
Abstract
Several laboratories have carried out molecular dynamics (MD) simulations of arginine interactions with lipid bilayers and found that the energetic cost of placing arginine in lipid bilayers is an order of magnitude greater than observed in molecular biology experiments in which Arg-containing transmembrane helices are inserted across the endoplasmic reticulum membrane by the Sec61 translocon. We attempt here to reconcile the results of the two approaches. We first present MD simulations of guanidinium groups alone in lipid bilayers, and then, to mimic the molecular biology experiments, we present simulations of hydrophobic helices containing single Arg residues at different positions along the helix. We discuss the simulation results in the context of molecular biology results and show that the energetic discrepancy is reduced, but not eliminated, by considering free energy differences between Arg at the interface and at the center of the model helices. The reduction occurs because Arg snorkeling to the interface prevents Arg from residing in the bilayer center where the energetic cost of desolvation is highest. We then show that the problem with MD simulations is that they measure water-to-bilayer free energies, whereas the molecular biology experiments measure the energetics of partitioning from translocon to bilayer, which raises the fundamental question of the relationship between water-to-bilayer and water-to-translocon partitioning. We present two thermodynamic scenarios as a foundation for reconciliation of the simulation and molecular biology results. The simplest scenario is that translocon-to-bilayer partitioning is independent of water-to-bilayer partitioning; there is no thermodynamic cycle connecting the two paths.
Similar content being viewed by others

Introduction
Voltage-gated ion channels open and close in response to changes in transmembrane (TM) electric potential (Hille 2001). The opening and closing of the ion-conducting pore is coupled to conformational changes in the voltage-sensing domains, which contain the highly charged S4 helix (Tombola et al. 2006). The charge in the S4 helix is carried by basic amino acids, the majority being arginine. Crystal structures have suggested that at least some of the arginine side chains in the S4 helix are exposed to lipids in the surrounding membrane (Jiang et al. 2003; Lee et al. 2005; Long et al. 2005, 2007). This observation raises the question, how can charged arginine side chains be accommodated in a lipid bilayer?
The presence of a nonpolar, fluid hydrocarbon interior is one of the defining features of a lipid bilayer. The transfer of a charged amino acid side chain such as arginine from water to the interior of a membrane is expected to be unfavorable, primarily because of the high energetic cost of dehydrating the charge. Indeed, continuum electrostatics calculations that model the membrane as a slab of low dielectric material (e.g., oil) sandwiched by a high dielectric material (e.g., water) predict that the energetic penalty of moving a charge from water into a membrane is on the order of tens of kcal mol−1 (Parsegian 1969). On the basis of such calculations, Arg side chains were not expected to be exposed to lipids. To begin to understand how Arg side chains can be accommodated in membranes, it is necessary to take a more detailed look at the membrane.
The complete structure of a lipid bilayer, determined by joint refinement of X-ray and neutron scattering data, reveals that only roughly half of the bilayer, the innermost 3 nm or so, is occupied exclusively by hydrocarbon, while the remainder is a chemically heterogeneous mixture of lipid polar groups and water molecules (Wiener and White 1992). Consequently, the bilayer–water interface is not an abrupt boundary between low and high dielectric media, but rather is a roughly 1.5 nm region over which the polarity smoothly decreases from the high polarity of water to the low polarity of liquid hydrocarbon (White and Wimley 1998). Thus, it is conceivable that the energetic cost of placing a charged side chain into a membrane could be greatly reduced if the side chain is located in a region of the membrane where it is solvated by water and lipid polar groups.
Indeed, Hessa et al. (2005b) demonstrated, using translocon-mediated insertion experiments, that a model S4 helix based on the KvAP voltage sensor (which has four equally spaced arginine residues) can be inserted into the endoplasmic reticulum membrane with the astonishingly low free energy penalty of 0.5 kcal mol−1. A subsequent study showed that the S4 helices of the Shaker and KAT1 channels also have relatively low free energies of translocon-mediated insertion (~1 kcal mol−1) (Zhang et al. 2007). The TM configuration assumed for an isolated S4 helix has been confirmed by oriented circular dichroism spectroscopy (Fernández-Vidal et al. 2006) and solid-state nuclear magnetic resonance measurements (Doherty et al. 2010). In an molecular dynamics (MD) simulation of the S4 helix in a TM configuration in a palmitoyl oleoyl phosphatidyl choline (POPC) bilayer, the membrane deformed in the vicinity of the helix, enabling the charged guanidinium groups of all the arginines along the length of the helix to be completely solvated (and presumably stabilized) by negatively charged lipid phosphodiester moieties and water molecules (Freites et al. 2005). Taken together, the MD simulation and translocon-mediated insertion studies suggest that it is structurally and energetically feasible to expose arginines to lipids, in large part due to solvation of the charged guanidinium group by water and lipid polar groups, particularly phosphates. A potential functional role for arginine–phosphate interactions was identified by Schmidt et al. (2006), who showed that KvAP channels function in a variety of membranes containing phospholipids, but not in membranes containing a variety of lipids (including negatively charged ones) lacking phosphodiester groups. Interactions between phospholipids and voltage-sensing domains were revealed explicitly in the 2.4 Å resolution crystal structure of a “paddle-chimera” channel in which the helix-turn-helix motif comprising the S3b and S4 helices in Kv1.2 has been replaced by the one from Kv2.1 (Long et al. 2007). In addition, it has been shown via a combination of solid-state nuclear magnetic resonance and neutron scattering that the S4 arginines of membrane-embedded voltage-sensing domains interact directly with water (Krepkiy et al. 2009).
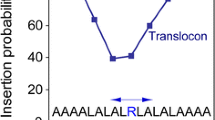
The experiments by Hessa et al. have also shown that translocon-mediated integration of TM helices into the endoplasmic reticulum membrane depends not only on the identity of these residues but also their position within the sequence. For example, accurate measurements of the apparent free energy of insertion (ΔG app) were found to be very sensitive to the location of an Arg residue on an otherwise hydrophobic helix, with ΔG app showing a monotonic increase upon moving the Arg from the ends of the helix to the middle, but the difference in ΔG app between the middle and ends of the helix, ~2.5 kcal mol−1, was remarkably small (Hessa et al. 2005b). This is in sharp contrast to continuum electrostatics models, which predict a large (tens of kcal mol−1) free energy penalty for inserting a charge throughout most of the membrane (Parsegian 1969).
Free energy profiles (also known as potentials of mean force, or PMFs) for translocating an Arg side chain (or analogs such as the guanidinium, methylguanidinium, or propylguanidinium ions) across lipid bilayers, computed from MD simulations by several groups using different sampling protocols and force fields (Dorairaj and Allen 2007; MacCallum et al. 2007; Johansson and Lindahl 2008, 2009; Li et al. 2008; Vorobyov et al. 2008), generally reproduce the shape of ΔG app versus Arg position in the sequence TM helix reported by Hessa et al. (2005b). There is, however, a great disparity in the energy scales: the free energy required to move Arg (or analogs) from the membrane–water interface, predicted by MD simulations, 15–20 kcal mol−1, is almost an order of magnitude larger than the change in ΔG app for moving Arg from the end to the middle of the sequence of a TM helix. In this paper, we attempt to resolve this discrepancy and reconcile the results of MD simulations and translocon-mediated insertion experiments.
We begin by reporting another PMF that is based on biased atomistic MD simulations of a guanidinium (Gdm) ion in a POPC lipid bilayer in excess water that, consistent with previous simulation studies, reproduces the shape of the free energy profile (ΔG app vs. Arg position in TM helix) reported by Hessa et al. (2005b). We observe that the Gdm ion remains solvated by water and lipid polar groups as it penetrates deeply into the hydrocarbon region. Then, to make closer contact with translocon-mediated insertion experiments, we have also performed MD simulations of 20-residue TM helices consisting of 19 leucines and one arginine, in a POPC bilayer. Counting from the first leucine on the N-terminal end, we have placed the single arginine in the 7th, 10th, and 13th positions. We find, for each position, that the Gdm group at the end of the Arg side chain snorkels toward the membrane–water interface, where it is localized within a narrow free energy well, and surrounded by the ubiquitous solvation shell consisting of water and lipid polar groups around the Gdm group in the Arg residues on the TM helices. As a consequence of Arg snorkeling, the Arg position in the sequence of a TM helix does not correspond with its preferred location in the bilayer. Thus, the PMFs computed from MD simulations should not be compared directly to ΔG app versus Arg position in sequence determined in the biological experiments. Accounting for snorkeling by mapping the Arg center-of-charge positions from the TM helix simulations onto the Gdm PMF improves agreement with translocon data, but still leaves a considerable discrepancy. We conclude the paper by providing arguments that the remaining discrepancy persists because the MD simulations and biological experiments consider different thermodynamic processes, specifically, that the translocon-mediated insertion experiments measure the free energy of transfer between the translocon and the membrane, and not the partitioning between water and bilayer modeled by the MD simulations.
Methods
Calculation of the Atomistic PMF for Guanidinium Ion Translocation
The PMF for moving the Gdm ion from the middle of a lipid bilayer into water was calculated by thermodynamic integration using the adaptive biasing force (ABF) method (Darve and Pohorille 2001; Rodriguez-Gomez et al. 2004) as implemented by Hénin and Chipot (2004) in the NAMD 2.6 software package (Phillips et al. 2005). Given a reaction coordinate ξ, the change in free energy between two states identified by ξ = ξa and ξ = ξb is given by
where \( \left\langle {F_{\xi } } \right\rangle_{{\xi^{*} }} \) denotes the mean-force along ξ computed at ξ = ξ* (see Darve and Pohorille (2001) and Rodriguez-Gomez et al. (2004) for details on the correct expression for \( \left\langle {F_{\xi } } \right\rangle_{{\xi^{*} }} \)). In the present case, ξ is the position (z), along the TM direction, of the Gdm ion center of mass measured with respect to the lipid bilayer center. In the ABF method, uniform sampling along the reaction coordinate is achieved by the application of an external force to the system that is equal and opposite to the running average of F ξ (Hénin and Chipot 2004; Rodriguez-Gomez et al. 2004):
where F ξ,i is the force on the reaction coordinate at the ith step of the n step MD simulation. The reaction coordinate was sampled from −30 to 30 Å in nine nonoverlapping windows, for 2 ns per window, and using a bin size of 0.1 Å.
The initial configuration used in the atomistic PMF calculations was derived from a simulation of two Gdm ions in the hydrocarbon core region of a POPC bilayer in excess water. The ions were placed in a symmetric configuration about the bilayer center, at z = ±5 Å along the TM direction, and restrained to those locations by fixing the position of the Gdm carbon atoms. A solvation shell involving water molecules and a few phospholipid molecules developed around each Gdm ion during the first few nanoseconds of the simulation, which was run for 34 ns to produce stable configurations of both solvation shells. A system with a single Gdm ion was constructed by choosing the solvation shell configuration that more closely resembled that of an isolated arginine (R18) in the simulation by Freites et al. (2005) of the S4 helix from the KvAP potassium channel. The selected Gdm ion and all of the molecules participating in the solvation shell were shifted so that the center of mass of the Gdm ion was at the bilayer center, and the other Gdm ion and its solvation shell were eliminated from the system. The resulting system was re-equilibrated for 2 ns, and the equilibrated configuration was used to initiate the PMF calculations.
Calculation of the Continuum PMF for Ion Translocation
The conventional continuum electrostatics model for estimating ion permeation in lipid membranes was introduced in the classic paper by Parsegian (1969). The lipid bilayer is constructed as a semi-infinite, homogeneous, and isotropic slab of thickness L with dielectric constant εM, surrounded by a homogeneous and isotropic medium representing water with dielectric constant εW. The electrostatic component of the ion free-energy, as a function of the ion position z, is given by
where a is the radius of the ion, which is assumed to be spherical, and \( \varepsilon = \varepsilon_{W} - \varepsilon_{M} /\varepsilon_{W} + \varepsilon_{M} \). The origin of the z-coordinate is placed at the bilayer center, and the reference state is bulk water. The first two terms on the right-hand side of Eq. 3 correspond to the ion self-energies in each medium, and they are given by the Born (1920) solvation formula. The remaining two terms account for the energy due to the polarization of the slab by a point charge at z. Here, only the leading term in the charge position is given. The exact solution to this problem has been reported several times in the literature (Anderson and Jackson 1974; von Kitzing and Soumpasis 1996; Allen and Hansen 2003). In the present membrane model, the large dielectric contrast implies ε ~ 1, which justifies considering only the leading term. Permeation estimates are usually performed by ignoring the dependence on the ion position, i.e., neglecting the second term in the square brackets in Eq. 3.
To calculate the continuum result reported in Fig. 1, we used Eq. 3 with εW = 89 and εM = 1 (the values reported for the lipid bilayer hydrocarbon core and bulk water, respectively, in a dipalmitoyl phosphatidylcholine (DPPC) simulation using the CHARMM force field; Stern and Feller 2003), L = 40 Å (i.e., the distance between the peaks in the phosphate distributions in the atomistic model) for 0 ≤ |z| ≤ L/2 − a, and took ΔG(z) = 0 for |z| ≥ L/2 + a. The two intervals of z were connected with a cubic spline. We considered the ion radius a as a free parameter to be determined by matching the ΔG at the center of the bilayer (z = 0) to the atomistic PMF. The atomistic value, ΔG(0) = 23 kcal mol−1, corresponds to a radius a = 5.7 Å. To put this value in the context of the atomistic model, consider that the average distance between C atom of the Gdm ion and the O atoms of the solvation partners (water molecules, phosphate and carbonyl lipid groups) is 5.0 Å. A similar consideration of the results reported by Wilson and Pohorille (1996) for Na+ in a glycerol mono oleate (GMO) bilayer suggests that charged groups buried in lipid bilayers should be assumed to be fully solvated in calculations of partition energetics.

The first five panels show snapshots of a guanidinium ion passing from bulk water to the middle of a phospholipid bilayer (Gdm blue; water in solvation shell red O, white H; lipid carbonyl orange; phosphocholines yellow; water purple). The ion center-of-mass distance from the center of the bilayer along the bilayer normal is indicated at the bottom of each panel. The rightmost panel shows the free energy profile (PMF) for the Gdm ion in a lipid bilayer computed by atomistic MD simulations (solid line), and the electrostatic free energy (dashed line) in a continuum model of the membrane as a rigid slab (dielectric constant εM = 1) with sharp interfaces at z = ±20 Å with semi-infinite regions of water (εW = 89), including both the ion self-energy contribution, given by the Born solvation formula, and that due to the polarization of the slab by the charge. The PMF was mapped from z = 0 (bilayer center) to z = 30 Å, where it was set to 0. Given the system symmetry, at equilibrium the PMF should be symmetric with respect to the center of the bilayer. We therefore reflected the PMF about z = 0 for clarity of presentation. In the continuum calculation we adjusted the radius of the charge to match the PMF at z = 0. The resulting value of 5.7 Å compares reasonably well with the average location of the first coordination shell (~5 Å from the Gdm carbon atom) (Color figure online)
MD Simulations of Arginine-Containing TM Polyleucine Helices
MD simulations were carried out on three poly-Leu TM helices containing a single Arg residue. The initial peptide conformations were constructed as ideal α-helices consisting of 19 Leu residues and one Arg residues with GGPG segments at each end (to correspond with the experiments of Hessa et al. (2005a, b). Arg was placed at either position 7, 10, or 13 in the poly-Leu segment. Specifically, the sequences used in the simulations were GGPGL6RL13GPGG, GGPGL9RL10GPGG, and GGPGL12RL7GPGG. The helices were placed in the middle of a pre-equilibrated lipid bilayer containing 280 POPC molecules in excess water with a single chloride counterion. After 15 ns equilibration, a production run of 15 ns was carried out for each system. For the GGPGL9RL10GPGG peptide, two simulations were run to sample positions of the Arg side chain above (z > 0, referred to as R10 up) and below (z < 0, referred to as R10 down) the center of the bilayer (z = 0).
Calculation of PMFs for Arginine in TM Polyleucine Helices
The PMFs for moving the center-of-charge of Arg side chains along z in TM GGPGL6RL13GPGG, GGPGL9RL10GPGG, and GGPGL12RL7GPGG helices were computed using the ABF method (Darve and Pohorille 2001; Rodriguez-Gomez et al. 2004) described above. The reaction coordinate ξ was taken as the z-component of the difference in the positions of the Arg Cζ and Cα atoms. The ABF calculations were initiated from the end of the corresponding equilibrium simulations of the Arg-containing poly-Leu helices. The ABF simulations were carried out in several (8 for R7 and R13, and 9 for each R10 calculation) 1-Å windows with sampling bins of 0.1 Å, with the windows overlapping by 0.1 Å such that there was one common bin between neighboring windows. Where the windows overlapped, the applied force in the corresponding bins was similar between windows, and we used the weighted average of the force in those bins to calculate the PMF. Because convergence in the ABF method is determined by uniform sampling, different windows required different amounts of sampling before achieving uniform sampling. We sampled between 3 and 9 ns per window, for an aggregate sampling time of 190 ns across all systems.
MD Simulation Protocols
All of the MD runs were conducted at a constant temperature of 300 K and a constant pressure of 1 atm using the NAMD software package (Phillips et al. 2005). The CHARMM22 force field (MacKerell et al. 1998) was used for the Gdm ion and the arginine-containing TM helical peptides, the CHARMM27 force field for the lipids (Feller and MacKerell 2000), and the TIP3P model for water (Jorgensen et al. 1983). The smooth particle mesh Ewald method (Darden et al. 1993) was used to calculate electrostatic interactions, and the short-range, real-space interactions were truncated at 11 Å by using a switching function. A multiple-time step algorithm (Grubmüller et al. 1991; Tuckerman and Berne 1992) was used to integrate the equations of motion with a time step of 4 fs for electrostatic forces, 2 fs for short-range nonbonded forces, and 1 fs for bonded forces. All bond lengths involving hydrogen atoms were held fixed using the SHAKE algorithm (Ryckaert et al. 1977). A Langevin dynamics scheme was used for thermostatting, and Nose–Hoover–Langevin pistons were used for pressure control (Martyna et al. 1994; Feller et al. 1995). Molecular graphics and simulation analyses were performed using the VMD 1.8.5 software package (Humphrey et al. 1996).
Results
Energetics of Guanidinium Ion Translocation Across a Lipid Bilayer
The PMF for translocation of the Gdm ion across a POPC bilayer computed by atomistic MD simulations (Fig. 1, rightmost panel, solid line) displays a shallow minimum as the ion is moved from bulk water into the membrane–water interface, followed by a monotonic increase as the ion is moved from the interface into the middle of the membrane. In contrast, the electrostatic free energy computed using the continuum model (Fig. 1, right, dashed line) shows a steep rise at the interface, followed by only a weak dependence on position within the membrane. The deficiency of the continuum model is not surprising, because the thermally disordered and chemically heterogeneous membrane–water interface spans roughly half the thickness of the membrane (Wiener and White 1992), and hence is poorly described by a sharp interface between uniform dielectric slabs.
The relatively slow rise in the atomistic PMF with extent of membrane penetration is qualitatively similar to the sequence dependence of ΔG app for translocon-mediated membrane insertion of an Arg residue on a hydrophobic TM helix (Hessa et al. 2005b). However, the energetic scales differ by roughly an order of magnitude: our PMF predicts a ~20 kcal mol−1 penalty for placing the Gdm ion in the middle of the membrane, while the translocon experiments suggest that the difference in free energy between an Arg residue at the end of the TM helix and one in the middle of the sequence is only ~2 kcal mol−1. This difference in scales will be discussed below.
Guanidinium Ion Solvation in a Lipid Bilayer
The deformability of the fluid lipid bilayer permits complete solvation of the Gdm ion throughout the membrane (Fig. 1, snapshots). In the middle of the bilayer, the solvation shell consists of water molecules, and lipid acyl chains, carbonyl, and phosphate groups (Fig. 2a, upper row). This configuration persists until the Gdm ion is in the head-group region of the lipid bilayer (the free energy minimum in Fig. 1), where the solvation shell consists primarily of water molecules and lipid phosphates (Fig. 2a, lower row). Finally, outside the lipid bilayer, the first solvation shell contains only water molecules. The perturbation of the lipid bilayer by the Gdm ion in the hydrocarbon region is localized and does not distort the lipid bilayer as a whole. When the ion leaves the hydrocarbon, no waters or lipid polar groups are left behind, and when it exits the head-group region and enters the bulk water, the distortion in the bilayer disappears completely (Fig. 1, snapshots).

a Predominant solvation shell structure of the Gdm ion represented by surfaces of equal number density for water O (red, 0.033 Å−3) and lipid phosphate O (yellow, 0.009 Å−3). The upper row corresponds to the ion in the interior of the membrane (0.0 ≤ |z| ≤ 2.5 Å), and the lower row to the region of the membrane–water interface (19.5 ≤ |z| ≤ 21.5 Å). b Average number of H-bonds formed by the Gdm ion with water molecules (red), lipid phosphate (yellow), and carbonyl groups (orange) as a function of the ion center of mass position. At any given instant of time, the Gdm ion forms 3–4 H bonds with water and/or lipid polar groups (Color figure online)
The structure of the Gdm ion solvation shell in the vicinity of the membrane–water interface (Fig. 2a) closely resembles that of the Gdm ion in aqueous solutions, as described by Mason et al. (2003a, b, 2005, 2006) on the basis of neutron scattering experiments and MD simulations. Although the ion is deep in the lipid acyl chains, its orientation tends to maximize contact between the nonpolar face of the ion and the lipid acyl chains, and hydrogen-bond configurations are constrained to the ion plane (Fig. 2a, upper). The Gdm ion forms four hydrogen bonds (H-bonds) throughout most of the membrane except for close to the bilayer center, where the number of H-bonds drops to three (Fig. 2b). Although the ion is located in the bilayer, on average, three H-bonds are formed with water molecules and one with either a phosphate or a carbonyl lipid group. This pattern changes when the ion is located at the free energy minimum (z ~ 20 Å) where, on average, two H-bonds are formed with water and two with phosphate groups. H-bonding interactions of the Gdm ion with water and phosphates stabilize the ion and reduce the free energy penalty for entering the membrane vs. the continuum model. Decomposition of an atomistic PMF for Arg translocation have shown that the stabilization due to Arg–water interactions continues to increase as the ion is moved to the center of the membrane; Arg–head group interactions also provide increasing stabilization until the ion reaches z ~ 10 Å, where Arg–head group interactions switch to being destabilizing (Dorairaj and Allen 2007).
Arginine Snorkeling and Solvation in TM Polyleucine Helices
To make closer contact with the translocon experiments, we have performed simulations of TM-helices consisting of 19 leucines and one arginine, flanked symmetrically by GPGG segments, in a POPC bilayer. Counting from the first leucine on the N-terminal end, we have placed the single arginine in the 7th, 10th, and 13th positions. Two initial configurations were used in the simulations of the system with Arg in the 10th position, one with R10 pointing in the direction z > 0 (up), and the other pointing toward z < 0 (down). In each case, we observe that the Arg side chain snorkels toward the membrane–water interface, and the bilayer deforms to produce the ubiquitous solvation shell consisting of water and phosphates around the guanidinium group (Fig. 3). In addition to the reorientation of the side chain, we also observed changes in the location of the peptide center-of-mass. The R7 and R10 peptide centers-of-mass shift between 1 and 2 Å toward the interface. The R13 peptide center-of-mass shifts approximately 4 Å, moving at times as much as 7 Å toward the interface. Despite the large displacement, the R13 peptide maintains a TM helical configuration throughout the simulation.

The four leftmost panels depict snapshots of equilibrated helical TM configurations of GGPGL6RL13GPGG (R7), GGPGL9RL10GPGG (R10), and GGPGL12RL7GPGG (R13) in a POPC bilayer. The peptide backbone is drawn as a gray ribbon, and the Arg side chains are colored blue. The lipids and water molecules are colored as in Fig. 1. Two initial conditions (“up” and “down”) were used for the R10 peptide, in which the Arg residue is in the middle of the sequence of the TM segment. The rightmost panel shows histograms of the position of the center of charge of the Arg centers of charge along the bilayer normal. In each peptide, the Arg residue snorkels toward the membrane–water interface to participate in interactions with lipid phosphate groups and water molecules (Color figure online)
An important consequence of the Arg snorkeling and the shift of the peptide center of mass is that the Arg position in the sequence does not correspond with the position of the center-of-charge in the membrane (Fig. 3, rightmost panel). For example, the histograms of the Arg location along the bilayer normal, plotted in Fig. 3, show that the center-of-charge of R10 is ~7 Å away from the middle of the bilayer, even though residue 10 is in the middle of the 20-residue TM segment of GGPGL9RL10GPGG. The centers-of-charge of R7 and R13 are also much closer the membrane–water interface than is implied by their positions in the sequence (7–8 residues from the ends of the TM segments of GGPGL6RL13GPGG, and GGPGL12RL7GPGG, respectively).
Energetics of Arginine Displacement in TM Helices
To verify that the equilibrium simulations sampled the preferred location of Arg in the polyleucine helices, we used the ABF method to calculate the PMFs for Arg displacement along the bilayer normal for each of the Arg-containing polyleucine peptides in helical TM configurations. The PMFs plotted in Fig. 4 all contain narrow wells, consistent with the sharp distributions in Fig. 3, with the positions of their minima coinciding with the locations of the peaks in the histograms. The steep rise on high |z| side of each PMF reflects the large energetic penalty of bond stretching that occurs upon over-extension of the Arg side chains.

PMFs as a function of the position of the Arg center of charge along the bilayer normal for helical TM configurations of the GGPGL6RL13GPGG (R7), GGPGL9RL10GPGG (R10), and GGPGL12RL7GPGG (R13) peptides in a POPC bilayer. Two initial conditions (“up” and “down”) were used for the R10 peptide, in which the Arg residue is in the middle of the sequence of the TM segment. The most stable locations (free energy minima) of the Arg centers of charge are coincident with the most probable locations in equilibrium MD simulations (Fig. 3)
Interactions of Arg Residues in Arg-Containing Polyleucine TM helices
As in the case of the bare Gdm ion, Arg residues at positions 7, 10, and 13 in the polyleucine TM helices are well-solvated. Although the simulations of each peptide were initiated with the Arg Gdm group exposed to the hydrocarbon core of the bilayer, a neighborhood of polar groups quickly developed to solvate the Gdm moiety. In the equilibrated portion of each simulation, the Gdm group is consistently involved in 3–4 H-bonds (Fig. 5). Similar to the bare Gdm ion, the solvation shell consists primarily of water and lipid phosphate groups, with occasional participation by lipid carbonyl groups (especially for the R7 peptide).

Time dependence of the number of H bonds between Arg side chains and water (red), and lipid phosphate (yellow) and carbonyl (yellow) groups in helical TM configurations of the GGPGL6RL13GPGG (R7), GGPGL9RL10GPGG (R10), and GGPGL12RL7GPGG (R13) peptides in a POPC bilayer. Two initial conditions (“up” and “down”) were used for the R10 peptide, in which the Arg residue is in the middle of the sequence of the TM segment. The Arg residue participates in 3–4 H bonds at all times in each case (Color figure online)
Comparison of MD Simulations with Translocon-Mediated Insertion Experiments
Our simulations of Arg-containing polyleucine helices demonstrate that the position of the Arg residue in the sequence of the TM polyleucine helix is not a good indicator of the Arg position within the membrane. For example, snorkeling of R10, which is in the middle of the sequence, places the center-of-charge of the Arg side chain roughly a third of the way toward the membrane–water interface relative to the center of the bilayer. Referring to the atomistic PMF as a function of Gdm position in the membrane (Fig. 1), the actual position of R10 (~7 Å) in the R10 peptide corresponds to a much lower (by ~10 kcal mol−1) free energy than the position of the center-of-charge (~0 Å) that would be assumed from the sequence without taking the snorkeling into account. Thus, in order to compare more accurately the results of MD simulations to those of the translocation-mediated insertion experiments, in Fig. 6a we map the most probable positions of the Arg center-of-charge in the simulations of the Arg-containing helices (Fig. 3) onto the atomistic PMF for Gdm translocation. For comparison, the results of the translocon experiments are plotted in Fig. 6b. It is evident from Fig. 6 that although the disparity between the MD and translocon results is greatly reduced when the Arg positions in polyleucine helices are mapped onto the Gdm ion PMF, a substantial discrepancy remains. For example, the difference in ΔG app from translocon data on R10 and R7 is 0.3 kcal mol−1, whereas the corresponding difference in ΔG PMF from the MD results is ~7 kcal mol−1; for R10 and R13, the difference in ΔG app is 0.9 kcal mol−1, which is substantially less than the ~6 kcal mol−1 difference in ΔG PMF. Thus, it appears that the MD simulations are still significantly overestimating the free energy penalty of placing an Arg residue in a membrane.

a The most probable positions of the Arg side chain centers of charge in the single Arg-containing polyleucine helices (colored circles) mapped onto the atomistic PMF for Gdm translocation across a POPC bilayer (solid black curve). b Free energies of insertion, ΔG app, from translocon-mediated insertion experiments on single-Arg-containing TM helices (Hessa et al. 2005b). The colored circles correspond to the simulated systems; the circles for R7 and R13 were placed by linear interpolation between the two neighboring points (Color figure online)
Discussion
Two results from our simulations of Gdm ion translocation across a lipid bilayer are particularly notable: the shape of the PMF curve and the free energy cost ΔG PMF of moving the Gdm ion from water to the center of the bilayer (Fig. 1). The shape of the atomistic PMF in Fig. 1 appears to be generic for small cation translocation across deformable membranes composed of lipids with uncharged head groups (Vorobyov et al. 2010). Similar PMFs have been computed by others using different simulation models and protocols for Na+ in GMO and dimyristoyl phosphatidylcholine (DMPC) bilayers (Wilson and Pohorille 1996; Tepper and Voth 2006), Arg side chain analogs in DPPC and dioleoyl phosphatidylcholine (DOPC) bilayers (MacCallum et al. 2007; Johansson and Lindahl 2008, 2009; Li et al. 2008; Vorobyov et al. 2008), and an Arg side chain attached to a polyalanine helix dragged through a DPPC membrane (Dorairaj and Allen 2007; Li et al. 2008). The cusp in the atomistic PMF at the center of the membrane in Fig. 1 is an artifact of symmetrizing the PMF. Careful sampling in the vicinity of the membrane center leads to a smooth PMF with only a very slight reduction in the height of the free energy barrier to ion translocation (Li et al. 2008). A recent simulation study showed that the shape and magnitude of the free energy barrier are not changed significantly upon moving from zwitterionic to either positively or negatively charged lipids, but the existence of an interfacial minimum is lipid charge dependent (Johansson and Lindahl 2009).
The shape of ΔG PMF approximates well the shape of the “biological” ΔG app curve determined by Hessa et al. (2005a, b) for the transfer of designed Arg-containing leucine-rich TM helices (H-helix) from the Sec61 translocon to the mammalian endoplasmic reticulum (ER) membrane (Fig. 6b). The shape differs dramatically from the curve computed using continuum electrostatics (Fig. 1, dashed curve). The difference in shape is due to the interactions of the Gdm group with bilayer interfacial water and phospholipid phosphates (Fig. 2), which are not accounted for in continuum calculations. Roughly speaking, the free energy rises linearly with distance from the hydrated interface. The magnitude of ΔG PMF at the bilayer center, however, agrees well with the continuum calculation, for a reasonable choice of the solvated ion radius in the latter.
The great disparity between magnitudes of ΔG PMF (~23 kcal mol−1) and ΔG app (~0.5 kcal mol−1) at the bilayer center has created considerable controversy (Shental-Bechor et al. 2006; Andersen 2007; Dorairaj and Allen 2007; Roux 2007). A common suggestion has been that the biological experiment does not accurately report free energies, because the engineered “H-helices” containing 19 residues interact with neighboring helices, translocon components, or other membrane proteins. These possibilities are ruled out by two recent studies. Meindl-Beinker et al. (2006) demonstrated that helix–helix interactions within the engineered Lep protein containing the H-helix contribute little to ΔG app. Similar experiments by Xie et al. (2007), carried out in Escherichia coli, yielded ΔG app values very similar to those observed in the mammalian ER. Whereas in the mammalian ER experiments leader peptidase (Lep) constructs containing the H-helix were targeted to Sec61 translocon, in the E. coli experiments H-helices were engineered into M13 procoat, which is targeted to the YidC translocase rather than by SecY (the bacterial equivalent of Sec61). The close agreement of the ΔG app values between the mammalian Sec61 and the E. coli YidC systems rules out significant roles for specific interactions between the H-helices and other membrane proteins.
How can the disparity between ΔG PMF and ΔG app be reconciled? The first point of reconciliation is that even though R10 is in the center of the H-helix sequence (Fig. 6b), its Gdm group is not physically in the center of the bilayer. Rather, the Arg side chain snorkels outward toward the hydrated bilayer interfaces in order to salt-bridge with the lipid phosphate groups of the locally distorted bilayer (Fig. 3). This snorkeling keeps the Gdm group away from the peak of the PMF, so that the appropriate ΔG PMF value is about 6–7 kcal mol−1, rather than 23 kcal mol−1 (Fig. 6a).
The second point is that the biological experiment does not involve the insertion of a single Arg residue into the bilayer from the aqueous phase. Instead, the translocon works with whole helices that contain other residues in addition to Arg, and the partitioning is between translocon and membrane rather than between membrane and water (see below). The H-helix in the Hessa et al. (2005a, b) experiments contained significant numbers of hydrophobic residues. The 19-residue Ala-based H-helices used for the experiments summarized in Fig. 6b contained 6 Leu residues that provided a favorable free energy to oppose the unfavorable free energy of the lone Arg. This number of Leu residues was chosen empirically in order to make ΔG app ≈ 0, which is the region of maximum sensitivity of the biological assay (Hessa et al. 2005a). Consequently, the peak magnitude in Fig. 6b is rather arbitrary. The most significant feature of the ΔG app curve is its shape, which is explained well by the simulation data. The real discrepancy between the simulation and biological data emerges by examining how much ΔG app changes as the Arg is shifted from an end position to the center of the sequence. The difference between the end and center free energies in the translocon data is 2.2 kcal mol−1, which is considerably smaller than the ~10 kcal mol−1 predicted by the simulation. How can this be explained?
One possible explanation for the discrepancy is simply that the force fields for the simulations are not accurate. Although the reasonable consistency between MD results obtained with different force fields (MacCallum et al. 2007; Johansson and Lindahl 2008, 2009; Li et al. 2008; Vorobyov et al. 2008) tends to rule out this possibility, the issue can only be addressed fully by constructing a physical experiment that exactly mimics the simulated process. The best experiment in the present context would be to measure partitioning of α-helices. Unfortunately, experiments that rely upon measurements of the partitioning free energies of nonpolar helices from water to bilayer have so far proven unfeasible (White and Wimley 1999; Wimley and White 2000a, b; Ladokhin and White 2004). The problem is that any helix sufficiently hydrophobic to partition directly across a membrane from water also strongly aggregates in water.
Another possible explanation is repositioning of the H-helix along the membrane normal in order to move the Arg closer to the interface (Dorairaj and Allen 2007). That is, during membrane insertion from the translocon the helix adopts a TM position that optimizes Arg–phosphate interactions. Figure 6 suggests that a shift of ~7 Å would be sufficient to reconcile the biological and simulation results. Aware of this possibility, Hessa et al. (2005a) carried out a second series of experiments (called pair-scans) in which two Arg residues were moved symmetrically from the helix ends to the helix center, the idea being that Arg–phosphate interactions operating simultaneously and symmetrically at the two ends of the helix would prevent repositioning. In this case, the H-helix contained 11 Leu residues in order to keep ΔG app ≈ 0. The change in ΔG app as the two Arg residues were moved from the helix ends to the center was the same as for the single-residue scan within experimental uncertainty. The similarity of the single-scan and pair-scan results indicates that helix repositioning is not the primary cause of the disparity between the position dependence of the biological and simulation free energies.
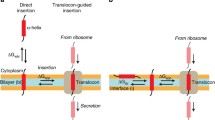
The most fundamental question bearing on the discrepancy between simulations and biological experiments is whether the biological experiment measures the free energy of H-helix insertion relative to water. Although it is true that the X-ray structure (Van den Berg et al. 2004) of a translocon and MD simulations (Gumbart et al. 2005; White and Von Heijne 2005a, b; Haider et al. 2006; Gumbart and Schulten 2007; Bondar et al. 2010) show that the translocon pore is filled with water in the closed state, the proteinaceous nature of the translocon makes it unlikely that the molecular environment of a transiting helix is equivalent to bulk water. The x-ray structure indicates that the entrance into the bilayer from the pore is formed by two TM “gate” helices (TM2b and TM7). When the gate is open, a transiting helix is assumed to sample, through thermal fluctuations, the environment of the pore and the lipid bilayer in the immediate vicinity. In other words, a transiting helix has the opportunity to partition from the pore into the membrane with some particular probability. The values of ΔG app determined in biological experiments (Hessa et al. 2005a, b, 2007; Xie et al. 2007) are assumed to measure this probability (White and Von Heijne 2005a, b; Von Heijne 2007; White 2007). From this perspective, one can construct the hypothetical thermodynamic process depicted in Fig. 7 for comparing the simulation and biological experiments.

Schematic depiction of processes involving helical peptide partitioning between water (w), bilayer (bi), and translocon (tl). The horizontal double arrows denote an equilibrium between translocon and bilayer, with a free energy change ΔG app. This is the process assumed to be probed by the translocon-mediated insertion experiments. The vertical double arrows denote an equilibrium between bilayer and water, with a free energy change ΔG wbi. This is the process considered in simulation studies of Arg (and analogs) insertion in membranes. The diagonal double arrows denote a hypothetical process involving equilibrium partitioning of the peptide between the translocon and water, with a free energy change ΔG wtl. The entry of the peptide into the translocon and subsequent secretion (denoted by vertical arrows labeled “enter” and “exit”) are likely nonequilibrium processes
Figure 7 raises fundamental questions. Is there a free energy difference ΔG wtl between a helix in the aqueous phase and in the translocon? Can a helix partition spontaneously between water and translocon, and does it matter? Said another way, does the translocon selection process depend upon the environment or state of the helix before or after it enters the translocon? There is little experimental evidence available to answer this question. We consider two scenarios. In the first scenario, chain passage is only weakly coupled to chain elongation. In the second scenario, there is tight energetic coupling between nascent chain elongation in the ribosome and chain passage through the translocon.
Scenario 1 assumes, in effect, that the transiting helix partitions between water and translocon rather than being pushed through as a result of energy expenditure. This sort of model was considered many years ago by Simon et al. (1992), who proposed that the nascent chain diffuses snakelike (reptates) through the translocon driven by Brownian motion with directionality imposed by the binding and unbinding of chaperonins on the trans side of the membrane that prevent net backsliding of the nascent chain. In other words, they proposed that translocation is driven by biased random fluctuations, referred to as the “Brownian ratchet” mechanism. This mechanism was formalized by Peskin et al. (1993) and expanded upon by Liebermeister et al. (2001) for the case of eukaryotic posttranslational insertion in which cytosolic (cis side) chaperones hand off unfolded chains to a translocon channel comprised of a supercomplex of Sec62/63 and Sec61. ATP-dependent BiP molecules on the trans-side impose directionality. Although it is not clear if this model is applicable to cotranslational insertion, it provides a useful counterpoint to Scenario 2 by making ΔG wtl important in translocation and insertion. In such a case, ΔG wtl = ΔG app + ΔG wbi, where ΔG wbi is the difference in free energy between the helix in water and the helix in the bilayer. Because ΔG app ≠ −ΔG wbi, scenario 1 requires generally that ΔG wtl ≠ 0, meaning that the interior of the translocon is not equivalent to bulk water. This means, in turn, that that the discrepancy between the ΔG PMF of simulations and the ΔG app of biological experiments cannot be resolved without measurements of ΔG wtl.
Scenario 2 makes three assumptions about an elongating nascent chain moving cotranslationally through the translocon: (1) The helix is driven at a steady rate through the translocon with sufficient energy from ribosomal GTP hydrolysis by elongation factors to overcome any free energy differences between water and translocon. (2) There is sufficient time for the transiting helix to come into equilibrium between the bilayer and the translocon. (Typically, nascent chains elongate at the rate of about 10 amino acids per second, meaning that a particular helix segment resides in the translocon for about 100 ms. This is long compared to the timescale of molecular fluctuations.) (3) The translocon/membrane partitioning process is independent of the disposition of a transiting helix before it enters and after it leaves the translocon. These assumptions imply that there are only two relevant equilibria, one between translocon and bilayer (ΔG app) and one between the bilayer and water (ΔG wbi). About all one can say in this case is that for a helix to enter the bilayer spontaneously from the translocon, ΔG app < 0; for it to stay in the bilayer, ΔG wbi < 0. What can be said about the relation between ΔG app and ΔG wbi in this scenario?
Hessa et al. (2007) have recently extended their earlier measurements (Hessa et al. 2005a) of ΔG app for the Sec61 translocon to include the position dependence of all amino acids and the dependence on sequence length. These measurements led to an equation for ΔG pred for predicting ΔG app for a given sequence. Application of the equation to sequences of proteins that are strictly secreted and those known to have a single TM span identified unambiguously the single-span proteins as those with ΔG pred < 0 and the secreted proteins as those with ΔG pred > 0 (compare the blue dots in Fig. 8 with the red dots along the ΔG pred axis). Jayasinghe et al. (2001) showed that the experimentally determined Wimley–White whole-residue octanol hydrophobicity scale (White and Wimley 1999) detects with high accuracy the TM helices of helix-bundle membrane proteins of known 3D structure. ΔG WW values for the secreted and single-span membrane proteins of Hessa et al. (2007) are plotted against ΔG pred in Fig. 8. The correlation between the two free energies is strong (R = 0.87), consistent with translocon selection of TM helices being a physicochemical process. The fitted line through the points is described by ΔG WW = −8.79 + 2.56ΔG pred. That is, the ΔG WW scale predicts that many of the secreted sequences would be stable in the membrane. This seems unreasonable. But if the curve were translated upward by 8.79 kcal mol−1 (Fig. 8, dashed line), both scales would generally agree on which sequences are inserted and which are secreted. Does it make sense to do this?

Correlation of free energies of insertion, ΔG pred, from a biological hydrophobicity scale including position dependence (Hessa et al. 2007) with predictions, ΔG WW, derived from the Wimley–White whole-residue octanol hydrophobicity scale (White and Wimley 1999) for single-span TM helices (blue dots) and proteins known to be secreted (red dots). The thick solid line is the fit through the points described by ΔG WW = −8.79 + 2.56ΔG pred (kcal mol−1). The dashed line is the fitted line shifted upward by 8.79 kcal mol−1 (Color figure online)
The reason that the WW scale was found to be an accurate predictor of TM helices is that, unlike side chain-only hydrophobicity scales, it accounts for the high thermodynamic cost of dehydrating the H-bonded peptide bonds of α-helices, estimated by Jayasinghe et al. (2001) to be 1.15 kcal mol−1 per residue for a polyglycine helix. The value was validated by assuming that all TM helices of helix-bundle proteins are independently stable in the membrane. However, we now know that this is not the case (Hessa et al. 2007), suggesting that the dehydration penalty of Jayasinghe et al. is too low. If the penalty were increased by 0.44 kcal mol−1 per residue to 1.59 kcal mol−1, the −8.79 kcal mol−1 offset would disappear, thus bringing the modified WW scale into agreement with the ΔG app scale in terms discriminating between secreted and inserted helices. This would imply that ΔG WW ≈ 2.6ΔG app. This factor goes a long way toward reconciling the position-dependent discrepancy between ΔΔG app (2.2 kcal mol−1) and ΔΔG PMF (~10 kcal mol−1) for moving an Arg residue from the end to the middle of a TM helix. Dividing the simulation free energy value by 2.6 yields 3.8 kcal mol−1. The physical principle underlying the scale factor is not entirely clear, but is likely related to the chemistry of the interior space of the translocon.
The free energy of transfer ΔG WW for nonpolar residues is a manifestation of the hydrophobic effect, which can be characterized by an atomic solvation parameter (σ) of −23 cal mol−1 Å−2 (Wimley et al. 1996) (water to nonpolar phase). On the other hand, for partitioning into the complex phospholipid interface, the value is −12 cal mol−1 Å−2 (Wimley and White 1996), which suggests that the atomic solvation parameter is a useful indicator of the “polarity” of the interface. Simplistically, the factor of 2.6 may be a measure of the polarity of the translocon interior. If that is the case, then the σ for the translocon would be −23/2.6 = −8.8 cal mol−1 Å−2. The transfer free energy to bilayer center for leucine relative to alanine with a difference in accessible surface area of 70 Å2 (Wimley et al. 1996), would be −1.61 kcal mol−1 from water and −0.62 kcal mol−1 from the translocon. The measured ΔΔG app of leucine relative to alanine is −0.66 kcal mol−1. But does such a relation help us understand ΔG app for arginine? Wimley and White (1996) plotted the phospholipid interface hydrophobicity values against ΔG WW and found the data to be described well by a linear curve with a slope of 0.5, irrespective of side chain polarity. There were, of course, fluctuations around the curve, but they were, overall, surprisingly small; arginine fell very close to the line, suggesting the usefulness of the solvation parameter as means of judging the apparent polarity of the interface. This implies the possible usefulness of a solvation parameter for describing the polarity of the translocon.
The simplest measure of polarity is the dielectric constant (ε), but Damodaran and Song (1986) have shown that solubility free energies of amino acid side chains relative to water are little affected by solvent dielectric constant. For example, they found water-to-solvent transfer free energy of leucine to be 2.15 kcal mol−1 for ethanol (ε = 25) and 2.55 kcal mol−1 for N-methylacetamide (ε = 191). The values for arginine were 0.784 and 0.624 kcal mol−1, respectively. Damodaran and Song suggested that the free energies of transfer in all cases were determined by the properties of water (e.g., the hydrophobic effect) rather than the properties of the organic solvent. This implies, for both the phospholipid bilayer interface and the translocon interior, that the state of the water in those complex environments determines the apparent solvation parameter.
References
Allen R, Hansen JP (2003) Electrostatic interactions of charges and dipoles near a polarizable membrane. Mol Phys 101:1575–1585
Andersen OS (2007) Perspectives on membrane protein insertion, protein–bilayer interactions, and amino acid side hydrophobicity. J Gen Physiol 129:351–352
Anderson JE, Jackson HW (1974) Membrane–water partition coefficients of ions. Calculated effects of membrane thickness. J Phys Chem 78:2259–2262
Bondar AN, del Val C, Freites JA, Tobias DJ, White SH (2010) Dynamics of SecY translocons with translocation-defective mutations. Structure 18:847–857
Born M (1920) Volumen und Hydratationswarme der Ionen. Z Phys A 1:45–48
Damodaran S, Song KB (1986) The role of solvent polarity in the free energy of transfer of amino acid side chains from water to organic solvents. J Biol Chem 261:7220–7222
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092
Darve E, Pohorille A (2001) Calculating free energies using average force. J Chem Phys 115:9169–9183
Doherty T, Su Y, Hong M (2010) High-resolution orientation and depth of insertion of the voltage-sensing S4 helix of a potassium channel in lipid bilayers. J Mol Biol 401:642–652
Dorairaj S, Allen TW (2007) On the thermodynamic stability of a charged arginine side chain in a transmembrane helix. Proc Natl Acad Sci USA 104:4943–4948
Feller SE, MacKerell AD Jr (2000) An improved empirical potential energy function for molecular simulations of phospholipids. J Phys Chem B 104:7510–7515
Feller SE, Zhang Y, Pastor RW, Brooks BR (1995) Constant pressure molecular dynamics simulation: the Langevin piston method. J Chem Phys 103:4613–4621
Fernández-Vidal M, Castro-Román F, White SH (2006) Membrane insertion of a S4 potassium channel voltage sensor: an experimental study. Biophys J 90:241a
Freites JA, Tobias DJ, von Heijne G, White SH (2005) Interface connections of a transmembrane voltage sensor. Proc Natl Acad Sci USA 102:15059–15064
Grubmüller H, Heller H, Windemuth A, Schulten K (1991) Generalized Verlet algorithm for efficient molecular dynamics simulations with long-range interactions. Mol Simul 6:121–142
Gumbart J, Schulten K (2007) Structural determinants of lateral gate opening in the protein translocon. Biochemistry 46:11147–11157
Gumbart J, Wang Y, Aksimentiev A, Tajkhorshid E, Schulten K (2005) Molecular dynamics simulations of proteins in lipid bilayers. Curr Opin Struct Biol 15:423–431
Haider S, Hall BA, Sansom MSP (2006) Simulations of a protein translocation pore: SecY. Biochemistry 45:13018–13024
Hénin J, Chipot C (2004) Overcoming free energy barriers using unconstrained molecular dynamics simulations. J Chem Phys 121:2904–2914
Hessa T, Kim H, Bihlmaier K, Lundin C, Boekel J, Andersson H, Nilsson IM, White SH, von Heijne G (2005a) Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433:377–381
Hessa T, White SH, von Heijne G (2005b) Membrane insertion of a potassium channel voltage sensor. Science 307:1427
Hessa T, Meindl-Beinker NM, Bernsel A, Kim H, Sato Y, Lerch-Bader M, Nilsson I, White SH, von Heijne G (2007) The molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450:1026–1030
Hille B (2001) Ion channels of excitable membranes. Sinauer Associates, Sunderland, MA
Humphrey W, Dalke W, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38
Jayasinghe S, Hristova K, White SH (2001) Energetics, stability, and prediction of transmembrane helices. J Mol Biol 312:927–934
Jiang YX, Lee A, Chen JY, Ruta V, Cadene M, Chait BT, MacKinnon R (2003) X-ray structure of a voltage-dependent K+ channel. Nature 423:33–41
Johansson ACV, Lindahl E (2008) Position-resolved free energy of solvation for amino acids in lipid membranes from molecular dynamics simulations. Proteins 70:1332–1344
Johansson AC, Lindahl E (2009) The role of lipid composition for insertion and stabilization of amino acids in membranes. J Chem Phys 130:185101-1–185101-8
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935
Krepkiy D, Mihailescu M, Freites JA, Schow EV, Worcester DL, Gawrisch K, Tobias DJ, White SH, Swartz KJ (2009) Structure and hydration of membranes embedded with voltage-sensing domains. Nature 462:473–479
Ladokhin AS, White SH (2004) Interfacial folding and membrane insertion of a designed helical peptide. Biochemistry 43:5782–5791
Lee SY, Lee A, Chen J, MacKinnon R (2005) Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc Natl Acad Sci USA 102:15441–15446
Li L, Vorobyov I, Allen TW (2008) Potential of mean force and pKa profile calculation for a lopid membrane-exposed arginine side chain. J Phys Chem 112:9574–9587
Liebermeister W, Rapoport TA, Heinrich R (2001) Ratcheting in post-translational protein translocation: a mathematical model. J Mol Biol 305:643–656
Long SB, Campbell EB, MacKinnon R (2005) Crystal structure of a mammalian voltage-dependent Shaker-family K+ channel. Science 309:897–903
Long SB, Tao X, Campbell EB, Mackinnon R (2007) Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450:376–382
MacCallum JL, Bennett WFD, Tieleman DP (2007) Partitioning of amino acid side chains into lipid bilayers: results from computer simulations and comparison to experiment. J Gen Physiol 129:371–377
MacKerell AD Jr, Bashford D, Bellott M, Dunbrack RL Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE III, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102:3586–3616
Martyna GJ, Tobias DJ, Klein ML (1994) Constant-pressure molecular-dynamics algorithms. J Chem Phys 101:4177–4189
Mason PE, Neilson GW, Barnes AC, Enderby JE, Brady JW, Saboungi ML (2003a) Neutron diffraction studies on aqueous solutions of glucose. J Chem Phys 119:3347–3353
Mason PE, Neilson GW, Dempsey CE, Barnes AC, Cruickshank JM (2003b) The hydration structure of guanidinium and thiocyanate ions: implications for protein stability in aqueous solution. Proc Natl Acad Sci USA 100:4557–4561
Mason PE, Dempsey CE, Neilson GW, Brady JW (2005) Nanometer-scale ion aggregates in aqueous electrolyte solutions: guanidinium sulfate and guanidinium thiocyanate. J Phys Chem B 109:24185–24196
Mason PE, Neilson GW, Kline SR, Dempsey CE, Brady JW (2006) Nanometer-scale ion aggregates in aqueous electrolyte solutions: guanidinium carbonate. J Phys Chem B 110:13477–13483
Meindl-Beinker NM, Lundin C, Nilsson I, White SH, Von Heijne G (2006) Asn- and Asp-mediated interactions between transmembrane helices during translocon-mediated membrane protein assembly. EMBO Rep 7:1111–1116
Parsegian A (1969) Energy of an ion crossing a low dielectric membrane: solutions to four relevant electrostatic problems. Nature 221:844–846
Peskin CS, Odell GM, Oster GF (1993) Cellular motions and thermal fluctuations: the Brownian ratchet. Biophys J 65:316–324
Phillips JC, Braun B, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802
Rodriguez-Gomez D, Darve E, Pohorille A (2004) Assessing the efficiency of free energy calculation methods. J Chem Phys 120:3563–3578
Roux B (2007) Lonely arginine seeks friendly environment. J Gen Physiol 130:233–236
Ryckaert JP, Ciccotti G, Berendsen HJC (1977) Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23:327–341
Schmidt D, Jiang QX, MacKinnon R (2006) Phospholipids and the origin of cationic gating charges in voltage sensors. Nature 444:775–779
Shental-Bechor D, Fleishman SJ, Ben-Tal N (2006) Has the code for protein translocation been broken? Trends Biochem Sci 31:192–196
Simon SM, Peskin CS, Oster GF (1992) What drives the translocation of proteins? Proc Natl Acad Sci USA 89:3770–3774
Stern HA, Feller SE (2003) Calculation of the dielectric permittivity profile for a nonuniform system: application to a lipid bilayer simulation. J Chem Phys 118:3401
Tepper HL, Voth GA (2006) Mechanisms of passive ion permeation through lipid bilayers: insights from simulations. J Phys Chem B 110:21327–21337
Tombola F, Pathak MM, Isacoff EY (2006) How does voltage open an ion channel? Annu Rev Cell Dev Biol 22:23–52
Tuckerman M, Berne BJ (1992) Reversible multiple time scale molecular dynamics. J Chem Phys 97:1990–2001
Van den Berg B, Clemons WM Jr, Collinson I, Modis Y, Hartmann E, Harrison SC, Rapoport TA (2004) X-ray structure of a protein-conducting channel. Nature 427:36–44
Von Heijne G (2007) Formation of transmembrane helices in vivo—is hydrophobicity all that matters? J Gen Physiol 129:353–356
von Kitzing E, Soumpasis DM (1996) Electrostatics of a simple membrane model using Green’s functions formalism. Biophys J 71:795–810
Vorobyov I, Li L, Allen TW (2008) Assessing atomistic and coarse-grained force fields for protein–lipid interactions: the formidable challenge of an ionizable side chain in a membrane. J Phys Chem 112:9588–9602
Vorobyov I, Bekker B, Allen TW (2010) Electrostatics of deformable lipid membranes. Biophys J 98:2904–2913
White SH (2007) Membrane protein insertion: the biology–physics nexus. J Gen Physiol 129:363–369
White SH, Von Heijne G (2005a) Do protein–lipid interactions determine the recognition of transmembrane helices at the ER translocon? Biochem Soc Trans 33:1012–1015
White SH, von Heijne G (2005b) Transmembrane helices before, during, and after insertion. Curr Opin Struct Biol 15:378–386
White SH, Wimley WC (1998) Hydrophobic interactions of peptides with membrane interfaces. Biochim Biophys Acta 1376:339–352
White SH, Wimley WC (1999) Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct 28:319–365
Wiener MC, White SH (1992) Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of X-ray and neutron diffraction data. III. Complete structure. Biophys J 61:434–447
Wilson MA, Pohorille A (1996) Mechanism of unassisted ion transport across membrane bilayers. J Am Chem Soc 118:6580–6587
Wimley WC, White SH (1996) Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol 3:842–848
Wimley WC, White SH (2000a) Designing transmembrane a-helices that insert spontaneously. Biochemistry 39:4432–4442
Wimley WC, White SH (2000b) Determining the membrane topology of peptides by fluorescence quenching. Biochemistry 39:161–170
Wimley WC, Creamer TP, White SH (1996) Solvation energies of amino acid sidechains and backbone in a family of host–guest pentapeptides. Biochemistry 35:5109–5124
Xie K, Hessa T, Seppälä S, Rapp M, Von Heijne G, Dalbey RE (2007) Features of transmembrane segments that promote the lateral release from the translocase into the lipid phase. Biochemistry 46:15153–15161
Zhang L, Sato Y, Hessa T, von Heijne G, Lee JK, Kodama I, Sakaguchi M, Uozumi N (2007) Contribution of hydrophobic and electrostatic interactions to the membrane integration of the Shaker K+ channel voltage sensor domain. Proc Natl Acad Sci USA 104:8263–8268
Acknowledgments
This research was supported in part by NSF grant CHE-0750175 to DJT, NIH grants GM746373 to SHW and GM86685 to SHW and DJT, and by National Research Service Award 5 T15 LM00744 from the National Library of Medicine to EVS and JAF. This work was also supported by grants from the European Research Council (ERC-2008-AdG 232648), the Swedish Foundation for Strategic Research, the Swedish Research Council, and the Swedish Cancer Foundation to G.v.H. NAMD and VMD were developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Schow, E.V., Freites, J.A., Myint, P.C. et al. Arginine in Membranes: The Connection Between Molecular Dynamics Simulations and Translocon-Mediated Insertion Experiments. J Membrane Biol 239, 35–48 (2011). https://doi.org/10.1007/s00232-010-9330-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00232-010-9330-x