
Abstract
Aims/hypothesis
GIP(6-30)Cex-K40[Pal] has been characterised as a fatty-acid-derived gastric inhibitory polypeptide (GIP) inhibitor that can induce pancreatic beta cell rest by diminishing the incretin effect. We investigated its therapeutic efficacy with and without the glucagon-like peptide-1 (GLP-1) beta cell cytotropic agent liraglutide.
Methods
The therapeutic efficacy of GIP(6-30)Cex-K40[Pal] alone, and in combination with liraglutide, was determined in C57BL/KsJ db/db mice using a sequential 12 h administration schedule.
Results
GIP(6-30)Cex-K40[Pal] was devoid of cAMP-generating or insulin-secretory activity, and inhibited GIP-induced cAMP production and insulin secretion. GIP(6-30)Cex-K40[Pal] also inhibited GIP-induced glucose-lowering and insulin-releasing actions in mice. Dose- and time-dependent studies in mice revealed that 2.5 nmol/kg GIP(6-30)Cex-K40[Pal], and 0.25 nmol/kg liraglutide, imparted distinct biological effects for 8–12 h post administration. When GIP(6-30)Cex-K40[Pal] (2.5 nmol/kg) and liraglutide (0.25 nmol/kg) were administered sequentially at 12 h intervals (at 08:00 and 20:00 hours) to db/db mice for 28 days, mice treated with GIP(6-30)Cex-K40[Pal] (08:00 hours) and liraglutide (20:00 hours) displayed pronounced reductions in circulating glucose and insulin. Both oral and intraperitoneal glucose tolerance and glucose-stimulated plasma insulin concentrations were improved together with enhanced insulin sensitivity. The expression of genes involved in adipocyte lipid deposition was generally decreased. The other treatment modalities, including GIP(6-30)Cex-K40[Pal] (08:00 and 20:00 hours), liraglutide (08:00 and 20:00 hours) and liraglutide (08:00 hours) combined with GIP(6-30)Cex-K40[Pal] (20:00 hours), also imparted beneficial effects but these were not as prominent as those of GIP(6-30)Cex-K40[Pal] (08:00 hours) and liraglutide (20:00 hours).
Conclusion/interpretation
These data demonstrate that periods of beta cell rest combined with intervals of beta cell stimulation benefit diabetes control and should be further evaluated as a potential treatment option for type 2 diabetes.
Similar content being viewed by others


Introduction
Type 2 diabetes is characterised by pancreatic beta cell destruction, which results in an increased insulin-secretory demand on remaining beta cells [1, 2]. This results in further progressive loss of beta cell mass and function, ultimately leading to exogenous insulin becoming the mainstay therapy [3]. Conventional beta cell stimulatory glucose-lowering drugs, such as sulfonylureas, induce a reduction in beta cell mass over time [4]. Thus, the beneficial effects of extended periods of beta cell rest have been considered as a suitable approach to preserve long-term beta cell function and improve overall enduring glycaemic control [1].
In this context, inhibition of gastric inhibitory polypeptide (GIP) receptor signalling has been shown to improve metabolic control and glycaemic status in animal models of obesity-related diabetes [5–7]. Nonetheless, the principally recognised biological function of GIP is as an incretin hormone that potentiates glucose-induced insulin secretion [8]. Therefore, a likely aspect of the beneficial effects of GIP receptor blockade could be related to the induction of pancreatic beta cell rest [9, 10]. Although GIP receptor inhibitors have already been described [10–13], some doubt still exists regarding their capacity to detrimentally inhibit the naturally occurring incretin effect following feeding in humans [5]. Unlike conventional sulfonylureas glucagon-like peptide-1 (GLP-1) stimulates insulin secretion in a glucose-dependent fashion and this helps to preserve beta cell mass and function [14]. It follows that co-administration of GLP-1 coupled with appropriate periods of beta cell rest induced by administration of a GIP receptor inhibitor could represent a useful therapeutic approach for type 2 diabetes. Consequently, we have attempted to replicate this scenario, through staggered administration of GIP(6-30)Cex-K40[Pal] and the clinically approved GLP-1 mimetic liraglutide. GIP(6-30)Cex-K40[Pal] is a novel effective GIP inhibitor based on GIP(6-30) that is known to bind, but not activate, the GIP receptor [15].
Following in vitro and in vivo characterisation of GIP(6-30)Cex-K40[Pal], we determined the most appropriate doses of GIP(6-30)Cex-K40[Pal] and liraglutide that would allow separate once-daily administration of each, with little overlap of biological actions. This facilitated a twice-daily (at 08:00 and 20:00 hours) dosing regimen in C57BL/KsJ db/db mice, where beta cells would be stimulated for 8–12 h by liraglutide and then be rested for the remaining 8–12 h of the day through subsequent GIP(6-30)Cex-K40[Pal] administration. Notably, akin to the human setting, db/db mice exhibit a gradual age-dependent loss of beta cell mass, which culminates in islet degeneration, hyperglycaemia and overt diabetes [16]. Here we demonstrate the positive metabolic effects of combined GIP(6-30)Cex-K40[Pal] and liraglutide therapy on circulating glucose concentrations, glucose tolerance and insulin sensitivity. These effects were particularly apparent when GIP(6-30)Cex-K40[Pal] was administered during the light cycle, coupled with liraglutide injection during the dark cycle.
Methods
Peptides
Peptides were purchased from GL Biochem (Shanghai, China). GIP(6-30)Cex-K40[Pal] was synthesised on a native GIP(6-30) backbone with subsequent C-terminal extension (Cex) of the nine C-terminal amino acid residues of exendin(1-39). A C-16 fatty acid moiety (palmitate [Pal]) was attached to an additional Lys40 residue via a γ-glutamyl spacer. Peptides were characterised in-house using matrix-assisted laser desorption/ionisation–time of flight (MALDI-ToF) MS, as described previously [13]. The structures of native GIP and GIP(6-30)Cex-K40[Pal] are given in electronic supplementary material (ESM) Table 1.
In vitro cAMP production
Chinese Hamster Lung (CHL) cells transfected with the human GIP receptor were used [13]. This cell line was a gift from. B. Thorens (University of Lausanne, Switzerland [13]). For experimental conditions please see ESM Methods for further details. cAMP generation was measured using a Parameter cAMP assay (R&D Systems, Abingdon, UK) according to the manufacturer’s instructions.
In vitro insulin release studies
Clonal BRIN-BD11 cells were used, the characteristics of which have been described elsewhere [17]. This cell line was established in-house after electrofusion of RINm5F cells with New England Deaconess Hospital rat pancreatic islet cells [17]. For experimental conditions please see ESM Methods for further details. Insulin was measured by radioimmunoassay [18].
Animals
Acute and persistent dose-dependency studies were carried out in male NIH Swiss mice. Longer-term experiments were conducted with male C57BL/KsJ db/db mice. For further details of mouse age and maintenance please see ESM Methods.
Acute in vivo studies
The effects of GIP(6-30)Cex-K40[Pal] (25 nmol/[kg body weight]; i.p.) on glucose homeostasis and insulin secretion in the presence of native GIP (25 nmol/kg; i.p.) were examined in combination with glucose administered by i.p. injection (18 mmol/kg) in non-fasted normal mice (n = 6). Persistent dose-dependent effects were examined in a separate experimental series. Non-fasted mice (n = 6) received an i.p. injection of glucose (18 mmol/kg) in combination with native GIP (25 nmol/kg), 8, 12 or 24 h following administration of either GIP(6-30)Cex-K40[Pal] (0.25, 2.5, 12.5 or 25 nmol/kg) or saline vehicle (0.9% wt/vol. NaCl [154 mmol/l]). To assess the bioactivity of liraglutide, non-fasted mice (n = 6) received an i.p. injection of glucose (18 mmol/kg) 8, 12 or 24 h following administration of either liraglutide (0.25, 2.5, 12.5 or 25 nmol/kg) or saline vehicle.
Subchronic in vivo studies
The db/db mice (n = 8) received twice-daily injections (at 08:00 and 20:00 hours) for 28 days, corresponding to the following five treatment groups: (1) saline vehicle administered at 08:00 and 20:00 hours; (2) GIP(6-30)Cex-K40[Pal] administered at 08:00 and 20:00 hours; (3) liraglutide administered at 08:00 and 20:00 hours; (4) GIP(6-30)Cex-K40[Pal] administered at 08:00 hours combined with liraglutide administration at 20:00 hours and (5) liraglutide administered at 08:00 hours combined with GIP(6-30)Cex-K40[Pal] administered at 20:00 hours. In all cases, GIP(6-30)Cex-K40[Pal] was given at a dose of 2.5 nmol/kg and liraglutide at 0.25 nmol/kg. Energy intake, body weight, non-fasting plasma glucose and plasma insulin concentrations were measured 6 days before the study and at 2–5 day intervals during the study. On day 28, glucose tolerance (18 mmol/kg, oral and i.p.) and insulin sensitivity (15 U/kg) tests were carried out in separate groups of fasted (18 h) and non-fasted mice, respectively. These tests and other blood collections were performed at 08:00 hours, and daily injections were withheld until after blood collection. After the animals were killed, pancreatic tissue was collected for measurement of insulin following extraction using acid ethanol (5 ml/g), as described previously [19]. For details of further terminal analyses please see ESM Methods.
Real-time RT-PCR
Adipose tissue mRNA extraction and cDNA conversion were carried out using Qiazol lysis reagent (Qiagen, Manchester, UK) and superscript II reverse transcriptase – RNase H kit (Invitrogen, Paisley, UK) following the manufacturer’s instructions. For PCR reaction mixture and conditions please see ESM Methods for further details.
Biochemical analysis
From the cut tip of the tail vein of conscious mice, blood samples were collected into fluoride/heparin microcentrifuge tubes (Sarstedt, Numbrecht, Germany) at the times indicated in the Figures. Samples were immediately centrifuged (Beckman microcentrifuge; Beckman Instruments, High Wycombe, UK) for 30 s at 13,000 g and resulting plasma was stored at −20°C until analysis. Plasma and pancreatic insulin levels were determined by an insulin radioimmunoassay as described previously [18]. Plasma glucose was assayed by an automated glucose oxidase procedure using a Beckman Glucose Analyser II (Beckman Instruments, Galway, Ireland).
Statistical analysis
For gene expression studies (Table 2) and pancreatic insulin content (Fig. 4d) all samples were numbered and blinded. For acute and subchronic metabolic tests, there was no blinding. All results are expressed as means ± SEM and data were compared using unpaired t test and one-way ANOVA, followed by the Student–Newman–Keuls post hoc test. AUCs were calculated using the trapezoidal rule with baseline subtraction (PRISM 5; GraphPad Software, San Diego, CA, USA). Statistical significance between groups of data was considered if p < 0.05.
Results
In vitro cAMP production and insulin secretion
Native GIP evoked a concentration-dependent increase in cAMP production in GIP receptor-transfected cells (Fig. 1a). In contrast, GIP(6-30)Cex-K40[Pal] did not affect basal cAMP generation (Fig. 1a). However, when incubated with stimulatory native GIP (10−7 mol/l), GIP(6-30)Cex-K40[Pal] significantly (p < 0.001) inhibited GIP-induced cAMP production (Fig. 1b). The IC50 value for GIP(6-30)Cex-K40[Pal] in this in vitro system was 3.1 × 10−9 mol/l. In agreement, GIP(6-30)Cex-K40[Pal] did not stimulate insulin release above basal control levels (Fig. 1c) and it significantly (p < 0.01 to p < 0.001) inhibited GIP-induced insulin secretion with an IC50 value of 4.9 × 10−11 mol/l (Fig. 1d). In addition, administration of GIP(6-30)Cex-K40[Pal] abolished the glucose-lowering action of native GIP in normal mice (Fig. 1e) and also abolished GIP-induced elevation in plasma insulin concentrations (p < 0.05) (Fig. 1f).

Effects of GIP(6-30)Cex-K40[Pal] on cAMP production, insulin secretion and GIP-mediated glucose-lowering and insulin response. (a, b) GIP receptor-transfected cells were exposed to various concentrations of GIP(6-30)Cex-K40[Pal] alone (a) or in the presence of native GIP (10−7 mol/l) (b) for 60 min (n = 4). cAMP production was measured using ELISA. Black circles, native GIP; white triangles, GIP(6-30)Cex-K40[Pal] alone (a) or in the presence of native GIP (b). (c, d) BRIN-BD11 cells were exposed to a range of concentrations (10−12 to 10−6 mol/l) of GIP(6-30)Cex-K40[Pal] alone (c) or in the presence of native GIP (10−7 mol/l) (d) at 5.6 mmol/l glucose during 20 min incubations (n = 8). Black bars, glucose control; white bars, native GIP; light-grey bars, GIP(6-30)Cex-K40[Pal] alone (c) or in the presence of native GIP (d). (e, f) Plasma glucose (e) and insulin (f) concentrations in mice were measured before and after i.p. injection of glucose alone (18 mmol/kg) and in combination with GIP, or glucose plus GIP together with GIP(6-30)Cex-K40[Pal] (each at 25 nmol/kg). AUC values are shown in insets. Black circles and black bars, glucose control; white triangles and white bars, glucose in combination with native GIP; inverted black triangles and light-grey bars, glucose in combination with native GIP and GIP(6-30)Cex-K40[Pal]. Data are expressed as means ± SEM for four to eight separate observations. *p < 0.05 and ***p < 0.001 compared with respective control; † p < 0.05, †† p < 0.01 and ††† p < 0.001 compared with native GIP
Persistent dose-dependent in vivo effects of GIP(6-30)Cex-K40[Pal] and liraglutide
When administered to normal mice 8 h before an injection of native GIP plus glucose, GIP(6-30)Cex-K40[Pal], at doses of 25, 12.5 and 2.5 nmol/kg, significantly (p < 0.05 to p < 0.001) reduced or completely abolished the glucose-lowering and insulin-secretory actions of 25 nmol/kg native GIP (Table 1). However, when administered at a dose of 0.25 nmol/kg, the glucose-lowering and insulin-secretory actions of GIP were still apparent (Table 1). Interestingly, when injected 8 h previously, liraglutide evoked prominent and significant (p < 0.05 to p < 0.001) glucose-lowering and insulinotropic actions at all doses examined (Table 1). In addition, when administered 12 h before the glucose challenge all doses of liraglutide, barring 0.25 nmol/kg, were effective in lowering plasma glucose levels (p < 0.001) and elevating insulin concentrations (p < 0.01 to p < 0.001) compared with saline control (Table 1). With regards to GIP(6-30)Cex-K40[Pal] administered 12 h before the glucose challenge, only doses of 25 and 12.5 nmol/kg were effective (p < 0.001) in annulling the biological action of native GIP (Table 1). When injected 24 h beforehand, only the highest dose of GIP(6-30)Cex-K40[Pal] tested, 25 nmol/kg, was effective in thwarting the beneficial glucose-lowering and insulin-releasing actions of GIP (Table 1). Liraglutide induced clear (p < 0.05 to p < 0.001) glucose-lowering effects at doses of 25, 12.5 and 2.5 nmol/kg, when administered 24 h previously (Table 1). However, only doses of 25 and 12.5 nmol/kg liraglutide had significant (p < 0.001) overall beneficial insulin-secretory effects when injected 24 h previously (Table 1).
Subchronic effects of GIP(6-30)Cex-K40[Pal] and liraglutide, alone and in combination, on body weight, energy intake, plasma glucose and insulin in db/db mice
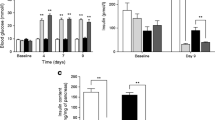
Twice-daily injection of GIP(6-30)Cex-K40[Pal] or liraglutide, alone and in combination, significantly (p < 0.05 to p < 0.01) lowered body-weight gain at various observation points over the treatment period (Fig. 2a). Indeed, on day 28 body-weight gain was significantly (p < 0.05 to p < 0.01) reduced in all treatment groups, barring liraglutide in the morning and GIP(6-30)Cex-K40[Pal] in the evening (Fig. 2a). Percentage body fat was significantly reduced in all treatment groups by day 28 (Fig. 2b). All treatment regimens, barring twice-daily injection of GIP(6-30)Cex-K40[Pal], significantly (p < 0.05 to p < 0.01) reduced energy intake on at least one observation day (Fig. 2c). Non-fasting plasma glucose levels were significantly (p < 0.05 to p < 0.001) reduced in all treatment groups on days 26 and 28, except in those mice given liraglutide in the morning and GIP(6-30)Cex-K40[Pal] in the evening (Fig. 2d). Mice treated with GIP(6-30)Cex-K40[Pal] in the morning (08:00 hours) and liraglutide in the evening (20:00 hours) had reduced (p < 0.05 to p < 0.001) circulating glucose levels from day 8 onwards (Fig. 2d). Non-fasting plasma insulin concentrations were also significantly lowered (p < 0.05 to p < 0.001) in this group of mice at all but one observation point (Fig. 2e). All other treatment groups had reduced (p < 0.05) insulin concentrations on only one or two of the nine observation days during the treatment period (Fig. 2e).

Effects of GIP(6-30)Cex-K40[Pal] and liraglutide on body weight (a), fat content (b), cumulative energy intake (c), plasma glucose (d) and plasma insulin (e) in db/db mice. Variables were measured at 3–4 day intervals for 6 days before and 4 weeks during (indicated by black horizontal bar) treatment with twice-daily (08:00 and 20:00 hours) injection of GIP(6-30)Cex-K40[Pal] (2.5 nmol/kg) and liraglutide (0.25 nmol/kg). Black circles and black bars, saline controls; white triangles and white bars, GIP(6-30)Cex-K40[Pal] (08:00 hours) and liraglutide (20:00 hours); inverted black triangles and light-grey bars, GIP(6-30)Cex-K40[Pal] (08:00 and 20:00 hours); white squares and dark-grey bars, liraglutide (08:00 hours) and GIP(6-30)Cex-K40[Pal] (20:00 hours); black triangles and hatched bars, liraglutide (08:00 and 20:00 hours). Values are expressed as means ± SEM for eight mice. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with saline control
Subchronic effects of GIP(6-30)Cex-K40[Pal] and liraglutide, alone and in combination, on glucose tolerance and plasma insulin responses in db/db mice
Glucose levels were significantly (p < 0.05 to p < 0.01) reduced at 30, 60 and 105 min post i.p. injection of glucose in mice treated with GIP(6-30)Cex-K40[Pal] at 08:00 hours combined with liraglutide at 20:00 hours (Fig. 3a). This was accompanied by a significantly (p < 0.001) reduced overall glycaemic excursion (Fig. 3a inset). Notably, none of the other treatment regimens reduced the overall glycaemic excursion (Fig. 3a inset). Indeed, the GIP(6-30)Cex-K40[Pal] morning and liraglutide evening group had significantly (p < 0.05 to p < 0.01) decreased overall AUC values compared with all other treatment groups (Fig. 3a). Corresponding glucose-induced insulin concentrations revealed a similar situation, with GIP(6-30)Cex-K40[Pal] treatment in the morning and liraglutide treatment in the evening clearly being the most effective treatment modality (Fig. 3b). Moreover, AUC measures were significantly (p < 0.05 to p < 0.01) elevated in these mice when compared with db/db mice treated twice daily with saline control or GIP(6-30)Cex-K40[Pal] and those treated with liraglutide in the morning and GIP(6-30)Cex-K40[Pal] in the evening (Fig. 3b). Similar observations were also made during an oral glucose challenge at the end of the study (Fig. 3c, d). As such, GIP(6-30)Cex-K40[Pal] treatment in the morning coupled with liraglutide administration in the evening significantly (p < 0.05 to p < 0.01) decreased overall glucose levels compared with controls and all other mice that received liraglutide therapy (Fig. 3c). An identical scenario was observed in terms of overall oral glucose-stimulated insulin concentrations (Fig. 3d).

Effects of GIP(6-30)Cex-K40[Pal] and liraglutide on glucose and insulin responses to exogenous intraperitoneally and orally administered glucose in db/db mice. Glucose (18 mmol/kg) was administered by i.p. injection (a, b) or oral gavage (c, d) at t = 0 min, following 28 days’ treatment with twice-daily (08:00 and 20:00 hours) injection of GIP(6-30)Cex-K40[Pal] (2.5 nmol/kg) and liraglutide (0.25 nmol/kg). Overall plasma glucose and insulin AUC values are shown in insets. Black circles and black bars, saline controls; white triangles and white bars, GIP(6-30)Cex-K40[Pal] (08:00 hours) and liraglutide (20:00 hours); inverted black triangles and light-grey bars, GIP(6-30)Cex-K40[Pal] (08:00 and 20:00 hours); white squares and dark-grey bars, liraglutide (08:00 hours) and GIP(6-30)Cex-K40[Pal] (20:00 hours); black triangles and hatched bars, liraglutide (08:00 and 20:00 hours). Values are expressed as means ± SEM for eight mice. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with saline control; † p < 0.05 and †† p < 0.01 compared with GIP(6-30)Cex-K40[Pal] (08:00 hours) and liraglutide (20:00 hours) group
Subchronic effects of GIP(6-30)Cex-K40[Pal] and liraglutide, alone and in combination, on insulin sensitivity, HOMA-IR and pancreatic insulin content in db/db mice
At the end of the study, only those mice that were administered GIP(6-30)Cex-K40[Pal] in the morning together with liraglutide in the evening had significantly (p < 0.05) reduced individual plasma glucose levels at 30 and 60 min following exogenous insulin injection (Fig. 4a). However, the overall glucose-lowering response to insulin was significantly (p < 0.05 to p < 0.001) augmented in all treatment groups, except those mice given liraglutide in the morning and GIP(6-30)Cex-K40[Pal] in the evening, when compared with saline controls (Fig. 4b). HOMA-IR was also significantly (p < 0.05 to p < 0.001) decreased in all treatment groups when compared with saline control (Fig. 4c). In addition, all groups, barring those given liraglutide in the morning and GIP(6-30)Cex-K40[Pal] in the evening, had significantly (p < 0.05 to p < 0.001) reduced pancreatic insulin content at the end of the study (Fig. 4d).

Effects of GIP(6-30)Cex-K40[Pal] and liraglutide on insulin sensitivity, HOMA-IR and pancreatic insulin stores in db/db mice. (a, b) Insulin (15 U/kg) was administered by i.p. injection at t = 0 min, following 28 days treatment with twice-daily (08:00 and 20:00 hours) injection of GIP(6-30)Cex-K40[Pal] (2.5 nmol/kg) and liraglutide (0.25 nmol/kg). Plasma glucose concentration (a) and overall plasma glucose AUC values for 0–60 min (b) are shown. (c) HOMA-IR values calculated from fasting glucose and insulin concentrations. (d) Pancreatic insulin content was measured on day 28. Black circles and black bars, saline controls; white triangles and white bars, GIP(6-30)Cex-K40[Pal] (08:00 hours) and liraglutide (20:00 hours); inverted black triangles and light-grey bars, GIP(6-30)Cex-K40[Pal] (08:00 and 20:00 hours); white squares and dark-grey bars, liraglutide (08:00 hours) and GIP(6-30)Cex-K40[Pal] (20:00 hours); black triangles and hatched bars, liraglutide (08:00 and 20:00 hours). Values are expressed as means ± SEM for eight mice. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with saline control
Subchronic effects of GIP(6-30)Cex-K40[Pal] and liraglutide, alone and in combination, on adipose tissue gene expression in db/db mice
Twice-daily administration of GIP(6-30)Cex-K40[Pal] significantly (p < 0.05 to p < 0.01) reduced Gipr, Fatp, Lpl and Acc-1 (also known as Acc1) mRNA expression in db/db mice when compared with administration of saline control (Table 2). Similar twice-daily treatment with liraglutide reduced mRNA expression of Gipr (p < 0.001), Glp1r (p < 0.01), Gcgr (p < 0.05), Lpl (p < 0.001), Dgat1 (p < 0.001), Acc-1 (p < 0.01) and Hsl (p < 0.05) compared with the expression in control db/db mice (Table 2). However, administration of GIP(6-30)Cex-K40[Pal] at 08:00 hours followed by liraglutide at 20:00 hours markedly reduced the transcription of Gipr (p < 0.05), Gcgr (p < 0.001), Insr (p < 0.05), Lpl (p < 0.01) and Fatp (p < 0.01), while increasing (p < 0.01) transcription of Hsl (Table 2). The reciprocal treatment regimen of liraglutide at 08:00 hours followed by GIP(6-30)Cex-K40[Pal] at 20:00 hours had nominal effects on adipocyte gene expression, with significantly decreased (p < 0.01) Glp1r mRNA expression and increased (p < 0.05) Insr expression (Table 2). None of the treatment regimens affected expression of Glut4 (Slc2a4).
Discussion
Agents that directly induce pancreatic beta cell rest may also have additional beneficial extrapancreatic actions on glucose metabolism beyond their direct inhibitory effects on beta cells [1], which is undoubtedly the case for GIP(6-30)Cex-K40[Pal] [5]. However, uncertainty remains regarding the therapeutic promise of compromised GIP receptor signalling in man. We have evaluated the therapeutic efficacy of combined, but separated by 12 h, daily administration of GIP(6-30)Cex-K40[Pal] and liraglutide in C57/BL6J db/db mice.
We developed GIP(6-30)Cex-K40[Pal] as a novel GIP inhibitor. This peptide analogue is not susceptible to dipeptidyl peptidase-4 degradation because it lacks the N-terminal amino acids Tyr1-Ala2. Moreover, addition of the nine-amino-acid C-terminal extension of exendin1-39 (Cex), together with lipidation at Lys40, aids stability, reduces clearance and enhances in vivo activity [20–22]. GIP(6-30)Cex-K40[Pal] did not stimulate cAMP production or insulin secretion in vitro. Importantly, GIP(6-30)Cex-K40[Pal] dose-dependently compromised GIP-induced elevation of cellular cAMP production and insulin secretion. This indicates that GIP(6-30)Cex-K40[Pal] retains GIP-receptor-binding affinity and actively competes with the native peptide [15]. GIP(6-30)Cex-K40[Pal] also inhibited the glucose-lowering and insulin-secretory effects of native GIP in normal mice.
To discover the most appropriate treatment regimen for subchronic evaluation in db/db mice, we embarked on a series of acute dose- and time-dependent studies in normal mice pertaining to the biological actions of GIP(6-30)Cex-K40[Pal] and liraglutide. Ideally, the scenario to be created is one where beta cells would be rested for 12 h by GIP(6-30)Cex-K40[Pal], followed by stimulation for 12 h with liraglutide. Development of a specific assay to directly measure GIP(6-30)Cex-K40[Pal] as well as liraglutide in plasma would have been useful in enabling determination of pharmacokinetic data. However, consistent with the prolonged biological action profile of fatty-acid-derived peptides [19], both GIP(6-30)Cex-K40[Pal] and liraglutide displayed prominent bioactivity 24 h post administration at a dose of 25 nmol/kg. Our studies revealed that GIP(6-30)Cex-K40[Pal] should be employed at a dose of 2.5 nmol/kg, and liraglutide at 0.25 nmol/kg, in order to achieve a biological action profile greater than 8 h but less than 12 h. Accordingly, these respective doses were employed for the subsequent 28 day subchronic study in db/db mice.
In accordance with the established satiating effect of GLP-1 receptor activation [14], all db/db mice treated with liraglutide for 28 days exhibited marked decreases in food intake. In addition, there were notable reductions in body-weight gain in all mice, independent of energy intake. Thus, as noted previously, reduction in body-weight gain with GIP receptor blockade is directly related to the extent of GIP receptor annulment induced [6], regardless of feeding behaviour [5]. This points towards a possible effect on adipose tissue leading to increased energy dissipation [5]. Consistent with this, there was a clear decrease in the percentage of body-fat mass with all treatment regimens. Reduced adiposity would be expected to indirectly improve the action of insulin, and all treatment groups had improved HOMA-IR values. However, detailed assessment of metabolic rate and locomotor activity would be useful to demonstrate increased energy expenditure, as observed previously in mice with diminished GIP activity [6, 14]. Nonetheless, all mice, barring those given liraglutide at 08:00 hours and GIP(6-30)Cex-K40[Pal] at 20:00 hours, exhibited significantly improved hypoglycaemic responses to exogenous insulin. Indeed, this same group of db/db mice had unchanged pancreatic insulin stores, whereas all other treatment groups had markedly reduced stores, indicative of reduced insulin demand.
Of the treatment modalities employed, the most effective was clearly GIP(6-30)Cex-K40[Pal] administered at 08:00 hours coupled with liraglutide treatment at 20:00 hours. Thus, beta cell rest was induced during the light phase when mice were less likely to consume food, with beta cell stimulation during the more active dark phase [23]. For example, this group of mice had markedly reduced glucose concentrations in the non-fasting state, and during glucose tolerance tests, when compared with all other groups. In addition, circulating insulin levels were distinctly lower, insulin sensitivity and HOMA-IR were improved and glucose-stimulated insulin concentrations elevated. This is characteristic of the beneficial effects associated with extended periods of beta cell rest [24]. As such, prolonged periods of rest would clearly allow chronically overstimulated beta cells to replenish the immediately secretable insulin granule pool [25]. Such actions, together with the positive effects of GLP-1 receptor activation on beta cell stimulus–secretion coupling, survival and growth [14], are potential mediators of the observed beneficial effects. Other factors known to occur during extended periods of beta cell rest, such as enhanced activity of beta cell glucokinase [26] or improved hepatic glucose handling [27], may also contribute to the observed effects. The enhancement of insulin sensitivity is also likely to reflect upregulation of insulin receptors consequent to alleviation of hyperinsulinaemia [28].
Analysis of the effects of the treatments on adipose tissue gene expression also indicates substantial effects at the molecular level. Twice-daily administration of GIP(6-30)Cex-K40[Pal] decreased expression of Gipr together with Fatp, Lpl and Acc-1, genes involved in promoting lipogenesis. In contrast, twice-daily administration of liraglutide decreased expression of genes encoding GLP-1, GIP and glucagon receptors (Glp1r, Gipr, Gcgr) together with downregulation of genes associated with lipogenesis (Lpl, Dgat1, Acc-1) and lipolysis (Hsl). Such effects may be consequent to negative effects on GIP and glucagon receptor gene expression. However, it is clear from the present data that administration of GIP(6-30)Cex-K40[Pal] in the morning followed by liraglutide in the evening induced an impressive pattern of gene expression with similarities between, and differences from, each monotherapy. Thus, expression of receptors for GIP, glucagon and insulin were suppressed along with decreased expression of Lpl and Fatp, which provide a source of NEFA for adipocyte lipogenesis. Hsl was increased concomitantly, suggesting that a decrease in metabolic flux through pathways leading to triacylglycerol formation was accompanied by increased lipolysis and diversion of NEFA to other tissues for metabolism. Interestingly, this scenario has strong parallels with the demonstration of increased fat oxidation in liver and skeletal muscle under conditions of diminished GIP activity [5]. In contrast, the reciprocal treatment regimen had minimal effects on adipocyte genes, being limited to decreased expression of receptors for GLP-1 and insulin. Furthermore, this treatment resulted in no significant differences in non-fasting plasma glucose and insulin concentrations, glucose tolerance, insulin sensitivity or pancreatic insulin content when these variables were compared with those in saline-treated control mice. Thus, beta cell stimulation during the inactive light phase, combined with beta cell rest during the nocturnal phase, was unable to improve many aspects of the metabolic dysregulation. This undoubtedly reveals the important clinical relevance of our findings, which are likely to be directly related to the timing of injections, diurnal cycle and feeding behaviour [23].
In conclusion, the present study has demonstrated clear beneficial metabolic effects of combined GIP(6-30)Cex-K40[Pal] and liraglutide therapy. Importantly, benefits where chiefly apparent when GIP(6-30)Cex-K40[Pal] was administered during the light cycle, coupled with liraglutide injection during the dark cycle. Thus, although the current trend is to develop dual or triple agonist for diabetes [29–32], treatment modalities that incorporate sustained periods of beta cell rest combined with suitable intervals of beta cell stimulation appear to have significant therapeutic potential for diabetes. As such, appropriate combined, but non-simultaneous, administration of a GIP receptor inhibitor and GLP-1 receptor mimetic should be further investigated for therapeutic efficacy in other animal models of diabetes and type 2 diabetes in humans.
Abbreviations
- Cex:
-
C-terminal extension
- GIP:
-
Gastric inhibitory polypeptide
- GLP-1:
-
Glucagon-like peptide-1
- Pal:
-
Palmitate
References
Brown RJ, Rother KI (2008) Effects of beta-cell rest on beta-cell function: a review of clinical and preclinical data. Pediatr Diabetes 9:14–22
Hansen JB, Arkhammar PO, Bodvarsdottir TB, Wahl P (2004) Inhibition of insulin secretion as a new drug target in the treatment of metabolic disorders. Curr Med Chem 11:1595–1615
King A (2013) Integrating advances in insulin into clinical practice: advances in insulin formulations. J Fam Pract 62:S9–S17
Maedler K, Carr RD, Bosco D, Zuellig RA, Berney T, Donath MY (2005) Sulfonylurea induced beta-cell apoptosis in cultured human islets. J Clin Endocrinol Metab 90:501–506
Irwin N, Flatt PR (2009) Evidence for beneficial effects of compromised gastric inhibitory polypeptide action in obesity-related diabetes and possible therapeutic implications. Diabetologia 52:1724–1731
Nasteska D, Harada N, Suzuki K et al (2014) Chronic reduction of GIP secretion alleviates obesity and insulin resistance under high fat diet condition. Diabetes 63:2332–2343
de Toro-Martín J, Fernández-Millán E, Lizárraga-Mollinedo E et al (2014) Predominant role of GIP in the development of a metabolic syndrome-like phenotype in female Wistar rats submitted to forced catch-up growth. Endocrinology 155:3769–3780
Drucker DJ (2007) The role of gut hormones in glucose homeostasis. J Clin Invest 117:24–32
Skak K, Gotfredsen CF, Lundsgaard D, Hansen JB, Sturis J, Markholst H (2004) Improved beta-cell survival and reduced insulitis in a type 1 diabetic rat model after treatment with a beta-cell-selective K(ATP) channel opener. Diabetes 53:1089–1095
Gault VA, Irwin N, Green BD et al (2005) Chemical ablation of gastric inhibitory polypeptide receptor action by daily (Pro3)GIP administration improves glucose tolerance and ameliorates insulin resistance and abnormalities of islet structure in obesity-related diabetes. Diabetes 54:2436–2446
McClean PL, Irwin N, Hunter K, Gault VA, Flatt PR (2008) (Pro3)GIP[mPEG]: novel, long-acting, mPEGylated antagonist of gastric inhibitory polypeptide for obesity-diabetes (diabesity) therapy. Br J Pharmacol 155:690–701
Nakamura T, Tanimoto H, Mizuno Y, Tsubamoto Y, Noda H (2012) Biological and functional characteristics of a novel low-molecular weight antagonist of glucose-dependent insulinotropic polypeptide receptor, SKL-14959, in vitro and in vivo. Diabetes Obes Metab 14:511–517
Pathak V, Gault VA, Flatt PR, Irwin N (2014) Antagonism of gastric inhibitory polypeptide (GIP) by palmitoylation of GIP analogues with N- and C-terminal modifications improves obesity and metabolic control in high fat fed mice. Mol Cell Endocrinol 401:120–129
Baggio LL, Drucker DJ (2007) Biology of incretins: GLP-1 and GIP. Gastroenterology 132:2131–2157
Gelling RW, Coy DH, Pederson RA et al (1997) GIP(6-30amide) contains the high affinity binding region of GIP and is a potent inhibitor of GIP1-42 action in vitro. Regul Pept 69:151–154
Coleman DL, Hummel KP (1967) Studies with the mutation, diabetes, in the mouse. Diabetologia 3:238–248
McClenaghan NH, Barnett CR, O’Harte FP, Flatt PR (1996) Mechanisms of amino acid-induced insulin secretion from the glucose-responsive BRIN-BD11 pancreatic B cell line. J Endocrinol 151:349–357
Flatt PR, Bailey CJ (1981) Abnormal plasma glucose and insulin responses in heterozygous lean (ob/+) mice. Diabetologia 20:573–577
Irwin N, Clarke GC, Green BD et al (2006) Evaluation of the antidiabetic activity of DPP IV resistant N-terminally modified versus mid-chain acylated analogues of glucose-dependent insulinotropic polypeptide. Biochem Pharmacol 72:719–728
Gault VA, McClean PL, Cassidy RS, Irwin N, Flatt PR (2007) Chemical gastric inhibitory polypeptide receptor antagonism protects against obesity, insulin resistance, glucose intolerance and associated disturbances in mice fed high-fat and cafeteria diet. Diabetologia 50:1752–1762
Simonsen L, Holst JJ, Madsen K, Deacon CF (2013) The C-terminal extension of exendin 4 provides additional metabolic stability when added to GLP-1, while there is minimal effect of truncating exendin-4 in anaesthetized pigs. Regul Pept 10:17–21
Tartarkiewicz K, Hargrove DM, Jodka CM et al (2014) A novel long-acting glucose-dependent insulinotropic peptide analogue: enhanced efficacy in normal and diabetic rodents. Diabetes Obes Metab 16:75–85
Albrecht U, Oster H (2001) The circadian clock and behavior. Behav Brain Res 125:89–91
Alemzadeh R, Tushaus KM (2004) Modulation of adipoinsular axis in prediabetic zucker diabetic fatty rats by diazoxide. Endocrinology 145:5476–5484
Ritzel RA, Hansen JB, Veldhuis JD, Butler PC (2004) Induction of beta-cell rest by a Kir6.2/SUR1-selective K(ATP)-channel opener preserves beta-cell insulin stores and insulin secretion in human islets cultured at high (11 mM) glucose. J Clin Endocrinol Metab 89:795–805
Kim WH, Lee JW, Suh YH et al (2005) Exposure to chronic high glucose induces β-cell apoptosis through decreased interaction of glucokinase with mitochondria: downregulation of glucokinase in pancreatic β-cells. Diabetes 54:2602–2611
Alemzadeh R, Holshouser S, Massey P, Koontz J (2002) Chronic suppression of insulin by diazoxide alters the activities of key enzymes regulating hepatic gluconeogenesis in Zucker rats. Eur J Endocrinol 146:871–879
Karpe F, Dickmann JR, Frayn KN (2011) Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes 60:2441–2449
Bhat VK, Kerr BD, Vasu S, Flatt PR, Gault VA (2013) A DPP-IV-resistant triple-acting agonist of GIP, GLP-1 and glucagon receptors with potent glucose-lowering and insulinotropic actions in high-fat-fed mice. Diabetologia 56:1417–1424
Dalbøge LS, Almholt DL, Neerup TS, Vrang N, Jelsing J, Fosgerau K (2014) The novel GLP-1-gastrin dual agonist ZP3022 improves glucose homeostasis and increases β-cell mass without affecting islet number in db/db mice. J Pharmacol Exp Ther 350:353–360
Finan B, Yang B, Ottaway N et al (2015) A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med 21:27–36
Gault VA, Bhat VK, Irwin N, Flatt PR (2013) A novel glucagon-like peptide-1 (GLP-1)/glucagon hybrid peptide with triple-acting agonist activity at glucose-dependent insulinotropic polypeptide, GLP-1, and glucagon receptors and therapeutic potential in high fat-fed mice. J Biol Chem 288:35581–35591
Contribution statement
NI, VAG and PRF conceived the study, participated in the analysis and interpretation of data, drafted the manuscript and revised it critically for intellectual content. VP and SV participated in the analysis and interpretation of data and drafted the manuscript and revised it critically for intellectual content. All authors approved the final version of the manuscript. VP is the guarantor of this work.
Duality of interest
NI, VAG and PRF are named on patents filed by the University of Ulster for exploitation of incretin-based drugs and other peptide therapeutics. All other authors declare that there is no duality of interest associated with their contribution to this manuscript.
Funding
These studies were supported by the SAAD Trading and Contracting Company, a research grant from Diabetes UK and University of Ulster selective research funding.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 41 kb)
ESM Table 1
(PDF 146 kb)
Rights and permissions
About this article

Cite this article
Pathak, V., Vasu, S., Gault, V.A. et al. Sequential induction of beta cell rest and stimulation using stable GIP inhibitor and GLP-1 mimetic peptides improves metabolic control in C57BL/KsJ db/db mice. Diabetologia 58, 2144–2153 (2015). https://doi.org/10.1007/s00125-015-3653-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3653-1