
Abstract
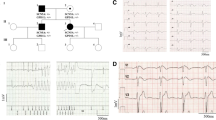
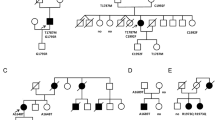
Brugada syndrome (BrS) is characterized by ST-segment elevation in the right precordial leads and is associated with increased risk of sudden cardiac death. We have recently reported families with BrS and SCN5A mutations where some affected members do not carry the familial mutation. We evaluated the involvement of additional genetic determinants for BrS in an affected family. We identified three distinct gene variants within a family presenting BrS (5 individuals), cardiac conduction defects (CCD, 3 individuals) and shortened QT interval (4 individuals). The first mutation is nonsense, p.Q1695*, lying within the SCN5A gene, which encodes for NaV1.5, the α–subunit of the cardiac Na+ channel. The second mutation is missense, p.N300D, and alters the CACNA1C gene, which encodes the α–subunit CaV1.2 of the L-type cardiac Ca2+ channel. The SCN5A mutation strictly segregates with CCD. Four out of the 5 BrS patients carry the CACNA1C variant, and three of them present shortened QT interval. One of the BrS patients carries none of these mutations but a rare variant located in the ABCC9 gene as well as his asymptomatic mother. Patch-clamp studies identified a loss-of-function of the mutated CaV1.2 channel. Western-blot experiments showed a global expression defect while increased mobility of CaV1.2 channels on cell surface was revealed by FRAP experiments. Finally, computer simulations of the two mutations recapitulated patient phenotypes. We report a rare CACNA1C mutation as causing BrS and/or shortened QT interval in a family also carrying a SCN5A stop mutation, but which does not segregate with BrS. This study underlies the complexity of BrS inheritance and its pre-symptomatic genetic screening interpretation.







Similar content being viewed by others

References
Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP Jr, Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu LF, Haïssaguerre M, Schimpf R, Borggrefe M, Wolpert C (2007) Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115:442–449. doi:10.1161/CIRCULATIONAHA.106.668392
Behr ER, Casey A, Sheppard M, Wright M, Bowker TJ, Davies MJ, McKenna WJ, Wood DA (2007) Sudden arrhythmic death syndrome: a national survey of sudden unexplained cardiac death. Heart 93:601–605. doi:10.1136/hrt.2006.099598
Bellocq C, Wilders R, Schott JJ, Louérat-Oriou B, Boisseau P, Le Marec H, Escande D, Baró I (2004) A common antitussive drug, clobutinol, precipitates the long QT syndrome 2. Mol Pharmacol 66:1093–1102. doi:10.1124/mol.104.001065
Benson DW, Wang DW, Dyment M, Knilans TK, Fish FA, Strieper MJ, Rhodes TH, George AL Jr (2003) Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest 112:1019–1028. doi:10.1172/JCI18062
Berne P, Brugada J (2012) Brugada syndrome 2012. Circ J 76:1563–1571. doi:10.1253/circj.CJ-12-0717
Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, Verkerk AO, Schwartz PJ, Crotti L, Dagradi F, Guicheney P, Fressart V, Leenhardt A, Antzelevitch C, Bartkowiak S, Borggrefe M, Schimpf R, Schulze-Bahr E, Zumhagen S, Behr ER, Bastiaenen R, Tfelt-Hansen J, Olesen MS, Kääb S, Beckmann BM, Weeke P, Watanabe H, Endo N, Minamino T, Horie M, Ohno S, Hasegawa K, Makita N, Nogami A, Shimizu W, Aiba T, Froguel P, Balkau B, Lantieri O, Torchio M, Wiese C, Weber D, Wolswinkel R, Coronel R, Boukens BJ, Bézieau S, Charpentier E, Chatel S, Despres A, Gros F, Kyndt F, Lecointe S, Lindenbaum P, Portero V, Violleau J, Gessler M, Tan HL, Roden DM, Christoffels VM, Le Marec H, Wilde AA, Probst V, Schott JJ, Dina C, Redon R (2013) Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet 45:1044–1049. doi:10.1038/ng.2712
Brugada P, Brugada J (1992) Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol 20:1391–1396. doi:10.1016/0735-1097(92)90253-J
Burashnikov E, Pfeiffer R, Barajas-Martinez H, Delpón E, Hu D, Desai M, Borggrefe M, Häissaguerre M, Kanter R, Pollevick GD, Guerchicoff A, Laiño R, Marieb M, Nademanee K, Nam GB, Robles R, Schimpf R, Stapleton DD, Viskin S, Winters S, Wolpert C, Zimmern S, Veltmann C, Antzelevitch C (2010) Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 7:1872–1882. doi:10.1016/j.hrthm.2010.08.026
Chugh SS, Kelly KL, Titus JL (2000) Sudden cardiac death with apparently normal heart. Circulation 102:649–654. doi:10.1161/01.CIR.102.6.649
Clatot J, Ziyadeh-Isleem A, Maugenre S, Denjoy I, Liu H, Dilanian G, Hatem SN, Deschênes I, Coulombe A, Guicheney P, Neyroud N (2012) Dominant-negative effect of SCN5A N-terminal mutations through the interaction of Nav1.5 alpha-subunits. Cardiovasc Res 96:53–63. doi:10.1093/cvr/cvs211
Dolmatova E, Mahida S, Ellinor PT, Lubitz SA (2013) Genetic etiology and evaluation of sudden cardiac death. Curr Cardiol Rep 15:389. doi:10.1007/s11886-013-0389-8
Dolz-Gaitón P, Núñez M, Núñez L, Barana A, Amorós I, Matamoros M, Pérez-Hernández M, González de la Fuente M, Alvarez-López M, Macías-Ruiz R, Tercedor-Sánchez L, Jiménez-Jáimez J, Delpón E, Caballero R, Tamargo J (2013) Functional characterization of a novel frameshift mutation in the C-terminus of the Nav1.5 channel underlying a Brugada syndrome with variable expression in a Spanish family. PLoS One 8:e81493. doi:10.1371/journal.pone.0081493
Gima K, Rudy Y (2002) Ionic current basis of electrocardiographic waveforms: a model study. Circ Res 90:889–896. doi:10.1161/01.RES.0000016960.61087.86
Herfst LJ, Potet F, Bezzina CR, Groenewegen WA, Le Marec H, Hoorntje TM, Demolombe S, Baró I, Escande D, Jongsma HJ, Wilde AA, Rook MB (2003) Na+ channel mutation leading to loss of function and non-progressive cardiac conduction defects. J Mol Cell Cardiol 35:549–557. doi:10.1016/S0022-2828(03)00078-6
Hoshi M, Du XX, Shinlapawittayatorn K, Liu H, Chai S, Wan X, Ficker E, Deschênes I (2014) Brugada syndrome disease phenotype explained in apparently benign sodium channel mutations. Circ Cardiovasc Genet 7:123–131. doi:10.1161/CIRCGENETICS.113.000292
Hu D, Barajas-Martínez H, Terzic A, Park S, Pfeiffer R, Burashnikov E, Wu Y, Borggrefe M, Veltmann C, Schimpf R, Cai JJ, Nam GB, Deshmukh P, Scheinman M, Preminger M, Steinberg J, López-Izquierdo A, Ponce-Balbuena D, Wolpert C, Haïssaguerre M, Sánchez-Chapula JA, Antzelevitch C (2014) ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int J Cardiol 171:431–442. doi:10.1016/j.ijcard.2013.12.084
Iyer V, Mazhari R, Winslow RL (2004) A computational model of the human left-ventricular epicardial myocyte. Biophys J 87:1507–1525. doi:10.1529/biophysj.104.043299
Kobrinsky E, Tiwari S, Maltsev VA, Harry JB, Lakatta E, Abernethy DR, Soldatov NM (2005) Differential role of the alpha1C subunit tails in regulation of the CaV1.2 channel by membrane potential, beta subunits, and Ca2+ ions. J Biol Chem 280:12474–12485. doi:10.1074/jbc.M412140200
Letsas KP, Gavrielatos G, Efremidis M, Kounas SP, Filippatos GS, Sideris A, Kardaras F (2007) Prevalence of Brugada sign in a Greek tertiary hospital population. Europace 9:1077–1080. doi:10.1093/europace/eum221
Mezghrani A, Monteil A, Watschinger K, Sinnegger-Brauns MJ, Barrère C, Bourinet E, Nargeot J, Striessnig J, Lory P (2008) A destructive interaction mechanism accounts for dominant-negative effects of misfolded mutants of voltage-gated calcium channels. J Neurosci 28:4501–4511. doi:10.1523/JNEUROSCI.2844-07.2008
Neyroud N, Ziyadeh-Isleem A, Clatot J, Deschenes I, Coulombe A, Guicheney P (2014) Role of the N- and distal C-terminal domains in Nav1.5 alpha-subunit interaction. Cardiovasc Res 103(Suppl 1):S68. doi:10.1093/cvr/cvu091.54
Ng PC, Henikoff S (2001) Predicting deleterious amino acid substitutions. Genome Res 11:863–874. doi:10.1101/gr.176601
Page KM, Heblich F, Margas W, Pratt WS, Nieto-Rostro M, Chaggar K, Sandhu K, Davies A, Dolphin AC (2010) N terminus is key to the dominant negative suppression of Cav2 calcium channels: implications for episodic ataxia type 2. J Biol Chem 285:835–844. doi:10.1074/jbc.M109.065045
Pambrun T, Mercier A, Chatelier A, Patri S, Schott JJ, Le Scouarnec S, Chahine M, Degand B, Bois P (2014) Myotonic dystrophy type 1 mimics and exacerbates Brugada phenotype induced by Nav1.5 sodium channel loss-of-function mutation. Heart Rhythm 11:1393–1400. doi:10.1016/j.hrthm.2014.04.026
Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G, Tracy C (2013) HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 10:1932–1963. doi:10.1016/j.hrthm.2013.05.014
Probst V, Allouis M, Sacher F, Pattier S, Babuty D, Mabo P, Mansourati J, Victor J, Nguyen JM, Schott JJ, Boisseau P, Escande D, Le Marec H (2006) Progressive cardiac conduction defect is the prevailing phenotype in carriers of a Brugada syndrome SCN5A mutation. J Cardiovasc Electrophysiol 17:270–275. doi:10.1111/j.1540-8167.2006.00349.x
Probst V, Wilde AA, Barc J, Sacher F, Babuty D, Mabo P, Mansourati J, Le Scouarnec S, Kyndt F, Le Caignec C, Guicheney P, Gouas L, Albuisson J, Meregalli PG, Le Marec H, Tan HL, Schott JJ (2009) SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ Cardiovasc Genet 2:552–557
Ramensky V, Bork P, Sunyaev S (2002) Human non-synonymous SNPs: server and survey. Nucleic Acids Res 30:3894–3900. doi:10.1093/nar/gkf493
Risgaard B, Jabbari R, Refsgaard L, Holst AG, Haunsø S, Sadjadieh A, Winkel BG, Olesen MS, Tfelt-Hansen J (2013) High prevalence of genetic variants previously associated with Brugada syndrome in new exome data. Clin Genet 84:489–495. doi:10.1111/cge.12126
Schott JJ, Alshinawi C, Kyndt F, Probst V, Hoorntje TM, Hulsbeek M, Wilde AA, Escande D, Mannens MM, Le Marec H (1999) Cardiac conduction defects associate with mutations in SCN5A. Nat Genet 23:20–21. doi:10.1161/CIRCGENETICS.109.853374
Schulze-Bahr E, Eckardt L, Breithardt G, Seidl K, Wichter T, Wolpert C, Borggrefe M, Haverkamp W (2003) Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Hum Mutat 21:651–652. doi:10.1002/humu.9144
Six I, Hermida JS, Huang H, Gouas L, Fressart V, Benammar N, Hainque B, Denjoy I, Chahine M, Guicheney P (2008) The occurrence of Brugada syndrome and isolated cardiac conductive disease in the same family could be due to a single SCN5A mutation or to the accidental association of both diseases. Europace 10:79–85. doi:10.1093/europace/eum271
Tang ZZ, Liao P, Li G, Jiang FL, Yu D, Hong X, Yong TF, Tan G, Lu S, Wang J, Soong TW (2008) Differential splicing patterns of L-type calcium channel Cav1.2 subunit in hearts of Spontaneously Hypertensive Rats and Wistar Kyoto Rats. Biochim Biophys Acta 1783:118–130. doi:10.1016/j.bbamcr.2007.11.003
Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT (1995) SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80:805–811. doi:10.1016/0092-8674(95)90359-3
Acknowledgments
We are greatly indebted to the patients. We thank Dr Alexandre Duparc (Pôle cardiovasculaire et Métabolique de Cardiologie B, Hôpital de Rangueil, Toulouse, France), Stéphanie Chatel and Christine Fruchet for assistance in the family recruitment; Estelle Baron, Stéphanie Bonnaud, Laëtitia Duboscq-Bidot, Béatrice Leray, and Aurore Girardeau for technical assistance; Jean-Baptiste Chéron for his help in action potential modelling; and Philippe Hulin at Imaging Core Facility (MicroPICell, SFR François Bonamy, University of Nantes) for expert technical assistance with FRAP. We are also grateful to the Genomic Platform of Nantes (Biogenouest Genomics) for technical support, to Raluca Teusan and Pierre Lindenbaum for the bioinformatics support and to Anne Ponchaux, Jerome Buscail, Thierry Marsaud, Prof Stéphane Bézieau (Service de génétique médicale, Nantes) for their assistance in SCN5A sequencing. We are also grateful to the referral centre against inherited cardiac arrhythmias of Nantes and Rennes. This work was supported by the Leducq Foundation [CVD-05; Alliance Against Sudden Cardiac Death]; and by the French Ministry of Health (P.H.R.C.I. DGS2001/0248).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
D. M. Béziau and J. Barc are contributed equally to this work.
J.-J. Schott, V. Probst and I. Baró authors jointly directed this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Béziau, D.M., Barc, J., O’Hara, T. et al. Complex Brugada syndrome inheritance in a family harbouring compound SCN5A and CACNA1C mutations. Basic Res Cardiol 109, 446 (2014). https://doi.org/10.1007/s00395-014-0446-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-014-0446-5