopper C, Aschenbrenner-Kilian M, Evers M, Peng K, Cayla C, Hok P, Uszynski G (2012) Diversity arrays technology: a generic genome profiling technology on open platforms. Methods Mol Biol 888:67–89
Kroc M, Koczyk G, Święcicki W, Kilian A, Nelson MN (2014) New evidence of ancestral polyploidy in the Genistoid legume Lupinus angustifolius L. (narrow-leafed lupin). Theor Appl Genet 127(5):1237–1249
Kurlovich BS, Heinanen J (2002) Biological features. In: Kurlovich BS (ed) Lupins (geography, classification, genetic resources and breeding). OY International North Express, St. Petersburg, p 468
Lambers H, Clements JC, Nelson MN (2013) How a phosphorus-acquisition strategy based on carboxylate exudation powers the success and agronomic potential of lupines (Lupinus, Fabaceae). Am J Bot 100(2):263–288
Lasky JR, Upadhyaya HD, Ramu P, Deshpande S, Hash CT, Bonnette J, Juenger TE, Hyma K, Acharya C, Mitchell SE (2015) Genome-environment associations in sorghum landraces predict adaptive traits. Sci Adv 1(6):e1400218
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, Gore MA, Buckler ES, Zhang Z (2012) GAPIT: genome association and prediction integrated tool. Bioinformatics 28(18):2397–2399
Maxted N, Kell S, Ford-Lloyd B, Dulloo E, Toledo Á (2012) Toward the systematic conservation of global crop wild relative diversity. Crop Sci 52(2):774–785
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303
Nelson MN, Książkiewicz M, Rychel S, Besharat N, Taylor CM, Wyrwa K, Jost R, Erskine W, Cowling WA, Berger JD, Berger J, Batley J, Weller J, Naganowska B, Wolko B (2017) The loss of vernalization requirement in narrow-leafed lupin is associated with a deletion in the promoter and de-repressed expression of a Flowering Locus T (FT) homologue. New Phytol 213(1):220–232
Nevado B, Atchison GW, Hughes CE, Filatov DA (2016) Widespread adaptive evolution during repeated evolutionary radiations in New World lupins. Nat Commun. https://doi.org/10.1038/ncomms12384
Parks DH, Mankowski T, Zangooei S, Porter MS, Armanini DG, Baird DJ, Langille MG, Beiko RG (2013) GenGIS 2: geospatial analysis of traditional and genetic biodiversity, with new gradient algorithms and an extensible plugin framework. PLoS One 8(7):e69885
Parmesan C, Yohe G (2003) A globally coherent fingerprint of climate change impacts across natural systems. Nature 421(6918):37–42
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38(8):904–909
Pulido F, Berthold P (2010) Current selection for lower migratory activity will drive the evolution of residency in a migratory bird population. Proc Natl Acad Sci 107(16):7341–7346
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, De Bakker PI, Daly MJ (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3):559–575
R Core Team (2016) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Raj A, Stephens M, Pritchard JK (2014) fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197(2):573–589
Roberts N, Moreno A, Valero-Garcés BL, Corella JP, Jones M, Allcock S, Woodbridge J, Morellón M, Luterbacher J, Xoplaki E (2012) Palaeolimnological evidence for an east–west climate see-saw in the Mediterranean since AD 900. Glob Planet Change 84:23–34
Sansaloni C, Petroli C, Jaccoud D, Carling J, Detering F, Grattapaglia D, Kilian A (2011) Diversity Arrays Technology (DArT) and next-generation sequencing combined: genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. In: BMC proceedings, 2011, vol 5, Supplementary 7. BioMed Central, p P54
Seymour M, Kirkegaard JA, Peoples MB, White PF, French RJ (2012) Break-crop benefits to wheat in Western Australia—insights from over three decades of research. Crop Pasture Sci 63(1):1–16
Stapley J, Reger J, Feulner PG, Smadja C, Galindo J, Ekblom R, Bennison C, Ball AD, Beckerman AP, Slate J (2010) Adaptation genomics: the next generation. Trends Ecol Evol 25(12):705–712
Thompson JD (2005) Plant evolution in the Mediterranean, 1st edn. Oxford University Press, Oxford, UK
Turner TL, Bourne EC, Von Wettberg EJ, Hu TT, Nuzhdin SV (2010) Population resequencing reveals local adaptation of Arabidopsis lyrata to serpentine soils. Nat Genet 42(3):260–263
Vincent H, Wiersema J, Kell S, Fielder H, Dobbie S, Castañeda-Álvarez NP, Guarino L, Eastwood R, Leόn B, Maxted N (2013) A prioritized crop wild relative inventory to help underpin global food security. Biol Conserv 167:265–275
VSN International (2011) GenStat for windows 14th edition. http://GenStat.co.uk. Accessed 1 Dec 2016
Weigel D, Nordborg M (2015) Population genomics for understanding adaptation in wild plant species. Annu Rev Genet 49:315–338
Wolko B, Clements JC, Naganowska B, Nelson MN, Yang H (2011) Lupinus. In: Kole C (ed) Wild crop relatives: genomic and breeding resources: legume crops and forages. Springer, Berlin, Heidelberg, pp 153–206
WorldClim (2006) WorldClim—global climate data. http://www.worldclim.org/. Accessed 1 Dec 2016
Yoder JB, Stanton-Geddes J, Zhou P, Briskine R, Young ND, Tiffin P (2014) Genomic signature of adaptation to climate in Medicago truncatula. Genetics 196(4):1263–1275
Acknowledgements
MMD gratefully acknowledges support by the Australian Government through an Endeavour Postgraduate Scholarship. We thank Dr. Aneeta Pradhan for assistance in growing wild accessions and Dr. Maria Pazos-Navarro for extracting DNA from these accessions. DArTseq genotyping was supported by Grain Research and Development Corporation Grants UWA00151 and UWA00147 to MNN and KBS, respectively. This work was supported by resources provided by the Pawsey Supercomputing Centre with funding from the Australian Government and the Government of Western Australia. This research was supported by use of the Nectar Research Cloud, a collaborative Australian research platform supported by the National Collaborative Research Infrastructure Strategy (NCRIS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical statement
The authors declare that this study complies with the current laws of the countries in which the experiments were performed.
Conflict of interest
The authors declare that MNN is on the editorial board of Theoretical and Applied Genetics. The authors declare no other potential conflicts of interest.
Additional information
Communicated by Henry T. Nguyen.
Electronic supplementary material
Below is the link to the electronic supplementary material.
122_2017_3045_MOESM4_ESM.pptx
Principal Component Analysis (PCA) for 142 accessions of wild narrow-leafed lupin colour-coded by phylogeny results (east/west Mediterranean) (PPTX 214 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mousavi-Derazmahalleh, M., Bayer, P.E., Nevado, B. et al. Exploring the genetic and adaptive diversity of a pan-Mediterranean crop wild relative: narrow-leafed lupin. Theor Appl Genet 131, 887–901 (2018). https://doi.org/10.1007/s00122-017-3045-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-017-3045-7