
Abstract
DNA gyrase is a type II topoisomerase that can introduce negative supercoils into DNA at the expense of ATP hydrolysis. It is essential in all bacteria but absent from higher eukaryotes, making it an attractive target for antibacterials. The fluoroquinolones are examples of very successful gyrase-targeted drugs, but the rise in bacterial resistance to these agents means that we not only need to seek new compounds, but also new modes of inhibition of this enzyme. We review known gyrase-specific drugs and toxins and assess the prospects for developing new antibacterials targeted to this enzyme.
Similar content being viewed by others
Introduction
DNA topoisomerases are enzymes that catalyse changes in the topology of DNA (Bates and Maxwell 2005; Wang 2009), i.e., they can interconvert relaxed and supercoiled forms and introduce and remove catenanes and knots (Schoeffler and Berger 2008; Dong and Berger 2008). These enzymes are found in all cell types and are essential to survival. Due to their essential nature and to their mechanisms of action (see below), topoisomerases have become key drug targets both for antibacterial and anti-cancer chemotherapy (Pommier et al. 2010; Tse-Dinh 2007; Bradbury and Pucci 2008).
DNA topoisomerases can be divided into two types, I and II, depending on whether they catalyse reactions involving the transient breakage of one (type I) or both (type II) strands of DNA (Liu et al. 1980). All topoisomerases (topos) can relax supercoiled DNA, but only DNA gyrase (a type II enzyme) can also introduce negative supercoils in a reaction that requires the hydrolysis of ATP (Nollmann et al. 2007; Bates and Maxwell 2007). Whereas eukaryotic type II topos, such as human or yeast topo II, are large single-subunit enzymes (∼170 kDa) that are active as homodimers, prokaryotic enzymes, such as gyrase and its close relative topo IV, are composed of two subunits: A and B in the case of gyrase, which associate to form an A2B2 complex in the active enzyme (Champoux 2001). The best-studied gyrase is that from Escherichia coli, which has A and B subunits (GyrA and GyrB) of molecular weights 97 and 90 kDa, respectively (Reece and Maxwell 1991). Broadly speaking, the A subunit is involved with interactions with DNA, it contains the active-site tyrosine responsible for DNA cleavage, and the B subunit contains the ATPase active site. There is currently no high-resolution structure for the gyrase holoenzyme (A2B2), but several X-ray crystal structures exist for individual domains from various bacterial species (Corbett et al. 2004; Morais Cabral et al. 1997; Wigley et al. 1991; Edwards et al. 2009; Fu et al. 2009; Piton et al. 2010; Tretter et al. 2010; Hsieh et al. 2010), a fusion of a GyrB with a GyrA domain, with and without DNA (Bax et al. 2010; Schoeffler et al. 2010), and low resolution small-angle X-ray scattering structures of the GyrA and GyrB proteins (Costenaro et al. 2005; Costenaro et al. 2007), and intact gyrase (Baker et al. 2011). Taken together, these structures have given us a very good idea of the overall organisation of the A2B2 complex and how it introduces supercoils into DNA.
Mechanism of DNA supercoiling by DNA gyrase
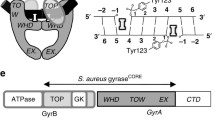
The usefulness of gyrase as a target of antibacterial agents stems from its mechanism of supercoiling (Schoeffler and Berger 2008; Nollmann et al. 2007). The details of this mechanism are still under investigation, but a model, generically known as the “two-gate mechanism” (Roca and Wang 1992, 1994), is strongly supported by biochemical and structural data. DNA gyrase possesses three interfaces that can be in an open or closed conformation (Fig. 1): the N-terminal domain of GyrB (referred to as the N-gate), the GyrA–GyrB–DNA interface, where the DNA is cleaved (referred to as the DNA gate), and the C-terminal area of coiled coils, which forms the C or exit gate (Fig. 1). The supercoiling reaction is thought to progress as follows: the DNA G (or gate) segment associates with the enzyme, at the interface of the N terminus of the GyrA dimer and the TOPRIM domain of GyrB (Bax et al. 2010; Morais Cabral et al. 1997), and DNA is wrapped around the enzyme in a right-handed supercoil of ∼130 base pairs (Orphanides and Maxwell 1994). Wrapping of DNA on the gyrase C-terminal domains facilitates a second segment (the transported or T segment) belonging to the same DNA molecule to reach the N gate, which is positioned over the G segment in preparation for strand passage (Heddle et al. 2004). Binding of ATP results in closure of the N gate and trapping of the T segment (Brino et al. 2000; Wigley et al. 1991). The enzyme cleaves the G segment forming DNA–phosphotyrosyl bonds 4 bp apart, thus creating a double-strand break and resulting in the covalent attachment of GyrA to the DNA. The T segment is passed through the open DNA gate and the broken G segment, and ultimately through the exit gate (Fig. 1). The passage of the T segment through the G segment (strand passage) is driven by the binding and hydrolysis of ATP. The hydrolysis of ATP and release of ADP opens the N gate and resets the enzyme for the next supercoiling cycle. One gyrase supercoiling cycle introduces two negative supercoils into the DNA molecule at the expense of 2 ATPs (Bates and Maxwell 2007). In the absence of ATP, gyrase can catalyse relaxation of negatively supercoiled DNA, essentially by the reverse mechanism (Gellert et al. 1977; Williams and Maxwell 1999b).

Gyrase mechanism (adapted from Costenaro et al. 2007). 1 Free states of the proteins and DNA. 2 Wrapping of the DNA around the enzyme presents the T segment over the G segment. 3 Upon ATP binding, GyrB dimerises, captures the T segment, and the G segment is transiently cleaved. 4 Hydrolysis of one ATP allows GyrB to rotate, the GyrA opening to widen and the transport of the T segment through the cleaved G segment. 5 Religation of the G segment results in the introduction of two negative supercoils into DNA; release of the T segment and hydrolysis of the second ATP resets the enzyme. Stars indicate the positions of the active-site residues for DNA cleavage, and the circle indicates the ATP-binding pocket. Colour code of the domains: GyrA59 (N-terminal domain; NTD), orange; GyrA-CTD, cyan; GyrB43 (NTD), blue; Toprim, red; Tail, green; G segment, black; T segment, purple. The approximate points of action of simocyclinone, novobiocin and ciprofloxacin are shown. N, D and E indicate the locations of the N gate, DNA gate and exit gate, respectively (reprinted with permission from Elsevier; Costenaro et al. 2007)
The gyrase supercoiling reaction is a complex process that is incompletely understood. However, through a combination of structural and mechanistic experiments, we currently have an overall understanding of this process (Fig. 1). What is clear is that the gyrase supercoiling cycle gives ample opportunity for disruption by inhibitors, which can, for example, interfere with DNA binding, DNA cleavage, strand passage and ATP hydrolysis. The exploitability of this reaction has, in part, made gyrase a very attractive target for antibacterial agents. In the following sections, we will summarise what is currently known about such agents and speculate on the possibilities for the development of new therapeutic agents that target gyrase.
Catalytic inhibitors of gyrase vs. gyrase poisons
Two main mechanisms are responsible for the antibacterial activity of drugs targeting gyrase. The first is the inhibition of the enzymatic activity of gyrase, and the second involves stabilisation of the covalent enzyme–DNA complex, or gyrase poisoning. Novobiocin, a compound that inhibits the ATPase activity of gyrase, is an example of a catalytic inhibitor (Fig. 3), and ciprofloxacin (Fig. 2) is an example of a cleavage-complex stabilising agent. Figure 1 gives examples of the proposed points of action of various inhibitors on the gyrase supercoiling reaction: simocyclinone prevents DNA binding, ciprofloxacin interrupts DNA cleavage-religation and stabilises the cleavage complex, and novobiocin inhibits the ATPase activity. It is cleavage-complex stabilisation that has been found to be the most effective mode of action of inhibitors of gyrase and other topoisomerases (Anderson and Osheroff 2001; Drlica et al. 2009). This is because relatively low occupancy of an inhibitor bound to its target can lead to sufficient protein-stabilised breaks in DNA to initiate a cascade of events that ultimately result in cell death (Drlica et al. 2009; Kohanski et al. 2010; Kohanski et al. 2007). In bacteria, this is thought to involve chromosome fragmentation, the induction of the SOS response, and the production of reactive oxygen species (Drlica et al. 2009; Kohanski et al. 2010; Kohanski et al. 2007). This mode of action is employed by the fluoroquinolones, such as ciprofloxacin, and is responsible for their success. Although ‘poisoning’ seems to be the ideal mode of action for compounds targeted to gyrase, the possibility of developing catalytic inhibitors as effective drugs should not be ruled out.

Quinolones. a Examples of fluoroquinolones and their precursor nalidixic acid. b Structure of the topo IV–fluoroquinolone complex (Laponogov et al. 2009). Upper: front view of the topo IV ParC55 and ParE30 proteins complexed with the G segment DNA and moxifloxacin, shown in cartoon representation. DNA is green, the TOPRIM domain (ParE30) is in yellow and the ParC55 is blue. The moxifloxacin molecules are shown in red. Lower: close up of the quinolone–topo IV cleavage complex in cartoon representation. The active site tyrosines (Tyr118) are in orange and the residues responsible for drug resistance upon mutation are shown in yellow (b is reproduced with permission from Nature Publishing Group; Laponogov et al. 2009)
Quinolones
Quinolones are by far the most successful antibacterials targeted to DNA gyrase. The compounds originated from nalidixic acid, a naphthyridone discovered by accident as a by-product during the synthesis of chloroquine (Fig. 2; Lesher et al. 1962). The first-generation quinolones, nalidixic acid and oxolinic acid have relatively weak antimicrobial activity, but the synthesis of the fluoroquinolones and their improvement over several generations, e.g., norfloxacin and ciprofloxacin (second generation), levofloxacin (third gen.) and moxifloxacin and gemifloxacin (fourth gen.), has led to a range of potent antibacterial compounds that have enjoyed enormous clinical and commercial success (Emmerson and Jones 2003; King et al. 2000; Oliphant and Green 2002).
Although gyrase is the target for quinolones in Gram-negative bacteria, topo IV can be the preferred target in some Gram-positive organisms (Hooper 2003), this being dependent on the particular organism and the quinolone (Ferrero et al. 1994; Pan and Fisher 1997). The indicator of primary target is the site of point mutations that confer quinolone resistance. This tends to be largely restricted to discrete regions of gyrA and gyrB (or parC and ParE, the equivalent topo IV genes) that are termed the quinolone-resistance-determining regions or QRDRs (Yoshida et al. 1991; Yoshida et al. 1990). Amino acids in these regions have subsequently been found to be important in the interaction with the drugs from X-ray crystal structures (see below).
In vivo, quinolones can have both bacteriostatic and bactericidal actions (Drlica et al. 2009; Drlica and Hooper 2003). The bacteriostatic effects are largely a consequence of replication-fork arrest by quinolone–gyrase (or quinolone–topo IV) complexes on DNA. Such events do not necessarily lead to DNA breakage and cell death. The lethal action of quinolones is proposed to involve two pathways, one that is dependent upon protein synthesis and one that can occur in its absence, for example, in the presence of the inhibitor chloramphenicol (Drlica et al. 2009; Drlica and Hooper 2003). First-generation compounds, such as nalidixic acid, are not lethal when protein synthesis is inhibited, whereas others, such as ciprofloxacin (a second gen. compound) can kill cells irrespective of protein synthesis. A consequence of quinolone-stabilised gyrase (or topo IV) complexes on DNA is chromosome fragmentation and ultimately cell death via a pathway that may involve active oxygen species (Dwyer et al. 2007; Kohanski et al. 2010; Kohanski et al. 2007).
Quinolones inhibit the DNA supercoiling and relaxation reactions of gyrase and can stabilise a covalent complex in which the GyrA protein is covalently attached to the 5′ ends of the broken DNA (Gellert et al. 1977; Sugino et al. 1977). From biochemical experiments, it was possible to deduce that quinolone action involved interactions with DNA, likely via intercalation, and interactions with the enzyme, likely to both subunits (Heddle and Maxwell 2002; Heddle et al. 2000). However, the details of these interactions have only been revealed very recently with the publications of X-ray structures of fragments of gyrase (and topo IV) complexed with DNA and quinolone drugs (Bax et al. 2010; Laponogov et al. 2010; Laponogov et al. 2009; Wohlkonig et al. 2010). These structures (e.g., Fig. 2b) show in detail how the drugs interact with the enzyme and DNA, and in so doing, disrupt the DNA cleavage-religation process. Although there are differences in detail concerning the specific interactions, which may be accounted for by the different sources of the enzyme, the different drugs employed, and/or the resolution of the structures, the overall mode of interaction is very similar and now gives us a very good idea as to how quinolones interact with gyrase (and topo IV) at the molecular level.
Success in crystallising gyrase–DNA–quinolone complexes has come from the construction of fusion proteins involving the C-terminal domain of GyrB (or ParE in the case of topo IV) with the N-terminal domain of GyrA (or ParC for topo IV). This generates a protein that retains catalytic activity, i.e., the ability to carry our DNA breakage-reunion and that emulates the structure of eukaryotic topo II (Berger et al. 1996). The structures solved (Bax et al. 2010; Laponogov et al. 2010; Laponogov et al. 2009; Wohlkonig et al. 2010) all show the DNA gate in the closed conformation, implying that opening of this gate is not a prerequisite for quinolone binding and DNA cleavage, consistent with earlier biochemical studies (Williams and Maxwell 1999a). The aromatic rings of the quinolone are found to be stacked against the DNA bases at the site of DNA cleavage, i.e., between the −1 and +1 base pairs, which straddle the site of DNA cleavage (Fig. 2b). It is easy to imagine how such an interaction could lead to misalignment of the DNA either side of the break and subsequent difficulty in religating the broken DNA. The exact interactions of the drugs with the proteins are not entirely clear as there are differences between the published structures. However, it is likely that these involve the Ser and Asp (or Glu) residues in GyrA that are commonly mutated in quinolone-resistant bacteria (e.g., Ser83 and Asp87 in E. coli) and a Mg2+ ion, consistent with biochemical studies (Sissi and Palumbo 2009).
These structures not only rationalise the activity and potency of current quinolones but will enable the design of new agents, whether quinolone or non-quinolone, that may overcome the problems of resistance encountered with existing agents.
Aminocoumarins
Aminocoumarins, also referred to as coumarins, are compounds containing a 3-amino-4,7-dihydroxycoumarin ring (Fig. 3). The aminocoumarins, novobiocin, clorobiocin and coumermycin A1, are natural products isolated from Streptomyces species; there are now also many derivatives made by genetic manipulation, metabolic engineering and mutasynthesis (Heide 2009b), and by chemical synthesis (Oblak et al. 2007). Simocyclinones also contain the aminocoumarin moiety but in addition contain an angucyclinone polyketide group (Schimana et al. 2000). Both classical aminocoumarins and simocyclinones are antibiotics that target DNA gyrase, but they do so in entirely different ways.


Aminocoumarins. a Classical aminocoumarins. b Upper: structure of the N-terminal domain of GyrB (GyrB43) complexed with an ATP analogue (Wigley et al. 1991); the protein is in cartoon representation with the two monomers in shades of green and shades of yellow; the nucleotide is in space-filling representation (reproduced with permission (Maxwell and Lawson 2003)). Lower: structure of the N-terminal sub-domain of GyrB (GyrB24) complexed with novobiocin (blue) showing the overlap with bound ATP (Lewis et al. 1996a). c Simocyclinone D8. d Upper: structure of the complex between simocyclinone D8 and the N-terminal domain of GyrA (GyrA59) (Edwards et al. 2009). The protein dimer is shown in blue in cartoon representation and the drug in yellow in stick representation (reprinted with permission from AAAS; Edwards et al. 2009) Lower: detail of the simocyclinone-binding pockets in GyrA showing two alternative modes of interaction (Edwards et al. 2009; Edwards et al. 2011). The blue subunit has the two simocyclinone pockets occupied by two different simocyclinone molecules; the brown subunit has a single simocyclinone molecule bridging both pockets (reprinted with permission from the American Chemical Society; Edwards et al. 2011)
Classical aminocoumarins
In many ways, aminocoumarins are regarded as the ‘Cinderellas’ of the gyrase inhibitors, particularly in comparison with fluoroquinolones. Although they bind tightly to gyrase, they have not enjoyed significant clinical success. The classical aminocoumarins share common structural features: a 3-amino-4, 7-dihydroxycoumarin structure, an l-noviosyl sugar and an aromatic acyl component attached to the amino group of the aminocoumarin moiety (Fig. 3; Heide 2009a). The difference between clorobiocin and novobiocin is the substitution of the methyl group at the 8′ position of the coumarin ring with a chlorine atom, and a 5-methyl-pyrrole-2-carboxyl group substitutes the carbamoyl group at the 3′ position of the noviose. Coumermycin A1 has coumarin rings attached to either side of a pyrrole group; a substituted sugar is attached to each of the coumarin rings, and a pyrrole group is attached to each noviose sugar.
Known since the 1950s for their inhibitory properties on nucleic acid synthesis in bacteria, it was found later, with the discovery of DNA gyrase, that aminocoumarins inhibit gyrase-catalysed supercoiling of DNA (Gellert et al. 1976). It was further shown that they inhibit the gyrase ATPase reaction by competing with ATP for binding to GyrB (Mizuuchi et al. 1978; Sugino and Cozzarelli 1980; Sugino et al. 1978). Their binding site lies in the 24-kDa amino-terminal sub-domain of GyrB (Gilbert and Maxwell 1994), a domain that does not bind ATP by itself; nonetheless, they overlap the ATP-binding site, thus preventing ATP binding (Lewis et al. 1996a).
The interactions of aminocoumarins with DNA gyrase are extremely well characterised; several high-resolution crystal structures exist (Lewis et al. 1996b; Maxwell and Lawson 2003). These include the 24-kDa N-terminal sub-domain of E. coli GyrB complexed with novobiocin (Holdgate et al. 1997; Lewis et al. 1996a) and clorobiocin (Tsai et al. 1997; Lafitte et al. 2002), and Thermus thermophilus GyrB43 complexed with novobiocin (Lamour et al. 2002). These crystal structures clearly show an overlap in the binding of ATP and the aminocoumarin, specifically the binding site of the noviose sugar and the adenine ring of ATP overlap (Fig. 3b). This illustrates how aminocoumarins can be competitive inhibitors of ATP hydrolysis by gyrase despite having limited structural resemblance to ATP.
Because of the overlap between the ATP- and aminocoumarin-binding sites in GyrB, resistance to aminocoumarins that maps to gyrB is limited to amino acids that do not significantly affect the supercoiling activity of gyrase, as this enzyme is essential to survival. Naturally occurring gyrB mutations to aminocoumarin resistance occur predominantly at Arg136 (E. coli numbering) (Contreras and Maxwell 1992; del Castillo et al. 1991; Holmes and Dyall-Smith 1991; Samuels et al. 1994), a residue that contacts the aminocoumarin ring of the drugs (Lewis et al. 1996a). A mutation to Gly164 (to Val) also confers resistance and also a temperature-sensitive phenotype (Contreras and Maxwell 1992). GyrB proteins bearing these mutations show loss of enzymic activity due to impaired ATPase activity (Contreras and Maxwell 1992; Gross et al. 2003). It is feasible to make site-directed mutations in GyrB at residues that make contact with the bound aminocoumarin. For example, mutations at Asp73 (to Asn) and Asn46 (to Asp and Leu) in E. coli GyrB have been made (Kampranis et al. 1999a), and, although the mutant GyrB proteins show reduced drug binding, they were catalytically inactive due to their failure to bind ATP. In other work, E. coli GyrB mutations at Asp73 (to Glu), Gly77 (to Ala and Ser), Ile78 (to Ala and Leu), and Thr165 (to Ala and Val) were found to confer resistance to novobiocin but with a concomitant loss in enzyme activity (Gross et al. 2003). Taken together, it appears that mutations conferring resistance to aminocoumarins tend to inactivate the enzyme due to their proximity to the ATPase active site. This feature is attractive when considering clinical antibacterials targeted to this site.
Simocyclinones
Simocyclinones are hybrid antibiotics containing both aminocoumarin and polyketide elements (Fig. 3c); there are a number of simocyclinone variants produced by Streptomyces antibioticus Tü 6040 that are mostly intermediates of simocyclinone D8 (SD8) (Theobald et al. 2000; Trefzer et al. 2002). SD8 has only modest antibacterial properties, it is most potent against Gram-positive organisms, such as Streptomyces viridochromogenes Tü 57, and largely ineffective against Gram-negative bacteria (Richter et al. 2010; Schimana et al. 2000), although it appears to have higher potency in clinical settings (Richter et al. 2010). Its target is bacterial gyrase (Edwards et al. 2009; Flatman et al. 2005) showing potency against both the E. coli and Staphylococcus aureus enzymes (Oppegard et al. 2009) but not against their topo IV counterparts. SD8 also has cytotoxic activity against human cells (Sadiq et al. 2009; Schimana et al. 2000), where its target appears to be human topoisomerase II (Flatman et al. 2006; Sadiq et al. 2009). The biosynthetic gene cluster of SD8 shares genes that are related to those found in the gene clusters of the classical aminocoumarins (Galm et al. 2002; Trefzer et al. 2002). Despite its relatedness to the classical aminocoumarins, SD8 does not target the ATPase binding site of GyrB, located within the N-terminal domain, but binds to the N-terminal domain of GyrA at the region that also binds DNA (Edwards et al. 2009; Flatman et al. 2006); evidence for a second binding site in the C-terminal domain of GyrB has also been found (Sissi et al. 2010). The action of SD8 on gyrase is to prevent DNA binding. This occurs via the interaction of the aminocoumarin and angucylic polyketide with two separate binding pockets in the N-terminal domain of GyrA (Edwards et al. 2009), i.e., SD8 is a ‘two-headed’ antibiotic. The crystal structure shows that four SD8 molecules stabilise a tetramer of GyrA, with one SD8 bridging two GyrA monomers (Fig. 3d). An alternative model in which a single SD8 bridges the two binding sites in the same GyrA subunit is supported by mass spectrometry data (Edwards et al. 2009; Edwards et al. 2011). This novel mechanism of action could be potentially exploited in the development of new antibacterial agents.
Perspectives on coumarins
Coumarins are potent inhibitors of gyrase in vitro, but their poor activity against Gram-negative bacteria, mammalian cytotoxicity and poor solubility prevent them from being clinically successful drugs. However, their study has shed light on mechanistic aspects of gyrase, and their mode of action could be emulated by other compounds having more attractive properties. Continued investigation of the coumarins could promote the development of new generations of non-quinolone, gyrase-targeted antibacterials. One intriguing question is how two sets of compounds, the classical aminocoumarins and the simocyclinones, which are evolutionarily related, target the same enzyme (gyrase) but bind at different subunits. A further twist to this story is the proposed existence of a simocyclinone-binding site in GyrB (Sissi et al. 2010), the significance of which is presently not clear.
Other small molecule inhibitors
From the foregoing discussion, it is clear that it is possible to develop small molecule inhibitors of bacterial gyrase that can be utilised in a clinical context. In terms of the complexity of the gyrase supercoiling reaction (Fig. 1), it is likely that we have probably really only just scratched the surface of the potential for targeting this enzyme. By analogy, there are a wide range of inhibitors of topo II that target the enzyme at various stages of its catalytic cycle (Larsen et al. 2003). Over the years, and particularly in the recent past, a host of small molecule inhibitors of gyrase have been reported in the literature. None of these have yet made it into clinical practice, but this is more likely to be a consequence of commercial rather than scientific considerations. Below, we review some of these compounds; the discussion is necessarily incomplete as the literature is peppered with possible agents active against gyrase.
Cyclothialidines
Cyclothialidines are cyclic peptides with antibacterial activity produced by streptomycetes (Goetschi et al. 1993). The compounds generally have poor antimicrobial activity only showing significant effects on eubacteria (Nakada et al. 1993; Goetschi et al. 1993). A number of analogues, some with improved activities, have been synthesised (Angehrn et al. 2004; Goetschi et al. 1993). Cyclothialidine (Ro 09-1437; Fig. 4) is produced by Streptomyces filipinensis and has been shown to target DNA gyrase specifically by binding to the B subunit and inhibiting the ATPase activity (Goetschi et al. 1993; Nakada et al. 1994; Oram et al. 1996). A crystal structure of the related compound GR122222X shows that, like novobiocin, the compound binds to the GyrB N-terminal domain at a site that overlaps with the ATP-binding site (Lewis et al. 1996a). Like the classical aminocoumarins, cyclothialidines are competitive inhibitors of the gyrase ATPase reaction by virtue of this overlap (Nakada et al. 1995; Oram et al. 1996). Mutants conferring resistance to cyclothialidines have been generated in S. aureus and found to map to the ATP-binding region of GyrB as expected (Stieger et al. 1996).

Other small molecule gyrase inhibitors
Cinodine
Cinodine is a compound produced by Nocardia species that belongs to the glycocinnamoylspermidine antibiotic class (Fig. 4) (Martin et al. 1978; Tresner et al. 1978); three different forms, β, γ1 and γ2 have been identified. It has been shown to inhibit bacterial DNA synthesis (Greenstein et al. 1981) and DNA supercoiling by Micrococcus luteus gyrase in vitro (Osburne et al. 1990). Cinodine-resistant mutants have been found to map to the gyrA gene (Osburne 1995), supporting the idea that gyrase is the target of cinodine in vivo.
Clerocidin
The diterpenoid clerocidin (Fig. 4) was isolated from the fungus Oidiodendron truncatum, it is known as a cytotoxic and antibacterial agent (Andersen and Rasmussen 1984) that stimulates in vitro DNA cleavage, mediated by eukaryotic DNA topo II (Kawada et al. 1991) and gyrase (McCullough et al. 1993). Clerocidin has been shown to promote in vitro cleavage by Streptococcus pneumoniae gyrase and topo IV (Pan et al. 2008; Richter et al. 2006). Clerocidin differs from other topoisomerase poisons in that it creates a break at the guanine immediately preceding the topoisomerase DNA-cleavage site. This break cannot be resealed by heating or salt treatment (Pan et al. 2008; Gatto et al. 2001). In the absence of topoisomerases, clerocidin is able to nick negatively supercoiled DNA and form adducts with the guanines in short unpaired oligos (Gatto et al. 2001). This reaction was shown to involve the attack by the clerocidin epoxide group of the guanine nitrogen at N7, leading to strand scission at the modified site (Gatto et al. 2001; Richter et al. 2003). Concerning the clerocidin structure, additional to the reactive epoxide function, the carbonyl group and oxirane ring have been shown to be essential for its activity (Richter et al. 2003). The replacement of the diterpenoid moiety by a naphthalene ring system was reported to have no effect on the reaction with the bases in the absence of a topoisomerase (Richter et al. 2003); however, it causes a loss of the clerocidin cleavage activity in presence of S. pneumoniae topo IV (Richter et al. 2006). With S. pneumoniae topo IV, clerocidin was still able to create strand breaks with quinolone-resistant ParC(S79F), but not with the cleavage-deficient ParC(Y118F) active-site mutant (Richter et al. 2006). Evidence suggests that during the formation of the topoisomerase–DNA cleavage complex, the DNA adopts a conformation favourable for the reaction of clerocidin with a guanidine occupying the −1 and/or +5 position of the cleavage site; alternatively, reversible cleavage will be favoured if a cytosine is present at one of these positions (Richter et al. 2006). Clerocidin possesses an unusual mode of action, but the compound itself is too toxic to be used as a drug; less toxic molecules able to mimic clerocidin activity would be of major interest.
Albicidin
Xanthomonas albilineans, the sugarcane scald pathogen, produces a family of antibiotics and phytotoxins known as albicidins. The major component of this mixture, albicidin, is bactericidal to E. coli and inhibits DNA synthesis (Birch and Patil 1985). Relatively little is known about the toxin, although it is thought to contain 38 C-atoms and several aromatic rings, and to be the product of a least two separate gene clusters in X. albilineans (Rott et al. 1996). Albicidin has been shown to be an inhibitor of E. coli and Arabidopsis thaliana DNA gyrase and to stabilise the cleavage complex in the presence of ATP (Hashimi et al. 2007). Although its precise mode of action is not yet clear, it appears to act in a manner distinct from other known gyrase inhibitors. X. albilineans gyrase is resistant to albicidin and a hybrid enzyme comprising the X. albilineans gyrase A subunit and the E. coli gyrase B subunit is active and also resistant, suggesting that GyrA is the target of albicidin (Hashimi et al. 2008). Currently, the albicidin structure is not known, but its apparently unique mode of action and the fact that it stabilises the cleavage complex make it an attractive candidate as a lead molecule for the development of new antibacterial agents.
New synthetic small molecules
There are a whole host of small molecules, sometimes referred to by the catch-all term of new bacterial topoisomerase inhibitors (NBTIs), which are antibacterial compounds not belonging to the quinolone chemical family but acting in a similar manner and not affected by quinolone-resistance mutations (Widdowson and Hennessy 2010). Researchers from GlaxoSmithKline have identified a potent DNA gyrase inhibitor, GSK299423 (Fig. 4) and solved its structure bound to S. aureus gyrase and DNA (Bax et al. 2010). The compound binds to the enzyme–DNA complex in the same region as the quinolone drug ciprofloxacin but at a distinct site. Unlike quinolones, GSK299423 does not stabilise the gyrase cleavage complex and induce double-strand breaks, but appears to induce single-stranded breaks in DNA (Bax et al. 2010), i.e., it appears to have a novel mode of action.
Other compounds that potentially fall into the NBTI class (Fig. 4) are a series of tetrahydroindazoles discovered by researchers at Johnson and Johnson (Gomez et al. 2007; Wiener et al. 2007), quinazoline-2,4-diones PD0305970 and PD0326448, from Pfizer (Huband et al. 2007), QPT-1, also from Pfizer (Miller et al. 2008), and the quinoline-based drug NXL-101 from Novexel (Black et al. 2008). It is not yet clear whether these compounds will enjoy the level of clinical success enjoyed by the fluoroquinolones.
Proteinaceous inhibitors
Aside from small molecule inhibitors of gyrase, there are a number of larger molecular weight protein-based inhibitors. These have distinct mechanisms of action that could, in principle, be emulated by small molecules to generate novel antimicrobial agents.
Microcin B17
Microcin B17 (MccB17; Fig. 5) is a 3.1-kDa glycine-rich peptide produced by enterobacteria carrying the pMccB17 plasmid (Davagnino et al. 1986). Its 3-D structure is not yet known, but a model has been suggested (Fig. 5b; Parks et al. 2007). The peptide is post-translationally modified to convert serine- and cysteine- residues into oxazole, thiazole, and oxazole–thiazole fused rings (Yorgey et al. 1994). MccB17 is active against many enterobacteria (Asensio et al. 1976) and has been shown to inhibit DNA replication, triggering the SOS response, and inducing DNA degradation and cell death (Herrero and Moreno 1986). Genetic and biochemical approaches have shown that the effect of MccB17 is linked to its ability to induce E. coli gyrase-mediated double-strand cleavage of DNA (Vizán et al. 1991). MccB17 is a relatively weak inhibitor of gyrase compared to quinolones and works via a different mechanism. The toxin does not completely inhibit the supercoiling and relaxation reaction of gyrase but merely slows them down by about threefold (Pierrat and Maxwell 2003). The effect of MccB17 on the DNA cleavage reaction in vitro is topology dependent: under supercoiling conditions, with relaxed DNA as substrate, MccB17 can only weakly stabilise the cleavage complex in the absence of ATP, but in its presence, cleavage is more efficiently stabilised. Under relaxation conditions, with a negatively supercoiled DNA as substrate, MccB17 can more efficiently stabilise the cleavage complex in the absence of ATP (Pierrat and Maxwell 2003). It has been shown that MccB17 does not require the DNA-wrapping (C-terminal) domain of GyrA or the ATPase (N-terminal) domain of GyrB (Pierrat and Maxwell 2005). This evidence, added to the fact that the only known MccB17-resistant mutation is at amino acid 751 of GyrB (W751R; Vizán et al. 1991), has led to the idea that the action of the toxin involves binding to the C-terminal domain of GyrB, preventing strand passage by trapping a transient enzyme intermediate (Pierrat and Maxwell 2005). Some quinolone-resistance mutations, GyrB (K447E) and GyrA (S83W), provide limited resistance to MccB17, but the CcdB-resistant mutant GyrA (R462C) is sensitive to MccB17 (Heddle et al. 2001); the significance of these data are presently not clear. The MccB17-producing strain contains three genes: mcbEFG that confers immunity to the host (Garrido et al. 1988); mcbEF that encodes a pump; and mcbG that encodes an immunity protein (San Millan et al. 1985) that is a pentapeptide repeat protein (see below). Interestingly, it has been shown that mcbEFG can confer resistance to fluoroquinolones (Lomovskaya et al. 1996).

Microcin B17. a Primary structure. b Proposed 3-D structure (adapted from (Parks et al. 2007) with permission from Elsevier). Letters A–F indicate the heterocycles, and key amino acids are numbered
Taken together, it seems that MccB17 binds to gyrase at a site that involves the C-terminal domain of GyrB and that this complex slows down DNA strand passage and stabilises the cleavage complex. It is likely that MccB17 recognises a conformation of the enzyme that is only revealed during the strand-passage process, the toxin perhaps binding to transiently exposed hydrophobic regions of the enzyme (Parks et al. 2007). This makes MccB17 an attractive prospect in relation to the development of novel antibacterials, as it stabilises the gyrase–DNA cleavage complex by a mechanism that is probably distinct from the quinolones. Unfortunately, due its unattractive physicochemical properties, it is inappropriate to be used as a drug; however, it is an attractive resource for designing novel drugs based on its particular heterocyclic structure and mechanism of action. The possibility of efficiently making MccB17 by wholly synthetic routes (Thompson et al. 2011; Videnov et al. 1996) raises this as a serious possibility.
CcdB
CcdB (Fig. 6) is a 11.7-kDa protein that is produced as a part of a two-component toxin–antitoxin system involved in the maintenance of the copy number of the F plasmid (Miki et al. 1984; Miki et al. 1992). CcdB is active as a dimer and is inactivated by CcdA (Loris et al. 1999; Madl et al. 2006); its intracellular target is DNA gyrase (Bernard and Couturier 1992; Bernard et al. 1993). CcdB kills bacteria by a mechanism similar to quinolones insofar as it involves stabilisation of the cleavage complex, although evidence shows that the interaction between CcdB and gyrase is significantly different. The mutation of Arg462 to Cys in GyrA, which confers gyrase resistance to CcdB (Bernard and Couturier 1992), is located in the central cavity of GyrA dimer, close to the exit gate and far from the QRDR (Morais Cabral et al. 1997); no cross-resistance between CcdB and quinolones has been observed (Bernard et al. 1993; Critchlow et al. 1997). Biochemical evidence shows that there are significant differences in the modes of action of quinolones and CcdB. CcdB-induced cleavage of DNA by gyrase requires nucleotides (ATP for linear or relaxed DNA and a non-hydrolysable nucleotide for negatively supercoiled DNA) and DNA templates of at least 160 bp (Critchlow et al. 1997), whereas quinolone-induced cleavage does not generally require ATP, and DNA substrates of ∼20 bp can be cleaved (Cove et al. 1997; Gmünder et al. 1997). In contrast to quinolones, CcdB is unable to mediate DNA cleavage with gyrases containing just the N-terminal domain of GyrA (GyrA64 associated with GyrB; Critchlow et al. 1997).

CcdB. a Model of gyrase inhibition by CcdB. The proteins and DNA come together (2) to form the holoenzyme with the G segment binding across the DNA gate (3). The DNA is wrapped around GyrA presenting the T segment over the G segment (4). Upon ATP binding, the GyrB monomers dimerise (5), the G segment is cleaved and the DNA gate opens. Either (6a) ‘top-down’ passage of the T segment occurs upon hydrolysis of a single ATP or (6b) CcdB accesses its binding site to stabilise the cleavage complex. A futile ATP hydrolysis/ATP binding cycle can occur (Kampranis et al. 1999b). Several cycles of (5) to (6a) transition may occur prior to CcdB binding. In the normal cycle, the DNA gate closes, the G segment is religated and the T segment passes out through the bottom gate, with hydrolysis of the second ATP resetting the enzyme (7) (reproduced from (Smith and Maxwell 2006) with permission from Oxford University Press.) b Complex of CcdB and GyrA14 and models: (a) Gyr-A14: CcdB complex viewed along its twofold axis. The two CcdB monomers are drawn in orange and red and the two GyrA14 monomers in two shades of blue. (b) Same as (a), but rotated 90°. (c) Model of closed GyrA59 (blue) with CcdB (green) docked onto it by superposition of the GyrA14:CcdB complex on the crystal structure of GyrA59. Residues Arg462 of both monomers of GyrA59 are highlighted. Strong steric overlaps (red) with the CAP domains are observed. (d) Model of the GyrA59:CcdB complex with GyrA59 in its open conformation. The open conformation of GyrA59 was modelled according to the corresponding yeast topoisomerase II structure (PDB entry 1BGW). In this model, there is no steric overlap between CcdB and GyrA59 (reproduced from (Dao-Thi et al. 2005) with permission from Elsevier)
A crystal structure of CcdB with an N-terminal fragment of GyrA (GyrA14) (Dao-Thi et al. 2005) suggests that the toxin binds to the GyrA dimer in the cavity between the two subunits, confirming the role of Arg462 in CcdB binding to GyrA (Fig. 6b). This residue makes key contacts with residues at the C-terminal end of CcdB. Modelling suggests that the CcdB dimer can be accommodated in the cavity within the dimer of the GyrA N-terminal domain, but only when the enzyme is in the DNA gate-open conformation (Fig. 6b; Dao-Thi et al. 2005), explaining why CcdB can only act when gyrase is carrying out strand-passage reactions (Fig. 6a; Smith and Maxwell 2006). Subsequent structural work with CcdA has shown that the antitoxin protein, CcdA, has an intrinsically disordered domain and that the protein binds to two partially overlapping binding sites in CcdB to relieve its inhibition of gyrase (De Jonge et al. 2009).
CcdB has a unique mode of action that could, in principle, be emulated by small molecules that could be developed as novel antibacterial agents. However, the large size of CcdB and its extensive contacts with GyrA make this a challenge. Nevertheless, CcdB derivatives, about half the size of the native protein, comprising fragments of CcdB connected by a 6-aminohexanoic acid flexible linker (Trovatti et al. 2008), have been reported to have inhibitory activity on gyrase supercoiling and topo IV relaxation, suggesting that it indeed may be possible to develop CcdB-derived agents.
ParE
ParE/ParD is another example of a toxin–antitoxin system where the toxin targets DNA gyrase (Jiang et al. 2002; Johnson et al. 1996). The ParE toxin is produced by the RK2 plasmid of E. coli and has been shown to inhibit DNA synthesis and to stabilise the gyrase–DNA cleavage complex. Studies on the ParE2 toxin from Vibrio cholerae have shown that its mode of inhibition of gyrase is quite distinct in that it requires ATP and inhibits supercoiling but not relaxation (Yuan et al. 2010). Moreover, although the antitoxin ParD2 prevents ParE2 toxicity, it cannot reverse it. The crystal structure of the ParD–ParE complex from Caulobacter crescentus has been solved and is found to be a hetero-tetramer, and the antitoxin (ParD) does not show the unfolded–folded transition seen with CcdA (Dalton and Crosson 2010); this was preceded by the determination of the solution structure of ParD by NMR (Oberer et al. 2007). There are significant similarities between CcdB and ParE (including a degree of sequence similarity), suggesting that ParE could be investigated in a similar way to what has been carried out with CcdB. ParD possesses features reminiscent of the exit gate of GyrA (Dalton and Crosson 2010); this structure could therefore form a useful start point for modelling the ParE–GyrA interaction.
Pentapeptide repeat proteins: Qnr and MfpA
Pentapeptide repeat proteins (PRPs) are a large (>500) family of prokaryotic and eukaryotic proteins that contain a tandemly repeated 5-amino acid motif (Vetting et al. 2006). Qnr is an example and is involved in plasmid-mediated fluoroquinolone resistance, originally found in Klebsiella pneumoniae (Martinez-Martinez et al. 1998). Qnr acts by protecting DNA gyrase and topoisomerase IV from quinolones, apparently by binding to the enzyme prior to the binding of DNA (Tran and Jacoby 2002; Tran et al. 2005a, b). This mode of action was confirmed by the structure of MfpA, a Qnr homologue from Mycobacterium tuberculosis, which shows that the protein adopts a ‘DNA-like’ structure containing a right-handed quadrilateral β-sheet composed of the pentapeptide repeats (Hegde et al. 2005). Modelling suggests that this motif could bind to the A subunit of gyrase, mimicking DNA (Fig. 7). The structure of a Qnr protein bound to GyrA has not yet been reported. The structure of a Qnr homologue from Enterococcus faecalis has also been published (Hegde et al. 2011) and shows an analogous structure to MfpA. Another PRP family member is McbG (San Millan et al. 1985), which is encoded on the MccB17-producing plasmids and confers resistance to MccB17, presumably by a mechanism similar to Qnr/MfpA.

Although Qnr/Mfp proteins confer protection from fluoroquinolones, they also inhibit gyrase-catalysed supercoiling (Hegde et al. 2011; Hegde et al. 2005), presumably by preventing the binding of the enzyme to DNA, a mode of action reminiscent of simocyclinones. It is currently unclear what the physiological role of these proteins is, but they have an intriguing mode of action, which could present possibilities for the rational design of novel antibacterials.
Other protein inhibitors of gyrase
GyrI (also known as SbmC and YeeB) is an 18-kDa E. coli protein that binds and inhibits DNA gyrase (Nakanishi et al. 1998). As SbmC, it was originally identified as a factor that protects cells from MccB17 (Baquero et al. 1995). GyrI binds to the gyrase holoenzyme (A2B2) with a higher affinity than to either subunit alone and a short stretch of eight amino acids from GyrI has also been shown to inhibit gyrase activity (Nakanishi et al. 1998). GyrI appears to act by preventing gyrase from binding DNA, thus inhibiting the enzyme but also protecting it from the action of MccB17 and CcdB (Chatterji and Nagaraja 2002). GyrI has been found to impart partial protection from quinolones and also negate the effects of the alkylating agents mitomycin C and N-methyl-N-nitroso-N′-nitroguanidine (Chatterji et al. 2003). A crystal structure of GyrI is available (Romanowski et al. 2002), but this does not give a clear idea of its molecular mode of action. However, the fact that peptides derived from GyrI also inhibit gyrase suggests that it ought to be possible to generate small molecules that emulate its activity.
Glutamate racemase (MurI) is an enzyme that converts L-glutamate to D-glutamate, which is an important step in bacterial cell wall synthesis; MurI was found to be an inhibitor of E. coli DNA gyrase (Ashiuchi et al. 2002). In Bacillus subtilis, there are two glutamate racemase enzymes, Glr and YrpC, and only YrpC was found to be a gyrase inhibitor (Ashiuchi et al. 2003). MurI proteins from M. tuberculosis and Mycobacterium smegmatis have also been shown to inhibit gyrase (Sengupta et al. 2008; Sengupta and Nagaraja 2008a; Sengupta et al. 2006) and over-expression protects cells from the action of the fluoroquinolone ciprofloxacin. Studies with mutants have shown that the racemisation and gyrase inhibitory functions of MurI are independent (Sengupta et al. 2008). It is not really clear why MurI has evolved this ‘moonlighting’ function of inhibiting gyrase, but it may provide new ideas for the development of antibacterials.
YacG is a small (∼7 kDa) protein from E. coli that contains a zinc-binding motif, which has been shown to inhibit gyrase by preventing DNA binding (Sengupta and Nagaraja 2008b); it does not affect topo IV or topo I. YacG acts by binding to the C-terminal domain of GyrB. Its 3-D structure has been determined by NMR (Ramelot et al. 2002).
Summary and perspectives
Fluoroquinolones have been the most successful antibacterial agents targeted to gyrase, but these compounds have been extensively explored both in terms of improving spectrum and potency, and overcoming bacterial resistance, and we may have reached the limits of what these compounds can provide. Therefore, we need to develop new types of compound and explore new modes of action. It is clear from the foregoing discussion that gyrase provides ample opportunities to develop new antibacterial agents. Screening chemical libraries has yielded new synthetic entities with gyrase-inhibiting properties, but it remains to be seen whether this approach and the compounds generated will achieve clinical success. Nonetheless, they introduce the possibilities of new scaffolds to explore in terms of new antibacterial drugs. A variety of natural products, small molecules and protein-based entities, have been identified as gyrase inhibitors. These are not generally directly usable as drugs, due to reasons such as toxicity and poor physicochemical properties, but they provide chemical diversity that is not readily achievable using chemical synthesis; they can also reveal novel modes of mechanism of inhibition of the enzyme. It seems that in our race to combat drug-resistant bacterial pathogens, the exploitation of DNA gyrase as an antibacterial target provides diverse and realistic possibilities.
References
Andersen NR, Rasmussen PR (1984) The constitution of clerocidin a new antibiotic isolated from Oidiodendron truncatum. Tetrahedron Letters 25(4):465–468
Anderson VE, Osheroff N (2001) Type II topoisomerases as targets for quinolone antibacterials: turning Dr.Jekyll into Mr. Hyde. Curr Pharm Des 7(5):337–353
Angehrn P, Buchmann S, Funk C, Goetschi E, Gmuender H, Hebeisen P, Kostrewa D, Link H, Luebbers T, Masciadri R, Nielsen J, Reindl P, Ricklin F, Schmitt-Hoffmann A, Theil FP (2004) New antibacterial agents derived from the DNA gyrase inhibitor cyclothialidine. J Med Chem 47(6):1487–1513
Asensio C, Pérez-Díaz JC, Martínez MC, Baquero F (1976) A new family of low molecular weight antibiotics from enterobacteria. Biochem Bioph Res Co 69(1):7–14
Ashiuchi M, Kuwana E, Yamamoto T, Komatsu K, Soda K, Misono H (2002) Glutamate racemase is an endogenous DNA gyrase inhibitor. J Biol Chem 277(42):39070–39073
Ashiuchi M, Kuwana E, Komatsu K, Soda K, Misono H (2003) Differences in effects on DNA gyrase activity between two glutamate racemases of Bacillus subtilis, the poly-gamma-glutamate synthesis-linking Glr enzyme and the YrpC (MurI) isozyme. FEMS Microbiol Lett 223(2):221–225
Baker NM, Weigand S, Maar-Mathias S, Mondragon A (2011) Solution structures of DNA-bound gyrase. Nucleic Acids Res 39(2):755–766
Baquero MR, Bouzon M, Varea J, Moreno F (1995) sbmC, a stationary-phase induced SOS Escherichia coli gene, whose product protects cells from the DNA replication inhibitor microcin B17. Mol Microbiol 18(2):301–311
Bates AD, Maxwell A (2005) DNA Topology. Oxford University Press, Oxford
Bates AD, Maxwell A (2007) Energy coupling in type ii topoisomerases: why do they hydrolyze atp? Biochemistry 46:7929–7941
Bax BD, Chan PF, Eggleston DS, Fosberry A, Gentry DR, Gorrec F, Giordano I, Hann MM, Hennessy A, Hibbs M, Huang J, Jones E, Jones J, Brown KK, Lewis CJ, May EW, Saunders MR, Singh O, Spitzfaden CE, Shen C, Shillings A, Theobald AJ, Wohlkonig A, Pearson ND, Gwynn MN (2010) Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466(7309):935–940
Berger JM, Gamblin SJ, Harrison SC, Wang JC (1996) Structure and mechanism of DNA topoisomerase II. Nature 379(6562):225–232
Bernard P, Couturier M (1992) Cell killing by the F plasmid CcdB protein involves poisoning of the DNA–topoisomerase II complex. J Mol Biol 226:735–745
Bernard P, Kézdy KE, Van Melderen L, Steyaert J, Wyns L, Pato ML, Higgins NP, Couturier M (1993) The F plasmid CcdB protein induces efficient ATP-dependent DNA cleavage by gyrase. J Mol Biol 234:534–541
Birch RG, Patil SS (1985) Preliminary characterization of an antibiotic produced by Xanthomonas albilineans which inhibits DNA synthesis in Escherichia coli. J Gen Microbiol 131(Pt 5):1069–1075
Black MT, Stachyra T, Platel D, Girard AM, Claudon M, Bruneau JM, Miossec C (2008) Mechanism of action of the antibiotic NXL101, a novel nonfluoroquinolone inhibitor of bacterial type II topoisomerases. Antimicrob Agents Chemother 52(9):3339–3349
Bradbury BJ, Pucci MJ (2008) Recent advances in bacterial topoisomerase inhibitors. Curr Opin Pharmacol 8(5):574–581
Brino L, Urzhumtsev A, Mousli M, Bronner C, Mitschler A, Oudet P, Moras D (2000) Dimerization of Escherichia coli DNA-gyrase B provides a structural mechanism for activating the ATPase catalytic center. J Biol Chem 275(13):9468–9475
Champoux JJ (2001) DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem 70:369–413
Chatterji M, Nagaraja V (2002) GyrI: a counter-defensive strategy against proteinaceous inhibitors of DNA gyrase. EMBO Rep 3(3):261–267
Chatterji M, Sengupta S, Nagaraja V (2003) Chromosomally encoded gyrase inhibitor GyrI protects Escherichia coli against DNA-damaging agents. Arch Microbiol 180(5):339–346
Contreras A, Maxwell A (1992) gyrB mutations which confer coumarin resistance also affect DNA supercoiling and ATP hydrolysis by Escherichia coli DNA gyrase. Molec Microbiol 6:1617–1624
Corbett KD, Shultzaberger RK, Berger JM (2004) The C-terminal domain of DNA gyrase A adopts a DNA-bending {beta}-pinwheel fold. Proc Natl Acad Sci U S A 101(19):7293–7298
Costenaro L, Grossmann JG, Ebel C, Maxwell A (2005) Small-angle X-ray scattering reveals the solution structure of the full-length DNA gyrase A subunit. Structure 13:287–296
Costenaro L, Grossmann JG, Ebel C, Maxwell A (2007) Modular structure of the full-length DNA gyrase B subunit revealed by small-angle X-ray scattering. Structure 15(3):329–339
Cove ME, Tingey AP, Maxwell A (1997) DNA gyrase can cleave short DNA fragments in the presence of quinolone drugs. Nucleic Acids Res 25(14):2716–2722
Critchlow SE, O’Dea MH, Howells AJ, Couturier M, Gellert M, Maxwell A (1997) The interaction of the F-plasmid killer protein, CcdB, with DNA gyrase: induction of DNA cleavage and blocking of transcription. J Mol Biol 273:826–839
Dalton KM, Crosson S (2010) A conserved mode of protein recognition and binding in a ParD–ParE toxin–antitoxin complex. Biochemistry 49(10):2205–2215
Dao-Thi MH, Van Melderen L, De Genst E, Afif H, Buts L, Wyns L, Loris R (2005) Molecular basis of gyrase poisoning by the addiction toxin CcdB. J Mol Biol 348(5):1091–1102
Davagnino J, Herrero M, Furlong D, Moreno F, Kolter R (1986) The DNA replication inhibitor microcin B17 is a forty-three-amino-acid protein containing sixty percent glycine. Proteins 1(3):230–238
De Jonge N, Garcia-Pino A, Buts L, Haesaerts S, Charlier D, Zangger K, Wyns L, De Greve H, Loris R (2009) Rejuvenation of CcdB-poisoned gyrase by an intrinsically disordered protein domain. Mol Cell 35(2):154–163
del Castillo I, Vizan J, Rodriguez-Sainz M, Moreno F (1991) An unusual mechanism for resistance to the antibiotic coumermycin A1. Proc Natl Acad Sci USA 88:8860–8864
Dong KC, Berger JM (2008) Structure and function of DNA topoisomerases. In: Rice PA, Correll CC (eds) Protein-nucleic acid interactions: structural biology. RSC Biomolecular Sciences. Royal Society of Chemistry, London
Drlica K, Hooper DC (2003) Mechanisms of quinolone action. In: Hooper DC, Rubinstein E (eds) Quinolone antimicrobial agents, 3rd edn. ASM Press, Washington DC, pp 19–40
Drlica K, Hiasa H, Kerns R, Malik M, Mustaev A, Zhao X (2009) Quinolones: action and resistance updated. Curr Top Med Chem 9(11):981–998
Dwyer DJ, Kohanski MA, Hayete B, Collins JJ (2007) Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol Syst Biol 3:91
Edwards MJ, Flatman RH, Mitchenall LA, Stevenson CE, Le TBK, Fiedler HP, McKay AR, Clarke TA, Buttner MJ, Lawson DM, Maxwell A (2009) A crystal structure of the bifunctional antibiotic, simocyclinone D8, bound to DNA gyrase. Science 326:1415–1418
Edwards MJ, Williams MA, Maxwell A, McKay AR (2011) Mass spectrometry reveals that the antibiotic simocyclinone D8 binds to DNA gyrase in a “bent-over” conformation: evidence of positive cooperativity in binding. Biochemistry 50(17):3432–3440
Emmerson AM, Jones AM (2003) The quinolones: decades of development and use. J Antimicrob Chemother 51(Suppl 1):13–20
Ferrero L, Cameron B, Manse B, Lagneaux D, Crouzet J, Famechon A, Blanche F (1994) Cloning and primary structure of Staphylococcus aureus DNA topoisomerase IV: a primary target of fluoroquinolones. Molec Microbiol 13:641–653
Flatman RH, Howells AJ, Heide L, Fiedler H-P, Maxwell A (2005) Simocyclinone D8: an inhibitor of DNA gyrase with a novel mode of action. Antimicrob Agents Chemother 49:1093–1100
Flatman RH, Eustaquio A, Li SM, Heide L, Maxwell A (2006) Structure–activity relationships of aminocoumarin-type gyrase and topoisomerase IV inhibitors obtained by combinatorial biosynthesis. Antimicrob Agents Chemother 50(4):1136–1142
Fu G, Wu J, Liu W, Zhu D, Hu Y, Deng J, Zhang XE, Bi L, Wang DC (2009) Crystal structure of DNA gyrase B′ domain sheds lights on the mechanism for T-segment navigation. Nucleic Acids Res 37(17):5908–5916
Galm U, Schimana J, Fiedler HP, Schmidt J, Li SM, Heide L (2002) Cloning and analysis of the simocyclinone biosynthetic gene cluster of Streptomyces antibioticus Tu 6040. Arch Microbiol 178(2):102–114
Garrido MC, Herrero M, Kolter R, Moreno F (1988) The export of the DNA replication inhibitor Microcin B17 provides immunity for the host cell. EMBO J 7:1853–1862
Gatto B, Richter S, Moro S, Capranico G, Palumbo M (2001) The topoisomerase II poison clerocidin alkylates non-paired guanines of DNA: implications for irreversible stimulation of DNA cleavage. Nucleic Acids Res 29(20):4224–4230
Gellert M, O’Dea MH, Itoh T, Tomizawa J (1976) Novobiocin and coumermycin inhibit DNA supercoiling catalyzed by DNA gyrase. Proc Natl Acad Sci USA 73:4474–4478
Gellert M, Mizuuchi K, O’Dea MH, Itoh T, Tomizawa J (1977) Nalidixic acid resistance: a second genetic character involved in DNA gyrase activity. Proc Natl Acad Sci USA 74:4772–4776
Gilbert EJ, Maxwell A (1994) The 24 kDa N-terminal sub-domain of the DNA gyrase B protein binds coumarin drugs. Molec Microbiol 12:365–373
Gmünder H, Kuratli K, Keck W (1997) In the presence of subunit A inhibitors DNA gyrase cleaves DNA fragments as short as 20 bp at specific sites. Nucleic Acids Res 25:604–611
Goetschi E, Angehrn P, Gmuender H, Hebeisen P, Link H, Masciadri R, Nielsen J (1993) Cyclothialidine and its congeners: a new class of DNA gyrase inhibitors. Pharmacol Ther 60:367–380
Gomez L, Hack MD, Wu J, Wiener JJ, Venkatesan H, Santillan A Jr, Pippel DJ, Mani N, Morrow BJ, Motley ST, Shaw KJ, Wolin R, Grice CA, Jones TK (2007) Novel pyrazole derivatives as potent inhibitors of type II topoisomerases. Part 1: synthesis and preliminary SAR analysis. Bioorg Med Chem Lett 17(10):2723–2727
Greenstein M, Speth JL, Maise WM (1981) Mechanism of action of cinodine, a glycocinnamoylspermidine antibiotic. Antimicrob Agents and Chemother 20:425–432
Gross CH, Parsons JD, Grossman TH, Charifson PS, Bellon S, Jernee J, Dwyer M, Chambers SP, Markland W, Botfield M, Raybuck SA (2003) Active-site residues of Escherichia coli DNA gyrase required in coupling ATP hydrolysis to DNA supercoiling and amino acid substitutions leading to novobiocin resistance. Antimicrob Agents Chemother 47(3):1037–1046
Hashimi SM, Wall MK, Smith AB, Maxwell A, Birch RG (2007) The phytotoxin albicidin is a novel inhibitor of DNA gyrase. Antimicrob Agents Chemother 51(1):181–187
Hashimi SM, Huang G, Maxwell A, Birch RG (2008) DNA gyrase from the albicidin producer Xanthomonas albilineans has multiple-antibiotic-resistance and unusual enzymatic properties. Antimicrob Agents Chemother 52(4):1382–1390
Heddle J, Maxwell A (2002) Quinolone-binding pocket of DNA gyrase: role of GyrB. Antimicrob Agents Chemother 46:1805–1815
Heddle JG, Barnard FM, Wentzell LM, Maxwell A (2000) The interaction of drugs with DNA gyrase: a model for the molecular basis of quinolone action. Nucleosides Nucleotides Nucleic Acids 19:1249–1264
Heddle JG, Blance SJ, Zamble DB, Hollfelder F, Miller DA, Wentzell LM, Walsh CT, Maxwell A (2001) The antibiotic microcin B17 is a DNA gyrase poison: characterisation of the mode of inhibition. J Mol Biol 307(5):1223–1234
Heddle JG, Mitelheiser S, Maxwell A, Thomson NH (2004) Nucleotide binding to DNA gyrase causes loss of DNA wrap. J Mol Biol 337(3):597–610
Hegde SS, Vetting MW, Roderick SL, Mitchenall LA, Maxwell A, Takiff HE, Blanchard JS (2005) A fluoroquinolone resistance protein from Mycobacterium tuberculosis that mimics DNA. Science 308:1480–1483
Hegde SS, Vetting MW, Mitchenall LA, Maxwell A, Blanchard JS (2011) Structural and biochemical analysis of the pentapeptide repeat protein EfsQnr, a potent DNA gyrase inhibitor. Antimicrob Agents Chemother 55(1):110–117
Heide L (2009a) The aminocoumarins: biosynthesis and biology. Nat Prod Rep 26(10):1241–1250
Heide L (2009b) Genetic engineering of antibiotic biosynthesis for the generation of new aminocoumarins. Biotechnol Adv 27(6):1006–1014
Herrero M, Moreno F (1986) Microcin B17 blocks DNA replication and induces the SOS system in Escherichia coli. J Gen Microbiol 132:393–402
Holdgate GA, Tunnicliffe A, Ward WHJ, Weston SA, Rosenbrock G, Barth PT, Taylor IWF, Pauptit RA, Timms D (1997) The entropic penalty of ordered water accounts for weaker binding of the antibiotic novobiocin to a resistant mutant of DNA gyrase: a thermodynamic and crystallographic study. Biochemistry 36:9663–9673
Holmes ML, Dyall-Smith ML (1991) Mutations in DNA gyrase result in novobiocin resistance in halophilic archaebacteria. J Bacteriol 173:642–648
Hooper DC (2003) Mechanisms of quinolone resistance. In: Hooper DC, Rubinstein E (eds) Quinolone antimicrobial agents, 3rd edn. ASM Press, Washington DC, pp 41–67
Hsieh TJ, Yen TJ, Lin TS, Chang HT, Huang SY, Hsu CH, Farh L, Chan NL (2010) Twisting of the DNA-binding surface by a {beta}-strand-bearing proline modulates DNA gyrase activity. Nucleic Acids Res 38(12):4173–4181
Huband MD, Cohen MA, Zurack M, Hanna DL, Skerlos LA, Sulavik MC, Gibson GW, Gage JW, Ellsworth E, Stier MA, Gracheck SJ (2007) In vitro and in vivo activities of PD 0305970 and PD 0326448, new bacterial gyrase/topoisomerase inhibitors with potent antibacterial activities versus multidrug-resistant Gram-positive and fastidious organism groups. Antimicrob Agents Chemother 51(4):1191–1201
Jiang Y, Pogliano J, Helinski DR, Konieczny I (2002) ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol Microbiol 44(4):971–979
Johnson EP, Strom AR, Helinski DR (1996) Plasmid RK2 toxin protein ParE: purification and interaction with the ParD antitoxin protein. J Bacteriol 178(5):1420–1429
Kampranis SC, Gormley NA, Tranter R, Orphanides G, Maxwell A (1999a) Probing the binding of coumarins and cyclothialidines to DNA gyrase. Biochemistry 38:1967–1976
Kampranis SC, Howells AJ, Maxwell A (1999b) The interaction of DNA gyrase with the bacterial toxin CcdB: evidence for the existence of two gyrase–CcdB complexes. J Mol Biol 293(3):733–744
Kawada S, Yamashita Y, Fujii N, Nakano H (1991) Induction of a heat-stable topoisomerase II–DNA cleavable complex by nonintercalative terpenoides, terpentecin and clerocidin. Cancer Res 51(11):2922–2925
King DE, Malone R, Lilley SH (2000) New classification and update on the quinolone antibiotics. Am Fam Physician 61(9):2741–2748
Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ (2007) A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130(5):797–810
Kohanski MA, Dwyer DJ, Collins JJ (2010) How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol 8(6):423–435
Lafitte D, Lamour V, Tsvetkov PO, Makarov AA, Klich M, Deprez P, Moras D, Briand C, Gilli R (2002) DNA gyrase interaction with coumarin-based inhibitors: the role of the hydroxybenzoate isopentenyl moiety and the 5′-methyl group of novobiose. Biochemistry 41:7217–7223
Lamour V, Hoermann L, Jeltsch JM, Oudet P, Moras D (2002) An open conformation of the Thermus thermophilus gyrase B ATP-binding domain. J Biol Chem 277(21):18947–18953
Laponogov I, Sohi MK, Veselkov DA, Pan X-S, Sawhney R, Thompson AW, McAuley KE, Fisher LM, Sanderson MR (2009) Structural insight into the quinolone–DNA cleavage complex of type IIA topoisomerases. Nat Struct Mol Biol 16(6):667–669
Laponogov I, Pan XS, Veselkov DA, McAuley KE, Fisher LM, Sanderson MR (2010) Structural basis of gate-DNA breakage and resealing by type II topoisomerases. PLoS ONE 5(6):e11338
Larsen AK, Escargueil AE, Skladanowski A (2003) Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol Ther 99(2):167–181
Lesher GY, Forelich EJ, Gruett MD, Bailey JH, Brundage RP (1962) 1,8-naphthyridine derivatives, a new class of chemotherapeutic agents. J Med Chem 5:1063–1065
Lewis RJ, Singh OMP, Smith CV, Skarynski T, Maxwell A, Wonacott AJ, Wigley DB (1996a) The nature of inhibition of DNA gyrase by the coumarins and the cyclothialidines revealed by X-ray crystallography. EMBO J 15:1412–1420
Lewis RJ, Tsai FT, Wigley DB (1996b) Molecular mechanisms of drug inhibition of DNA gyrase. Bioessays 18(8):661–671
Liu LF, Liu C-C, Alberts BM (1980) Type II DNA topoisomerases: enzymes that can unknot a topologically knotted DNA molecule via a reversible double-strand break. Cell 19:697–707
Lomovskaya O, Kawai F, Matin A (1996) Differential regulation of the mcb and emr operons of Escherichia coli: role of mcb in multidrug resistance. Antimicrob Agents Chemother 40(4):1050–1052
Loris R, Dao-Thi M-H, Bahassi EM, Van Melderen LV, Poortmans F, Liddington RC, Couturier M, Wyns L (1999) Crystal structure of CcdB, a topoisomerase poison from E. coli. J Mol Biol 285:1667–1677
Madl T, Van Melderen L, Mine N, Respondek M, Oberer M, Keller W, Khatai L, Zangger K (2006) Structural basis for nucleic acid and toxin recognition of the bacterial antitoxin CcdA. J Mol Biol 364(2):170–185
Martin JH, Kunstmann MP, Barbatschi F, Hertz M, Ellestad GA, Dann M, Redin GS, Dornbush AC, Kuck NA (1978) Glycocinnamoylspermidines, a new class of antibiotics. II. Isolation, physiocochemical and biological properties of LL-BM123beta, gamma1 and gamma2. J Antibiot (Tokyo) 31(5):398–404
Martinez-Martinez L, Pascual A, Jacoby GA (1998) Quinolone resistance from a transferable plasmid. Lancet 351(9105):797–799
Maxwell A, Lawson DM (2003) The ATP-binding site of type II topoisomerases as a target for antibacterial drugs. Curr Top Med Chem 3(1):283–303
McCullough JE, Muller MT, Howells AJ, Maxwell A, O’Sullivan J, Summerill RS, Parker WL, Wells JS, Bonner DP, Fernandes PB (1993) Clerocidin, a terpenoid antibiotic, inhibits bacterial DNA gyrase. J Antibiot 46:526–530
Miki T, Chang Z-T, Horiuchi T (1984) Control of cell division by sex factor F in Escherichia coli II. Identification of genes for inhibitor protein and trigger protein on the 42.84–43.6 F segment. J Mol Biol 174:627–646
Miki T, Park JA, Nagao K, Murayama N, Horiuchi T (1992) Control of segregation of chromosomal DNA by sex factor F in Escherichia coli. J Mol Biol 225:39–52
Miller AA, Bundy GL, Mott JE, Skepner JE, Boyle TP, Harris DW, Hromockyj AE, Marotti KR, Zurenko GE, Munzner JB, Sweeney MT, Bammert GF, Hamel JC, Ford CW, Zhong WZ, Graber DR, Martin GE, Han F, Dolak LA, Seest EP, Ruble JC, Kamilar GM, Palmer JR, Banitt LS, Hurd AR, Barbachyn MR (2008) Discovery and characterization of QPT-1, the progenitor of a new class of bacterial topoisomerase inhibitors. Antimicrob Agents Chemother 52(8):2806–2812
Mizuuchi K, O’Dea MH, Gellert M (1978) DNA gyrase: subunit structure and ATPase activity of the purified enzyme. Proc Natl Acad Sci USA 75:5960–5963
Morais Cabral JH, Jackson AP, Smith CV, Shikotra N, Maxwell A, Liddington RC (1997) Crystal structure of the breakage-reunion domain of DNA gyrase. Nature 388(6645):903–906
Nakada N, Shimada H, Hirata T, Aoki Y, Kamiyama T, Watanabe J, Arisawa M (1993) Biological characterization of cyclothialidine, a new DNA gyrase inhibitor. Antimicrob Agents Chemother 37:2656–2661
Nakada N, Gmünder H, Hirata T, Arisawa M (1994) Mechanism of inhibition of DNA gyrase by cyclothialidine, a novel DNA gyrase inhibitor. Antimicrob Agents Chemother 38:1966–1973
Nakada N, Gmünder H, Hirata T, Arisawa M (1995) Characterization of the binding site for cyclothialidine on the B subunit of DNA gyrase. J Biol Chem 270:14286–14291
Nakanishi A, Oshida T, Matsushita T, Imajoh-Ohmi S, Ohnuki T (1998) Identification of DNA gyrase inhibitor (GyrI) in Escherichia coli. J Biol Chem 273:1933–1938
Nollmann M, Crisona NJ, Arimondo PB (2007) Thirty years of Escherichia coli DNA gyrase: from in vivo function to single-molecule mechanism. Biochimie 89(4):490–499
Oberer M, Zangger K, Gruber K, Keller W (2007) The solution structure of ParD, the antidote of the ParDE toxin antitoxin module, provides the structural basis for DNA and toxin binding. Protein Sci 16(8):1676–1688
Oblak M, Kotnik M, Solmajer T (2007) Discovery and development of ATPase inhibitors of DNA gyrase as antibacterial agents. Curr Med Chem 14(19):2033–2047
Oliphant CM, Green GM (2002) Quinolones: a comprehensive review. Am Fam Physician 65(3):455–464
Oppegard LM, Hamann BL, Streck KR, Ellis KC, Fiedler HP, Khodursky AB, Hiasa H (2009) In vivo and in vitro patterns of the activity of simocyclinone D8, an angucyclinone antibiotic from Streptomyces antibioticus. Antimicrob Agents Chemother 53(5):2110–2119
Oram M, Dosanjh B, Gormley NA, Smith CV, Fisher LM, Maxwell A, Duncan K (1996) The mode of action of GR122222X, a novel inhibitor of bacterial DNA gyrase. Antimicrob Agents Chemother 40:473–476
Orphanides G, Maxwell A (1994) Evidence for a conformational change in the DNA gyrase–DNA complex from hydroxyl radical footprinting. Nucleic Acids Res 22:1567–1575
Osburne MS (1995) Characterization of a cinodine-resistant mutant of Escherichia coli. J Antibiot 48:1359–1361
Osburne MS, Maiese WM, Greenstein M (1990) In vitro inhibition of bacterial DNA gyrase by Cinodine, a glycocinnamoylspermidine antibiotic. Antimicrob Agents Chemother 34:1450–1452
Pan X-S, Fisher LM (1997) Targeting of DNA gyrase in Streptococcus pneumomiae by sparfloxacin: selective targeting of gyrase or topoisomerase IV by quinolones. Antimicrob Agents Chemotherap 41:471–474
Pan XS, Dias M, Palumbo M, Fisher LM (2008) Clerocidin selectively modifies the gyrase–DNA gate to induce irreversible and reversible DNA damage. Nucleic Acids Res 36(17):5516–5529
Parks WM, Bottrill AR, Pierrat OA, Durrant MC, Maxwell A (2007) The action of the bacterial toxin, microcin B17, on DNA gyrase. Biochimie 89:500–507
Pierrat OA, Maxwell A (2003) The action of the bacterial toxin microcin B17: insight into the cleavage-religation reaction of DNA gyrase. J Biol Chem 278:35016–35023
Pierrat OA, Maxwell A (2005) Evidence for the role of DNA strand passage in the mechanism of action of microcin B17 on DNA gyrase. Biochemistry 44:4204–4215
Piton J, Petrella S, Delarue M, Andre-Leroux G, Jarlier V, Aubry A, Mayer C (2010) Structural insights into the quinolone resistance mechanism of Mycobacterium tuberculosis DNA gyrase. PLoS ONE 5(8):e12245
Pommier Y, Leo E, Zhang H, Marchand C (2010) DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol 17(5):421–433
Ramelot TA, Cort JR, Yee AA, Semesi A, Edwards AM, Arrowsmith CH, Kennedy MA (2002) NMR structure of the Escherichia coli protein YacG: a novel sequence motif in the zinc-finger family of proteins. Proteins 49(2):289–293
Reece RJ, Maxwell A (1991) DNA gyrase: structure and function. CRC Crit Rev Biochem Mol Biol 26:335–375
Richter S, Gatto B, Fabris D, Takao K, Kobayashi S, Palumbo M (2003) Clerocidin alkylates DNA through its epoxide function: evidence for a fine tuned mechanism of action. Nucleic Acids Res 31(17):5149–5156
Richter SN, Leo E, Giaretta G, Gatto B, Fisher LM, Palumbo M (2006) Clerocidin interacts with the cleavage complex of Streptococcus pneumoniae topoisomerase IV to induce selective irreversible DNA damage. Nucleic Acids Res 34(7):1982–1991
Richter SN, Frasson I, Palumbo M, Sissi C, Palu G (2010) Simocyclinone D8 turns on against Gram-negative bacteria in a clinical setting. Bioorg Med Chem Lett 20(3):1202–1204
Roca J, Wang JC (1992) The capture of a DNA double helix by an ATP-dependent protein clamp: a key step in DNA transport by type II DNA topoisomerases. Cell 71:833–840
Roca J, Wang JC (1994) DNA transport by a type II DNA topoisomerase: evidence in favor of a two-gate mechanism. Cell 77:609–616
Romanowski MJ, Gibney SA, Burley SK (2002) Crystal structure of the Escherichia coli SbmC protein that protects cells from the DNA replication inhibitor microcin B17. Proteins 47(3):403–407
Rott PC, Costet L, Davis MJ, Frutos R, Gabriel DW (1996) At least two separate gene clusters are involved in albicidin production by Xanthomonas albilineans. J Bacteriol 178(15):4590–4596
Sadiq AA, Patel MR, Jacobson BA, Escobedo M, Ellis K, Oppegard LM, Hiasa H, Kratzke RA (2009) Anti-proliferative effects of simocyclinone D8 (SD8), a novel catalytic inhibitor of topoisomerase II. Invest New Drugs 28(1):20–25
Samuels DS, Marconi RT, Huang WM, Garon CF (1994) gyrB mutations in coumermycin A1-resistant Borrelia burgdorferi. J Bacteriol 176:3072–3075
San Millan JL, Hernandez-Chico C, Pereda P, Moreno F (1985) Cloning and mapping of the genetic determinants for microcin B17 production and immunity. J Bacteriol 163(1):275–281
Schimana J, Fiedler HP, Groth I, Sussmuth R, Beil W, Walker M, Zeeck A (2000) Simocyclinones, novel cytostatic angucyclinone antibiotics produced by Streptomyces antibioticus Tu 6040. I. Taxonomy, fermentation, isolation and biological activities. J Antibiot (Tokyo) 53(8):779–787
Schoeffler AJ, Berger JM (2008) DNA topoisomerases: harnessing and constraining energy to govern chromosome topology. Q Rev Biophys 41(1):41–101
Schoeffler AJ, May AP, Berger JM (2010) A domain insertion in Escherichia coli GyrB adopts a novel fold that plays a critical role in gyrase function. Nucleic Acids Res 38(21):7830–7844
Sengupta S, Nagaraja V (2008a) Inhibition of DNA gyrase activity by Mycobacterium smegmatis MurI. FEMS Microbiol Lett 279(1):40–47
Sengupta S, Nagaraja V (2008b) YacG from Escherichia coli is a specific endogenous inhibitor of DNA gyrase. Nucleic Acids Res 36(13):4310–4316
Sengupta S, Shah M, Nagaraja V (2006) Glutamate racemase from Mycobacterium tuberculosis inhibits DNA gyrase by affecting its DNA-binding. Nucleic Acids Res 34(19):5567–5576
Sengupta S, Ghosh S, Nagaraja V (2008) Moonlighting function of glutamate racemase from Mycobacterium tuberculosis: racemization and DNA gyrase inhibition are two independent activities of the enzyme. Microbiology 154(Pt 9):2796–2803
Sissi C, Palumbo M (2009) Effects of magnesium and related divalent metal ions in topoisomerase structure and function. Nucleic Acids Res 37(3):702–711
Sissi C, Vazquez E, Chemello A, Mitchenall LA, Maxwell A, Palumbo M (2010) Mapping simocyclinone D8 interaction with DNA gyrase: evidence for a new binding site on GyrB. Antimicrob Agents Chemother 54(1):213–220
Smith AB, Maxwell A (2006) A strand-passage conformation of DNA gyrase is required to allow the bacterial toxin, CcdB, to access its binding site. Nucleic Acids Res 34(17):4667–4676
Stieger M, Angehrn P, Wohlgensinger B, Gmünder H (1996) GyrB mutations in Staphylococcus aureus strains resistant to cyclothialidine, coumermycin, and novobiocin. Antimicrob Agents Chemother 40:1060–1062
Sugino A, Cozzarelli NR (1980) The intrinsic ATPase of DNA gyrase. J Biol Chem 255:6299–6306
Sugino A, Peebles CL, Kruezer KN, Cozzarelli NR (1977) Mechanism of action of nalidixic acid: purification of Escherichia coli nalA gene product and its relationship to DNA gyrase and a novel nicking-closing enzyme. Proc Natl Acad Sci USA 74:4767–4771
Sugino A, Higgins NP, Brown PO, Peebles CL, Cozzarelli NR (1978) Energy coupling in DNA gyrase and the mechanism of action of novobiocin. Proc Natl Acad Sci USA 75:4838–4842
Theobald U, Schimana J, Fiedler HP (2000) Microbial growth and production kinetics of Streptomyces antibioticus Tu 6040. Antonie Van Leeuwenhoek 78(3–4):307–313
Thompson RE, Jolliffe KA, Payne RJ (2011) Total synthesis of microcin b17 via a fragment condensation approach. Org Lett 13(4):680–683
Tran JH, Jacoby GA (2002) Mechanism of plasmid-mediated quinolone resistance. Proc Natl Acad Sci U S A 99(8):5638–5642
Tran JH, Jacoby GA, Hooper DC (2005a) Interaction of the plasmid-encoded quinolone resistance protein Qnr with Escherichia coli DNA gyrase. Antimicrob Agents Chemother 49(1):118–125
Tran JH, Jacoby GA, Hooper DC (2005b) Interaction of the plasmid-encoded quinolone resistance protein QnrA with Escherichia coli topoisomerase IV. Antimicrob Agents Chemother 49(7):3050–3052
Trefzer A, Pelzer S, Schimana J, Stockert S, Bihlmaier C, Fiedler HP, Welzel K, Vente A, Bechthold A (2002) Biosynthetic gene cluster of simocyclinone, a natural multihybrid antibiotic. Antimicrob Agents Chemother 46(5):1174–1182
Tresner HD, Korshalla JH, Fantini AA, Korshalla JD, Kirby JP, Goodman JJ, Kele RA, Shay AJ, Borders DB (1978) Glycocinnamoylspermidines, a new class of antibiotics. I. Description and fermentation of the organism producing the LL-BM123 antibiotics. J Antibiot (Tokyo) 31(5):394–397
Tretter EM, Schoeffler AJ, Weisfield SR, Berger JM (2010) Crystal structure of the DNA gyrase GyrA N-terminal domain from Mycobacterium tuberculosis. Proteins 78(2):492–495
Trovatti E, Cotrim CA, Garrido SS, Barros RS, Marchetto R (2008) Peptides based on CcdB protein as novel inhibitors of bacterial topoisomerases. Bioorg Med Chem Lett 18(23):6161–6164
Tsai FT, Singh OM, Skarzynski T, Wonacott AJ, Weston S, Tucker A, Pauptit RA, Breeze AL, Poyser JP, O’Brien R, Ladbury JE, Wigley DB (1997) The high-resolution crystal structure of a 24-kDa gyrase B fragment from E. coli complexed with one of the most potent coumarin inhibitors, clorobiocin. Proteins 28(1):41–52
Tse-Dinh YC (2007) Exploring DNA topoisomerases as targets of novel therapeutic agents in the treatment of infectious diseases. Infect Disord Drug Targets 7(1):3–9
Vetting MW, Hegde SS, Fajardo JE, Fiser A, Roderick SL, Takiff HE, Blanchard JS (2006) Pentapeptide repeat proteins. Biochemistry 45(1):1–10
Videnov G, Kaiser D, Brooks M, Jung G (1996) Synthesis of the DNA gyrase inhibitor microcin B17, a 43-peptie antibiotic with eight heterocycles in its backbone. Agnew Chem Int Ed Engl 35(13/14):1506–1508
Vizán J, Hernandex-Chico C, del Castillo I, Moreno F (1991) The peptide antibiotic microcin B17 induces double-strand cleavage of DNA mediated by E. coli DNA gyrase. EMBO J 10:467–476
Wang JC (2009) A journey in the world of DNA rings and beyond. Annu Rev Biochem 78:31–54
Widdowson K, Hennessy A (2010) Advances in structure-based drug design of novel bacterial topoisomerase inhibitors. Future Med Chem 2(11):1619–1622
Wiener JJ, Gomez L, Venkatesan H, Santillan A Jr, Allison BD, Schwarz KL, Shinde S, Tang L, Hack MD, Morrow BJ, Motley ST, Goldschmidt RM, Shaw KJ, Jones TK, Grice CA (2007) Tetrahydroindazole inhibitors of bacterial type II topoisomerases. Part 2: SAR development and potency against multidrug-resistant strains. Bioorg Med Chem Lett 17(10):2718–2722
Wigley DB, Davies GJ, Dodson EJ, Maxwell A, Dodson G (1991) Crystal structure of an N-terminal fragment of the DNA gyrase B protein. Nature 351:624–629
Williams NL, Maxwell A (1999a) Locking the DNA gate of DNA gyrase: investigating the effects on DNA cleavage and ATP hydrolysis. Biochemistry 38:14157–14164
Williams NL, Maxwell A (1999b) Probing the two-gate mechanism of DNA gyrase using cysteine cross-linking. Biochemistry 38:13502–13511
Wohlkonig A, Chan PF, Fosberry AP, Homes P, Huang J, Kranz M, Leydon VR, Miles TJ, Pearson ND, Perera RL, Shillings AJ, Gwynn MN, Bax BD (2010) Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat Struct Mol Biol 17(9):1152–1153
Yorgey P, Lee J, Kördel J, Vivas E, Warner P, Jebaratnam D, Kolter R (1994) Posttranslational modifications in microcin B17 define an additional class of DNA gyrase inhibitor. Proc Natl Acad Sci USA 91:4519–4523
Yoshida H, Bogaki M, Nakamura M, Nakamura S (1990) Quinolone resistance-determining region in the DNA gyrase gyrA gene of Escherichia coli. Antimicrob Agents Chemother 34:1271–1272
Yoshida H, Bogaki M, Nakamura M, Yamanaka LM, Nakamura S (1991) Quinolone resistance-determining region in the DNA gyrase gyrB gene of Escherichia coli. Antimicrob Agents Chemother 35:1647–1650
Yuan J, Sterckx Y, Mitchenall LA, Maxwell A, Loris R, Waldor MK (2010) Vibrio cholerae ParE2 poisons DNA gyrase via a mechanism distinct from other gyrase inhibitors. J Biol Chem 285(51):40397–40408
Acknowledgements
We thank David Lawson for comments on the manuscript. We acknowledge the financial support of BBSRC (UK), PBL (UK) and the John Innes Foundation; SK was supported in part by an EST early stage research scholarship.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Collin, F., Karkare, S. & Maxwell, A. Exploiting bacterial DNA gyrase as a drug target: current state and perspectives. Appl Microbiol Biotechnol 92, 479–497 (2011). https://doi.org/10.1007/s00253-011-3557-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3557-z