
Abstract
Cardiac disease is associated with deleterious emission of mitochondrial reactive oxygen species (mito-ROS), as well as enhanced oxidation and activity of the sarcoplasmic reticulum (SR) Ca2+ release channel, the ryanodine receptor (RyR2). The transfer of Ca2+ from the SR via RyR2 to mitochondria is thought to play a key role in matching increased metabolic demand during stress. In this study, we investigated whether augmented RyR2 activity results in self-imposed exacerbation of SR Ca2+ leak, via altered SR-mitochondrial Ca2+ transfer and elevated mito-ROS emission. Fluorescent indicators and spatially restricted genetic ROS probes revealed that both pharmacologically and genetically enhanced RyR2 activity, in ventricular myocytes from rats and catecholaminergic polymorphic ventricular tachycardia (CPVT) mice, respectively, resulted in increased ROS emission under β-adrenergic stimulation. Expression of mitochondrial Ca2+ probe mtRCamp1h revealed diminished net mitochondrial [Ca2+] with enhanced SR Ca2+ leak, accompanied by depolarization of the mitochondrial matrix. While this may serve as a protective mechanism to prevent mitochondrial Ca2+ overload, protection is not complete and enhanced mito-ROS emission resulted in oxidation of RyR2, further amplifying proarrhythmic SR Ca2+ release. Importantly, the effects of augmented RyR2 activity could be attenuated by mitochondrial ROS scavenging, and experiments with dominant-negative paralogs of the mitochondrial Ca2+ uniporter (MCU) supported the hypothesis that SR-mitochondria Ca2+ transfer is essential for the increase in mito-ROS. We conclude that in a process whereby leak begets leak, augmented RyR2 activity modulates mitochondrial Ca2+ handling, promoting mito-ROS emission and driving further channel activity in a proarrhythmic feedback cycle in the diseased heart.
Similar content being viewed by others
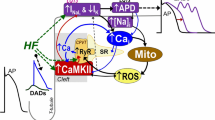


Introduction
Contraction of the heart is a high-energy demanding process. Most of the energy required for this process in the form of ATP is generated by mitochondria, which occupy ~ 35% of myocyte volume [56]. In ventricular myocytes (VMs), release of Ca2+ from the sarcoplasmic reticulum (SR) by ryanodine receptors (RyR2s) is critical in initiating muscle contraction and a major determinant of its strength [24]. During SR Ca2+ release, local transfer of Ca2+ to the mitochondrial matrix activates enzymes in the tricarboxylic acid cycle and drives the electron transport chain (ETC) to accelerate ATP production [52], thereby providing a link between Ca2+-dependent contraction and mitochondria metabolic output. It is well established that in cardiac disease such as heart failure (HF), abnormal mitochondrial function is often accompanied by increased emission of reactive oxygen species (ROS) [9]. Excessive mitochondria-derived ROS (mito-ROS) evokes profound changes in intracellular Ca2+ homeostasis [47]. Importantly, a mito-ROS-mediated increase in activity of RyR2 has been linked to the increased propensity for aberrant Ca2+ leak from the sarcoplasmic reticulum (SR), leading to diminished systolic Ca2+ transients and increased incidence of pro-arrhythmic diastolic Ca2+ waves [18]. However, the role of increased RyR2-mediated Ca2+ release on SR-mitochondria Ca2+ transfer and mito-ROS emission remains incompletely understood.
Mitochondrial ATP production and SR Ca2+ release are closely coupled processes during the ‘fight-or-flight’ response, whereby β-adrenergic receptors are stimulated due to an increased requirement of the heart to meet metabolic demand. A positive inotropic effect of the pathway includes augmentation of RyR2 activity, increasing the availability of cytosolic Ca2+ to activate the contractile machinery [7]. However, as β-adrenergic drive can exacerbate Ca2+ leak from RyR2 channels that are already hyperactive in cardiac disease, the detrimental effects of excessive diastolic Ca2+ leak are most obvious during this response. Leak of Ca2+ from the SR can precipitate to cause triggered activity and many life-threatening cardiac arrhythmias [8, 42, 68]. This is particularly evident in catecholaminergic polymorphic ventricular tachycardia (CPVT), a genetic disorder characterized by mutations in the RyR2 macromolecular complex, whereby ventricular tachycardia and fibrillation (VT/VF) often only present in patients after exercise or stress [30, 57].
It has also been suggested that during β-adrenergic stimulation, increased energy demand directly drives mito-ROS emission, thus enhancing aberrant SR Ca2+ leak via oxidized RyR2 [12,13,14]. When oxidative phosphorylation and electron flux via the ETC are accelerated to produce more ATP, a higher rate of electron slippage can occur, with the formation of mito-ROS as a consequence [9]. Enhancement of mitochondrial Ca2+ (mito-Ca2+) levels has previously been associated with increased mito-ROS production [27, 51, 59, 60], while in failing guinea pig hearts, others have observed decreased mito-Ca2+ levels in parallel with elevated mito-ROS emission [38, 46, 48]. We recently demonstrated that pharmacological enhancement of mito-Ca2+ accumulation leads to increased oxidation of RyR2 [32]. In the present study, we test the hypothesis that augmented RyR2-mediated SR Ca2+ leak feeds back on the oxidation status of RyR2 via mito-ROS, thus further exacerbating leak.
Using fluorescent indicators and genetically encoded spatially restricted ROS biosensors, we demonstrate that under conditions of both pharmacologically and genetically increased RyR2 activity, β-adrenergic stimulation by isoproterenol (ISO) increases ROS emission in VMs, both at the outer mitochondrial membrane and in close vicinity to RyR2 within the SR. Live cell imaging of mito-Ca2+ dynamics showed that acute enhanced RyR2-mediated Ca2+ leak resulted in diminished ability of mitochondria to accumulate and retain Ca2+. This led to oxidation of RyR2, further exacerbating SR Ca2+ leak. Importantly, augmented leak and the resulting decrease in Ca2+ transient amplitude, as well as the increase in incidence of proarrhythmic spontaneous Ca2+ waves (SCWs), were attenuated by scavenging of mito-ROS. Transfer of Ca2+ from the SR to mitochondria is essential for the enhancement of mito-ROS emission, as demonstrated by experiments with dominant-negative paralogs of the mitochondrial Ca2+ uniporter (MCU). Our results suggest that increased RyR2 activity disturbs mito-Ca2+ homeostasis and elevates mito-ROS emission, thus in a vicious feedback cycle exacerbates diastolic SR Ca2+ leak by increasing RyR2 oxidation, driving spontaneous Ca2+ release that is detrimental in cardiac disease.
Methods
Production of adenoviral constructs
The intra-mitochondrial Ca2+ biosensor mtRCamp1h was constructed by fusing cytochrome C oxidase subunit IV at the N-terminus of RCamp1h coding region [1], as described previously [32]. The outer-mitochondrial membrane H202 biosensor OMM-HyPer was constructed by fusing mAKAP1 followed by a linker to the N-terminus of the coding region of pC1-HyPer-3 [10], as described previously [32]. Oxidation in the SR was measured using the ERroGFP_iE biosensor [2], as described previously [32]. The pCMV G-CEPIA1er biosensor was a gift from Masamitsu Iino (Addgene plasmid # 58,215) [63].
Adenoviruses carrying biosensor constructs were generated utilizing the ViraPower Gateway expression system (Thermo Fisher Scientific, Waltham, MA, USA). Coding regions of described plasmids were cloned into the pENTR™ 1A entry vector and recombined into the pAd/CMV/V5-DEST™ destination vector by LR recombinase reaction. After sequence verification, destination vectors were digested with restriction endonuclease PacI, before transfection into HEK293A cells using Lipofectamine™ 2000 (Thermo Fisher Scientific) for viral production.
The MCU-DN adenovirus was kindly gifted by Dr. Jin O-Uchi, University of Minnesota Twin Cities.
Study animals
Control rats
Male Sprague–Dawley rats (controls) were obtained from Charles River Laboratories. Rats were aged 8–12 weeks. A total of 48 rats were used for the study.
Wild-type and calsequestrin-null mice
Male and female wild-type (WT) and calsequestrin2-null mutant mice in the C57BL/6 genetic background [37] were utilized in this study, and are referred to as CPVT mice. Mice were aged 8–14 months. A total of 16 mice were used for the study.
Myocyte isolation
Rat VM isolation
Ventricular myocytes were obtained by enzymatic digestion as previously described [32]. Rats were anesthetized with a lethal dose of sodium pentobarbital solution (120 mg/kg) before the heart was rapidly removed. Hearts were immersed in cold Ca2+-free Tyrode’s solution (in mmol/L: 140 NaCl, 5.4 KCl, 1.0 CaCl2, 1 MgCl2, 10 HEPES, 5.6 glucose, pH 7.3). Hearts were then mounted on a Langendorff apparatus and retrogradely perfused through the aorta with Tyrode’s solution containing collagenase II (Worthington Biochemical Corp.) at 37 °C for 16 min. Ventricles were minced and placed in a 37 °C water bath shaker in collagenase solution. Isolated VMs were then prepared for primary culture.
Mouse VM isolation
As previously described [31, 44], VMs from mice were also obtained by enzymatic digestion. Mice were anesthetized with isoflurane, hearts were rapidly excised, immersed in cold Ca2+-free Tyrode’s solution, and mounted on a Langendorff apparatus. Hearts were perfused with Tyrode’s solution containing liberase TH Research Grade enzyme (Roche) for 7 min. Ventricles were minced and single VMs were stabilized in perfusion solution containing BSA (20 mg/mL). Isolated VMs were plated onto laminin-coated glass coverslips in 24-well dishes in Tyrode’s solution containing 0.5 mmol/L Ca2+ and used immediately for imaging experiments.
Primary culture of rat VMs
For experiments with cultured control rat VMs, myocytes were plated onto laminin-coated glass coverslips in 24-well dishes in serum-free medium 199 (Thermo Fisher Scientific), supplemented with 25 mmol/L NaHCO3, 10 mmol/L HEPES, 5 mmol/L creatine, 5 mmol/L taurine, 10 μ/mL penicillin, 10 μg/mL streptomycin and 10 μg/mL gentamycin (pH 7.3). Any unattached cells were removed by replacing the medium after 1 h. Myocytes were infected with adenoviruses and were cultured at 37 °C in 95% air and 5% CO2 for 36–48 h before analysis. Rat VMs maintain structural integrity including T-tubule organization and electrical properties for at least the first 48 h of culture [3].
Confocal imaging
Laser scanning confocal imaging was performed using Leica SP8 dmi8 and Olympus Fluoview 1000 microscopes in x–y and linescan modes. Myocytes were paced via field stimulation using extracellular platinum electrodes.
Measurement of [Ca2+]SR using G-CEPIA1er
To directly assess SR Ca2+ concentration ([Ca2+]SR) and RyR2-mediated SR Ca2+ leak, intact VMs were infected and cultured with G-CEPIA1er virus on glass coverslips. After 36–48 h, VMs were perfused with Tyrode’s solution containing 1 mmol/L Ca2+. Isoproterenol (ISO, 50 nmol/L) was added to the solution to stimulate β-adrenergic receptors. Low-dose caffeine (200 µmol/L) was added to induce RyR2 leak. Myocytes were preincubated with specific mitochondrial superoxide scavenger mitoTEMPO (20 μmol/L; Millipore Sigma) for 10 min prior to experimentation, and was included in the perfusion solution. Biosensor G-CEPIA1er was excited using 488 nm line of argon laser and fluorescence emission was collected at 500–550 nm, measured in x–y mode at 400 Hz sampling rate. Resting VMs were exposed to sarco/endoplasmic reticulum Ca2+-ATPase (SERCa2a) inhibitor thapsigargin (10 μmol/L) after 5 min in ISO or ISO and caffeine, and fluorescence signal from G-CEPIA1er was monitored using confocal microscopy. The time constant of decay of G-CEPIA1er was used as a measure of the leak by fitting fluorescence data to a monoexponential function [4]. The SR Ca2+ store was depleted by application of high-dose caffeine (10 mmol/L) in Ca2+-free Tyrode’s solution.
Standard pacing protocol for measurements using biosensors and indicators in rat VMs
This standard pacing protocol was followed during assays using OMM-HyPer, ERroGFP_iE, mtRCamp1h, Fluo-3 and TMRM. Baseline myocyte fluorescence was recorded for 5 min (0–5 min of recording) under continuous perfusion with Tyrode’s solution containing 1 mmol/L Ca2+. Myocytes were field stimulated for 5 min at 2 Hz (5–10 min of recording). At 12 min, ISO (50 nmol/L) or ISO plus low-dose caffeine (200 µmol/L) was added and continuously perfused (12–17 min of recording). Next, VMs were paced for 5 min during drug perfusion (17–22 min of recording). Following cessation of pacing, fluorescence was recorded for an additional 5 min before any further treatment, as described in each assay below.
Measurement of oxidative stress using ERroGFP_iE and OMM-HyPer
Oxidative stress in intact VMs within the SR and at the OMM was assessed using ERroGFP_iE and OMM-HyPer biosensors, respectively. Myocytes were infected with viruses on glass coverslips and cultured for 36–48 h, before perfusion with Tyrode’s solution (1 mmol/L Ca2+). Biosensors were excited using 488 nm line of argon laser and fluorescence emission was collected at 500–550 nm wavelengths, measured in the x–y mode at 400 Hz sampling rate. The pacing protocol was followed as described above. Minimum fluorescence was obtained by application of ROS scavenger dithiothreitol (DTT, 5 mmol/L), and maximum fluorescence (Fmax) was obtained by application of deoxythymidine diphosphate (DTDP, 200 µmol/L). Data are presented as a percentage of ΔF/ΔFmax. where ΔF = F– Fmin and ΔFmax = Fmax–Fmin.
Measurement of mito-[Ca2+] using mtRCamp1h
To assess mito-Ca2+ handling in intact VMs, cells were infected with mtRCamp1h virus on glass coverslips and cultured for 36–48 h. Myocytes were perfused with Tyrode’s solution (1 mmol/L Ca2+). Biosensor mtRCamp1h was excited using 543 nm line of HeNe laser and fluorescence emission was collected at 560–660 nm wavelengths, measured in the x–y mode and 400 Hz sampling rate. The pacing protocol was followed as described above. After this protocol, VMs were washed in Ca2+-free Tyrode’s solution, before permeabilization with saponin (0.001%). The solution was replaced with an internal recording solution containing cytochalasin D (10 μmol/L) and Ca2+ buffer EGTA (2 mmol/L) to obtain minimum mtRCamp1h fluorescence. Maximum fluorescence was achieved by application of Ca2+ (20 μmol/L). Using the equation [Ca2+]m = Kd × (F – Fmin)/(Fmax – F), where Kd of mtRCamp1h = 1.3 µmol/L, fluorescence was converted to [Ca2+]m for each myocyte. Analysis parameters included peak mtRCamp1h [Ca2+]m (μmol/L), the time to peak amplitude (s), the first derivative of [Ca2+]m [62, 65] (nmol/L Ca2+ s−1) and the rate of decay (s−1).
Cytosolic Ca2+ imaging of rat VMs using Fluo-3 AM
To measure cytosolic Ca2+, cultured rat VMs were loaded with Fluo-3 AM (Invitrogen) at room temperature for 10 min in Ca2+-free Tyrode’s solution, followed by a 8 min wash in Tyrode’s solution containing 1 mmol/L Ca2+. Myocytes were perfused with Tyrode’s solution containing 1 mmol/L Ca2+ at room temperature during recordings. Fluo-3 AM was excited at 488 nm and fluorescence emission was collected at 500–550 nm wavelengths in line scan mode at 200 Hz sampling rate. The pacing protocol was followed as described above, with pacing for 5 min at 2 Hz, followed by the addition of either ISO (50 nmol/L) or ISO and low-dose caffeine (200 µmol/L), then further pacing for 5 min. For experiments with mitoTEMPO, VMs were preincubated with mitoTEMPO (20 μmol/L, Millipore Sigma) for 10 min prior to experimentation, and mitoTEMPO was included in the perfusion solution. Line scans were recorded during the last minute of pacing. Cytosolic Ca2+ transient amplitude is presented as ΔF/ΔF0, where F0 is basal fluorescence and ΔF = F–F0.
Cytosolic Ca2+ imaging of mouse VMs using Fluo-3 AM
To measure intracellular Ca2+ transient, intact VMs were loaded with Fluo-3 AM (Invitrogen) at room temperature for 20 min in Tyrode’s solution containing 0.5 mmol/L Ca2+, followed by a 20 min wash in Tyrode’s solution containing 1 mmol/L Ca2+. Myocytes were then perfused with Tyrode’s solution containing 2 mmol/L Ca2+ and ISO (100 nmol/L) at room temperature during Ca2+ transient recordings. For experiments with mitoTEMPO, VMs were preincubated with mitoTEMPO (20 μmol/L, Millipore Sigma) for 20 min prior to experimentation, and mitoTEMPO was included in the perfusion solution. Fluo-3 AM was excited at 488 nm and fluorescence emission was collected at 500–550 nm wavelengths in line scan mode at 200 Hz sampling rate. To test for the propensity of triggered activity, VMs were stimulated for 10 s at 0.5 Hz and the latency between the last pacing stimulus and the subsequent SCW was calculated. To assess SR Ca2+ load, high-dose caffeine (10 mmol/L) was applied at the end of the experiments. The data are presented as ΔF/F0, where F0 is the basal fluorescence and ΔF = F–F0.
Measurement of mitochondrial superoxide in mouse VMs using MitoSOX
The emission of mitochondrial superoxide was measured in isolated VMs in Tyrode’s solution containing 2 mmol/L Ca2+ using MitoSOX Red mitochondrial superoxide indicator (Thermo Fisher Scientific; 20 μmol/L, 20 min loading). The indicator was excited with 514 nm line of an argon laser and emission was collected at 560–660 nm, measured in the x–y mode. Fluorescence of MitoSOX was normalized to the maximum fluorescence signal obtained by application of DTDP (200 µmol/L).
Western blotting and assessment of RyR2 oxidation
List of antibodies used is present in Table 1
Immunofluorescence for biosensor colocalization in rat VMs
Cultured control rat VMs plated on laminin-coated glass coverslips were infected with G-CEPIAer or ERroGFP_iE adenoviruses and were cultured at 37 °C in 95% air and 5% CO2 for 36–48 h. Myocytes were then prepared for immunofluorescence by fixing with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100/PBS (pH 7.2) containing 1% BSA. Samples were then probed using RyR2 primary antibody. Secondary antibody used was goat anti‐mouse IgG (H + L) cross‐adsorbed secondary antibody Alexa Fluor 633.
Staining with MitoTracker Red for biosensor colocalization in rat VMs
Cultured control rat VMs plated on laminin-coated glass coverslips were infected with OMM-HyPer adenovirus and were cultured at 37 °C in 95% air and 5% CO2 for 36–48 h. Live VMs were then stained with MitoTracker Deep Red FM (Invitrogen, 500 nM) for 15 min, then washed in Tyrode’s solution prior to imaging. The indicator was excited with 546 nm line of an argon laser and emission was collected at 550–660 nm, measured in the x–y mode.
RyR2 immunoprecipitation and immunoblotting from rat VMs
Freshly isolated rat VMs were treated with isoproterenol (50 nmol/L),and caffeine (200 μmol/L) for 4 min prior to 2 Hz pacing for 1 min at room temperature. Cells were then immediately lysed in lysis buffer from Cell Signaling (Cat#9803S), supplemented with phosphatase (Calbiochem, Cat#524,625) and protease inhibitor cocktails (Sigma, Cat#P8340) as described previously (Terentyev et al. 2014).
RyR2 was immunoprecipitated from cell lysate using anti-RyR2 antibody (5 μL) in 0.5 mL RIPA buffer overnight at 4 °C. Samples were incubated with Protein A/G Plus-agarose beads (Santa Cruz cat # sc-2003) for 1 h at 4 °C and washed three times with RIPA buffer.
To determine the oxidation status of RyR2, the Oxidized Protein Western Blot Kit was used, whereby carbonyl groups of immunoprecipitated RyR2 were derivatized to 2,4 dinitrophenylhydrazone (DNP) by reaction with 2,4 dinitrophenylhydrazine. For control, we used the kit-provided Derivatization Control Solution. The DNP-RyR2 protein samples were separated on 4–20% Mini-PROTEAN TGX gels (Bio-Rad Laboratories, Cat#456–1094) and DNP-associated signal was assessed by the kit-provided anti-DNP rabbit primary antibody and anti-RyR2, followed by HRP-conjugated anti-rabbit goat secondary antibody and anti-mouse lgG(H + L),HRP secondary antibody.
Blots were developed with ECL (Bio-Rad Laboratories) and quantified using Image J (US National Institutes of Health) and Origin 8 software.
RyR2 immunoprecipitation and immunoblotting from mouse VMs
Hearts were rapidly excised from isoflurane-anesthetized mice and immersed in cold Ca2+-free Tyrode’s solution before mounting on a Langendorff apparatus. Hearts were then perfused with Tyrode’s solution containing 2 mmol/L Ca2+ for 15 min. The atria were removed and ventricles were homogenized using Tissue Tearer Model 985,370 in 1 mL of buffer containing: Tris–HCL (50 mmol/L, pH 7.4), NaCl (150 mmol/L) NaF (5 mmol/L), Na3VO4 (1 mmol/L), 0.5% Triton-X 100, protease inhibitor and phosphatase inhibitor. The homogenate was centrifuged at 12000xg for 15 min at 4 °C. RyR2 was immunoprecipitated from the supernatant (500 mL) using anti-RyR2 antibody (3 µL) for 2 h at 4 °C. The samples were then incubated with Protein A/G Plus-agarose beads (Santa Cruz, Cat#sc-2003) for 1 h at 4 °C and washed three times with RIPA buffer.
To determine the oxidation status of RyR2, the Oxidized Protein Western Blot Kit was used, whereby the carbonyl groups of immunoprecipitated RyR2 were derivatized to DNP by reaction with 2,4dinitrophenylhydrazine. Kit-provided Derivatization Control Solution was used as control. The DNP-RyR2 protein samples were separated on 4–20% Mini-PROTEAN TGX gels and DNP-associated signal was assessed by the kit-provided anti-DNP rabbit primary antibody and anti-RyR2, followed by HRP-conjugated anti-rabbit goat secondary antibody and anti-mouse lgG (H + L), HRP secondary antibody.
Blots were developed with ECL (Bio-Rad Laboratories) and quantified using Image J (US National Institutes of Healthand Origin 8 software.
Expression of MCU complex proteins
To determine MCU-DN expression, intact rat VMs were infected and cultured with MCU-DN virus on glass coverslips for 36–48 h. For the assessment of MCU/MCUb expression, freshly isolated mouse VMs from WT and CPVT hearts were obtained as described above.
Myocytes were lysed in lysis buffer from Cell Signaling (Cat#9803S), supplemented with phosphatase (Calbiochem, Cat#524,625) and protease inhibitor cocktails (Sigma, Cat#P8340) as described previously [36]. Samples (20–30 μg of proteins) were resolved on a 4–20% gel via SDS‐PAGE, transferred onto nitrocellulose membranes, and probed with antibodies specific for MCU complex proteins and subsequently probed with secondary antibody. Blots were developed with ECL (Bio‐Rad Laboratories) and quantified using ImageJ and Origin 8 software.
Statistics
All statistical analysis was performed using Origin 8 (OriginLab). Data are presented as mean ± standard error (SEM). Uppercase n(N) = number of animals, and lowercase n = number of VMs. Statistical significance between groups was calculated using Student’s t test (paired and unpaired), Fisher’s exact test and one-way ANOVA with Bonferroni post hoc test, where appropriate. A p value of less than 0.05 was considered significant.
Results
Caffeine-mediated enhancement of SR Ca2+ leak is reduced by mito-ROS scavenging
When used at low concentrations, caffeine acts as an agonist of RyR2. We first sought to demonstrate that low-dose caffeine (200 µmol/L) could reproduce the leaky SR phenotype observed in cardiac disease, using cultured VMs isolated from healthy rat hearts.
To assess the effects of caffeine on [Ca2+]SR, we utilized genetic probe G-CEPIAer, a GFP-based biosensor spatially restricted to the SR, with a Kd ~ 672 µmol/L for Ca2+ [63]. After generating adenovirus encoding the sensor, isolated VMs were infected and cultured 36–48 h prior to experimentation. When expressed in VMs, G-CEPIAer has a striated SR patterning in the VM, and overlaps with the signal produced by RyR2 antibody, demonstrating correct cellular localization (Fig. 1a). To demonstrate probe sensitivity, VMs were treated with thapsigargin and high-dose caffeine to deplete the SR Ca2+ store and obtain minimum fluorescence (Fig. 1b).

Mitochondrial ROS scavenging reduces caffeine-mediated SR Ca2+ leak in rat VMs. a Representative images of a cultured rat VM 48 h after adenoviral infection with G-CEPIAer intra-SR Ca2+ biosensor (left image, green). Using anti-RyR antibody, VMs were probed for RyR2 expression (center image, red). A merged image is shown on the right, indicating correct probe localization. b Representative images of infected VM treated with thapsigargin (20 µmol/L) and high-dose caffeine (20 mmol/L) to achieve minimal fluorescence, demonstrating G-CEPIAer sensitivity. c Representative time-dependent profiles (F/F0) of G-CEPIAer fluorescence from infected VMs. Myocytes were either treated with ISO (50 nmol/L), ISO and mitoTEMPO (MT, 20 μmol/L, 10 min pretreatment), ISO and caffeine (CAFF, 200 μmol/L), or ISO, CAFF and mitoTEMPO. Myocytes were exposed to SERCA inhibitor thapsigargin (TG, 10 μmol/L) after 1 min of recording, and the time constant of decay (τ, s) was calculated as a measure of SR Ca2+ leak. Signal was normalized to minimum fluorescence obtained by application of high-dose caffeine (10 mmol/L). The graph in d depicts mean data ± SEM for rate of decay (min−1). N = 5–9 animals, n = 14–26 VMs. *p < 0.05 vs. ISO group, **p < 0.05 vs. ISO + CAFF group, one-way ANOVA with Bonferroni post hoc test
To directly measure SR Ca2+ leak in VMs under β-adrenergic stimulation (ISO, 50 nmol/L), SR Ca2+ uptake via SERCa2a was inhibited by application of thapsigargin (20 µmol/L). G-CEPIAer fluorescence was normalized to minimal fluorescence signal obtained by application of high-dose caffeine (10 mmol/L) to empty the SR at the end of the experiment, as shown in the representative recordings (Fig. 1c). Application of low-dose caffeine to VMs induced a dramatic increase in SR leak rate (Fig. 1d).
To test whether the caffeine-induced acceleration of SR Ca2+ leak has a mito-ROS-dependent component, we tested whether the caffeine-mediated increase in RyR2 activity could be attenuated by scavenging of ROS with mitoTEMPO (20 µmol/L). Indeed, the increased rate of SR Ca2+ leak induced by caffeine was significantly attenuated by pretreatment of VMs with mitoTEMPO (Fig. 1c, d), indicative that increased mito-ROS emission may play a role in increased RyR2 activity. Pretreatment of VMs with mitoTEMPO did not have a significant effect on SR Ca2+ leak during stimulation with ISO alone.
Increased SR Ca2+ leak is associated with augmented ROS emission and RyR2 oxidation
Having demonstrated the protective effects of mito-ROS scavenging in VMs under conditions of enhanced SR Ca2+ leak, we next sought to visualize whether a caffeine-mediated increase in RyR2 activity leads to increased ROS emission. Using H202-sensitive fluorescent probe targeted to the outer mitochondrial membrane, OMM-HyPer [10, 32], ROS emission was assessed at the OMM of infected VMs (Fig. 2). Correct cellular localization of OMM-HyPer was confirmed by staining of mitochondria with far-red fluorescent indicator MitoTracker Red, as shown in Fig. 2a. Probe sensitivity is demonstrated in Fig. 2b, whereby VMs were treated with DTDP (200 µmol/L) to obtain maximum fluorescence.

Increased SR Ca2+ leak increases mitochondrial ROS emission in rat VMs. a Representative image of a cultured rat VM 48 h after adenoviral infection with OMM-HyPer mito-ROS biosensor (left image, green). Myocytes were stained with mitochondrial dye MitoTracker Red (center image, red). A merged image is shown on the right, indicating correct probe localization. b Representative images of infected VM treated with DTDP (200 µmol/L) to achieve maximal fluorescence, demonstrating OMM-HyPer sensitivity. c Representative traces of OMM-HyPer fluorescence from infected VMs. Myocytes were paced at 2 Hz for 5 min (black bars), and treated with ISO (50 nmol/L, left trace) or ISO and caffeine (CAFF, 200 µmol/L, right trace) before further pacing. The signal was normalized to minimum fluorescence obtained by application of DTT (5 mmol/L) and maximum fluorescence by application of DTDP (200 µmol/L). The graph in d depicts mean data ± SEM for maximum normalized fluorescence (%) after pacing and application of ISO, or application of ISO and CAFF. N = 3–4 animals, n = 7–16 VMs. *p < 0.05 vs. ISO group, two-sample Student’s t test
As described in the methods, a standard pacing protocol was followed for organelle-targeted biosensors. After 5 min of recording, VMs were paced at 2 Hz for 5 min to apply the workload. After 2 min of pacing cessation, ISO (50 nmol/L) or ISO and low-dose caffeine (200 µmol/L) was added, and perfused for 5 min, followed by an additional pacing train for 5 min. Representative fluorescence recordings are shown in Fig. 2c. Signal was normalized to minimal fluorescence obtained by application of DTT (5 mmol/L) and maximal fluorescence by application of DTDP (200 µmol/L). The application of ISO and caffeine significantly increased OMM-HyPer signal vs. ISO alone (Fig. 2d), indicative that increased RyR2 activity can lead to increased ROS emission from the mitochondria.
Mitochondria and the SR are closely situated in VMs, with bidirectional communication occurring between the two organelles [20, 25]. We therefore measured oxidative stress within the SR and in the vicinity of RyR2, using SR-targeted redox sensitive biosensor ERroGFP_iE [2, 32] (Fig. 3). The probe showed SR-like pattern of expression as evidenced by colocalization with RyR2 protein (Fig. 3a). Sensitivity of ERroGFP_iE is demonstrated in Fig. 3b, with application of DTDP to obtain maximal fluorescence. Similar results were obtained for those when using OMM-HyPer probe, whereby ISO plus caffeine treatment significantly increased ERroGFP_iE fluorescence vs. ISO alone (Fig. 3c, d), demonstrating that there is increased oxidative stress within the SR when RyR2 activity is enhanced.

Enhanced RyR2 activity in rat VMs leads to SR oxidative stress and RyR2 oxidation. a Representative images of a cultured rat VM 48 h after adenoviral infection with ERroGFP_iE oxidative stress biosensor (left image, green). Using anti-RyR antibody, VMs were probed for expression of RyR2 (center image, red). A merged image is shown on the right, indicating correct probe localization. b Representative images of infected VM treated with DTDP (200 µmol/L) to achieve maximal fluorescence, demonstrating ERroGFP_iE sensitivity. c Representative traces of ERroGFP_iE fluorescence from infected VMs. Myocytes were paced at 2 Hz for 5 min (black bars) and treated with ISO (50 nmol/L, left trace) or ISO and caffeine (CAFF, 200 µmol/L, right trace). Signal was normalized to minimum fluorescence obtained by application of DTT (5 mmol/L) and maximum fluorescence by application of DTDP (200 µmol/L). The graph in d depicts mean data ± SEM for maximum normalized fluorescence (%) after pacing and application of ISO, or application of ISO and CAFF. N = 3–4 animals, n = 15–16 VMs. *p < 0.05 vs. ISO group, two-sample Student’s t test. e Immunoprecipitated RyR2 from freshly isolated rat VMs was immunoblotted for oxidation using DNP antibody. Representative images of DNP and RyR2 immunoprecipitation signal from VMs without treatment (NO ISO), treated with ISO, or ISO and CAFF. The graph in f depicts quantification of normalized DNP signal (%). N = 5 animals. *p < 0.05 vs. NO ISO group, **p < 0.05 vs. ISO group, one-way ANOVA with Bonferroni post hoc test
To directly assess the oxidation status of RyR2 in VMs, the free thiol content of immunoprecipitated RyR2 was measured using the DNP antibody. Figure 3e, f demonstrates that treatment of VMs with ISO led to an increase in signal, but this was not significant. However, low-dose caffeine dramatically increased oxidation of RyR2 by sevenfold. This provides evidence that enhancement of RyR2 activity and SR Ca2+ leak can increase mito-ROS emission, increase deleterious oxidative stress within the SR and thus feedback on the redox status of the channel.
Mito-Ca2+ and mito-ROS emission is modulated by RyR2 activity
We next investigated whether Ca2+ transfer from the SR to the mitochondria is involved in mito-ROS production. Using mitochondrial-matrix targeted biosensor mtRCamp1h, with a Kd ~ 1.3 µmol/L for Ca2+ [1, 32], we examined whether caffeine-mediated SR Ca2+ leak modulated global free mito-Ca2+ concentration ([Ca2+]m) in rat VMs (Fig. 4). After infection and culture of isolated VMs with mtRCamp1h adenovirus, expression of the probe within the mitochondrial matrix is observed. Correct intracellular targeting of mtRCamp1h was confirmed by co-infection with mitochondrial-targeted GFP adenovirus (Fig. 4a). Probe sensitivity is demonstrated in Fig. 4b. Application of calcium buffer EGTA was used to obtain minimal fluorescence, while Ca2+ (20 µmol/L) was used to obtain maximal fluorescence of saponin-permeablized VMs.

Enhancement of RyR2 activity in rat VMs diminishes the ability of mitochondria to accumulate and retain Ca2+. a Representative image of a cultured rat VM 48 h after adenoviral infection with mtRCamp1h mitochondrial Ca2+ biosensor (left image, red), and mitochondrial-targeted GFP (mtGFP, center image, green). A merged image is shown on the right, indicating correct probe localization. b Representative images of infected VM treated with EGTA (2 mmol/L) to achieve minimal fluorescence, and Ca2+ (20 µmol/L) to achieve maximal fluorescence, demonstrating mtRCamp1h sensitivity. c Representative trace of mtRCamp1h fluorescence from infected VM. Myocytes were paced at 2 Hz for 5 min (black bars), before treatment with ISO (50 nmol/L), or ISO and caffeine (CAFF, 200 µmol/L) and further pacing. Myocytes were saponin permeabilized (SAP) before treatment with EGTA (2 mmol/L) to obtain minimum fluorescence, followed by Ca2+ (20 µmol/L) to obtain maximal fluorescence. d Representative traces of [Ca2+]m (µmol/L) during pacing in VMs treated with ISO (black line) or ISO and CAFF (red line). Graphs depict mean data ± SEM of ISO and ISO + CAFF groups for: e baseline [Ca2+]m (μmol/L); f peak [Ca2+]m (µmol/L); g time to peak [Ca2+]m (s); h the first derivative of [Ca2+]m, indicative of maximal Ca2+ influx rate into the mitochondria during pacing (nM Ca2+ s−1); and i the rate of decay (s−1). N = 4 animals per group, n = 9 VMs per group. *p < 0.05 vs. ISO group, two-sample Student’s t test
A representative recording is presented in Fig. 4c. A measurable increase in mtRCamp1h fluorescence occurs under conditions of enhanced workload, with field stimulation (pacing at 2 Hz, black bar). Fluorescence returns to baseline when field stimulation stops. Myocytes were paced using the standard 5 min, 2 Hz protocol before and after the application either ISO alone, or ISO and low-dose caffeine to induce SR Ca2+ leak. After this protocol, VMs were saponin permeabilized. Minimal (EGTA, 2 mmol/L) and maximal (Ca2+, 20 µmol/L) fluorescence signal was obtained, allowing for recalculation of signal to [Ca2+]m. As shown in Fig. 4d, [Ca2+]m increases during pacing, with significantly reduced peak [Ca2+]m when treated with ISO and low-dose caffeine, compared to ISO alone (Fig. 4f). The baseline [Ca2+]m of VMs in the experimental groups is unchanged (Fig. 4e). This is suggestive that SR Ca2+ leak reduces total mito-Ca2+ retention. Application of ISO and caffeine significantly reduced the time to peak amplitude (Fig. 4g) and increased the first derivative of [Ca2+]m (Fig. 4h). This indicates an increased maximal Ca2+ influx rate (nmol/L Ca2+ s−1) into the mitochondria during pacing. The rate of transient decay was significantly increased after caffeine treatment (Fig. 4i), demonstrating that caffeine also induces faster efflux of Ca2+ from the mitochondria that is evident when pacing stops.
We next sought to explore how enhanced RyR2 activity reduces the capacity of mitochondria to accumulate and retain Ca2+. Myocytes loaded with fluorescent Ca2+ indicator Fluo-3 were subjected to the standard 5 min, 2 Hz pacing protocol (Fig. 5). Line scans were recorded during the last minute of pacing (Fig. 5a). Cytosolic Ca2+ transient amplitude was significantly reduced in VMs treated with ISO and low-dose caffeine, compared to ISO alone (Fig. 5b), which may explain the differences in [Ca2+]m under these conditions. Pretreatment of VMs with mitoTEMPO attenuated the caffeine-mediated reduction in Ca2+ transient amplitude (Fig. 5a–b), indicative that enhanced mito-ROS is in part responsible for this effect.

Enhancement of RyR2 activity reduces cytosolic Ca2+ transient amplitude and mitochondrial membrane potential. a Representative Fluo-3 fluorescence (F/F0) images (upper images) and profiles (lower images) of ISO treated (50 nmol/L) rat VM undergoing standard pacing protocol (arrows) of 2 Hz for 5 min, before and after treatment with low-dose caffeine (CAFF, 200 µmol/L) or mitoTEMPO (20 µmol/L) and caffeine. Line scan images were recorded during the last minute of pacing. The graph in b depicts mean data ± SEM for Ca2+ transient amplitude (ΔF/F0). N = 2–4 animals, n = 12–15 VMs. *p < 0.05, one-way ANOVA with Bonferroni post hoc test. c Representative images of a rat VM before and after treatment with ISO. Myocyte ΔΨm was monitored by TMRM fluorescence. d Representative recording of changes in ΔΨm in response to pacing at 2 Hz (black bar) for 5 min, followed by application of ISO and further pacing. The signal was normalized to minimum fluorescence obtained by application of FCCP (50 μmol/L) and represented as percentage of baseline. e Representative images of a TMRM-stained rat VM before and after treatment with ISO and CAFF. f Representative recording of changes in ΔΨm in response to pacing and application of ISO and CAFF. The graph in g depicts mean data ± SEM of minimum fluorescence after application of ISO or ISO and CAFF, and pacing. N = 3–4 animals, n = 26 VMs per group. *p < 0.05 vs. ISO group, two-sample Student’s t test
Using cultured rat VMs stained with mitochondrial voltage-sensitive dye TMRM (20 μmol/L for < 1 min), we assessed whether increased RyR2 activity modifies mitochondrial membrane potential (ΔΨm). Images in Fig. 5c show a cultured VM before and after application of ISO. A representative trace is shown in Fig. 5d, whereby signal was normalized to minimum fluorescence obtained by the application of carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP, 50 μmol/L). Images in Fig. 5e show a cultured VM before and after application of ISO and low-dose caffeine, while a representative trace is shown in Fig. 5f. Treatment with ISO and caffeine significantly reduced TMRM fluorescence compared to treatment with ISO alone (Fig. 5g), suggestive that a reduction in ΔΨm may serve as a potential mechanism that contributes to [Ca2+]m reduction under conditions of enhanced RyR2 activity.
To examine whether mito-ROS emission seen in the presence of low-dose caffeine is a process dependent on SR-mitochondria Ca2+ transfer, we utilized genetic inhibition of mito-Ca2+ uptake by adenoviral overexpression of the dominant-negative MCU pore subunit, MCU-DN [21]. Overexpression of MCU-DN in VMs is indicated via western blot, with increased protein signal indicated by anti-MCU antibody (Fig. 6a, b). To confirm inhibition of mito-Ca2+ uptake with MCU-DN overexpression, we co-expressed the mtRCamp1h biosensor. As demonstrated in Fig. 6c, MCU-DN prevented an increase in mtRCamp1h signal usually observed during pacing, indicating an inhibition of Ca2+ influx into mitochondria. Co-expression of OMM-HyPer biosensor revealed no changes in OMM-HyPer signal in VMs stimulated with ISO alone (Fig. 6d). As demonstrated in Fig. 6e, expression of MCU-DN significantly reduced OMM-HyPer fluorescence of ISO and caffeine-treated VMs when mito-Ca2+ uptake is inhibited (Fig. 6e). This supports the hypothesis in conditions of enhanced RyR2 activity, increased mito-ROS emission is a process dependent on influx of Ca2+ into the mitochondria.

Inhibition of MCU Ca2+ uptake with MCU-DN reduces mito-ROS emission in rat VMs. a Representative western blots from VMs probed for MCU expression. The control lane (Ctrl) represents VMs infected with virus carrying an empty vector, and the MCU-DN lane represents VMs infected with virus carrying the dominant-negative MCU subunit. The graph in b depicts quantification of normalized MCU signal (%). N = 4 animals. *p < 0.05 vs. Ctrl group, two-sample Student’s t test. c Representative traces of mtRCamp1h fluorescence from control virus (black line) or MCU-DN virus (pink line) infected VMs. Myocytes were paced at 2 Hz for 5 min (black bar) after treatment with ISO (50 nmol/L) or ISO and low-dose caffeine (CAFF, 200 µmol/L). No mitochondrial Ca2+ uptake is observed with expression of MCU-DN. d Representative traces of OMM-HyPer fluorescence from infected VMs. Myocytes were paced at 2 Hz for 5 min (black bars), treated with ISO, and paced again at 2 Hz. The black line indicates control VM signal, while the pink line indicates MCU-DN infected VM. The signal was normalized to minimum fluorescence obtained by application of DTT (5 mmol/L) and maximum fluorescence by application of DTDP (200 µmol/L). The graph depicts mean data ± SEM for maximum normalized fluorescence (%) after pacing and application of ISO. N = 3–4 animals, n = 7–16 VMs. *p < 0.05 vs. Ctrl group, two-sample Student’s t test. e Representative traces of OMM-HyPer fluorescence from infected VMs. Myocytes were paced at 2 Hz for 5 min (black bars), treated with ISO and CAFF, and paced again at 2 Hz. The graph in e depicts mean data ± SEM for maximum normalized fluorescence (%) after pacing and application of ISO. N = 3 animals per group, n = 5–7 VMs. *p < 0.05 vs. Ctrl group, two-sample Student’s t test
Augmented activity of RyR2 in CPVT VMs can be attenuated by scavenging of mito-ROS
We next sought to determine whether chronic genetically evoked increase in SR Ca2+ leak modulates mito-ROS and the oxidation status of RyR2, amplifying SR Ca2+ leak. We used a mouse CPVT model where chronic RyR2-mediated leak is evoked by ablation of the SR Ca2+ buffering protein calsequestrin [37]. Freshly isolated WT and CPVT VMs were loaded with the fluorescent Ca2+ indicator Fluo-3 AM and cytosolic Ca2+ transients were recorded in the presence of β-adrenergic receptor agonist ISO (100 nmol/L), subjecting VMs to a burst-pace pause protocol (0.5 Hz, 20 s). Representative images of Ca2+ transients and Fluo-3 AM fluorescence profiles (ΔF/F0) are shown in Fig. 7a and b, respectively. At low stimulation frequency, the Ca2+ transient amplitude did not show significant differences between CPVT and WT VMs (Fig. 7c). However, the latency of SCW was significantly reduced in diseased VMs (Fig. 7d). A reduced SCW latency is attributable to abnormal activity of RyR2 and serves as an indicator for the propensity of arrhythmogenic Ca2+ release [5, 16]. The percentage of CPVT VMs exhibiting waves was also significantly increased in comparison to controls (Fig. 7e), while the SR Ca2+ content was reduced (Fig. 7f, g). Besides reduced buffering capacity of the SR devoid of major Ca2+ buffer calsequestrin, this may indicate the hyperactivity of the RyR2 channel complex. To demonstrate that increased SR Ca2+ leak is mediated at least in part by mito-ROS, we pre-treated CPVT VMs with ROS scavenger mitoTEMPO (20 µmol/L, 20 min). This significantly increased SCW latency and restored SR Ca2+ content to near WT levels, suggesting that ROS scavenging can partially stabilize proarrhythmic RyR2 activity in CPVT VMs, similar to the effects of mitoTEMPO in rat VMS acutely challenged with caffeine (Figs. 1, 5a, b).

Proarrhythmic SCWs in CPVT mice VMs with genetically increased RyR2 activity can be attenuated by mito-ROS scavenging. a Representative confocal line scan images of Ca2+ transients and b Fluo-3 fluorescence (F/F0) profiles of ISO treated (100 nmol/L) WT and CPVT mouse VMs undergoing 0.5 Hz pace-pause protocol (arrows) to induce SCWs. Myocytes from CPVT mice were pretreated with mitoTEMPO (20 µmol/L, 20 min, CPVT + MT group). Graphs in cand d depict mean data ± SEM for WT, CPVT and CPVT + MT groups of Ca2+ transient amplitude (ΔF/F0) and SCW latency (seconds), respectively. N = 3 WT, 5 CPVT animals, n = 12–16 VMs. *p < 0.05 vs. WT group, **p < 0.05 vs. CPVT group, one-way ANOVA with Bonferroni post hoc test. e Graph depicting the percentage (%) of cells exhibiting waves. N = 3 WT, 5 CPVT animals, n = 20–25 VMs. *p < 0.05 vs. WT group, **p < 0.05 vs. CPVT group, Fisher’s exact test with Freeman-Halton extension. f Representative Fluo-3 fluorescence profiles (F/F0) of caffeine-induced Ca2+ transients (10 mmol/L) used to determine SR Ca2+ content. The graph in g depicts mean data ± SEM of caffeine-sensitive SR Ca2+ content (ΔF/F0). N = 3 WT, 3 CPVT animals, n = 4–8 VMs. *p < 0.05 vs. WT group, one-way ANOVA with Bonferroni post hoc test
Using the mitochondria-specific ROS indicator MitoSOX, we show that ROS emission in CPVT VMs is significantly increased compared to controls (Fig. 8a, b). Furthermore, western blot revealed a significant increase in the oxidation status of RyR2 immunoprecipitated from diseased VMs (Fig. 8c, d), supporting the hypothesis that increased RyR2 activity in CPVT is partially modulated by leak-induced mito-ROS, as in rat VMs with a caffeine-induced increase in RyR2 activity (Figs. 2, 3).

Myocytes of CPVT mice have increased mito-ROS emission rates and RyR2 oxidation in comparison to controls. a Representative recording of ROS production measured with MitoSOX in ISO-treated (100 nmol/L) WT and CPVT VMs. Signal was normalized to maximum fluorescence obtained on application of DTDP (200 µmol/L). The ROS production rate (%) was calculated by normalization to the maximum rate of ROS emission observed on application of DTDP, and rates for these representative recordings are indicated. The graph in b depicts mean data ± SEM for the normalized ROS production rate of WT and CPVT VMs. N = 3 WT, 5 CPVT animals, n = 7–13 VMs. *p < 0.05 vs. WT group, paired Student’s t test. c Representative images of immunoprecipitated RyR2 from freshly isolated ISO-treated WT and CPVT VMs, immunoblotted for RyR2 signal and oxidation using DNP antibody. The graph in d depicts quantification of normalized DNP signal (%). N = 6 animals per group. *p < 0.05 vs. WT group, two sample Student’s t test. e Representative western blots from WT and CPVT VMs probed for MCUb and MCU expression. The graph in f depicts quantification of normalized MCUb/MCU signal (%). N = 5 animals per group. *p < 0.05 vs. Ctrl group, two-sample Student’s t test
Notably, CPVT does not cause massive functional changes at the whole heart and cellular levels [30], which would be expected if mitochondria function is substantially compromised. This points to the existence of compensatory mechanisms directed toward the reduction of SR-mitochondria Ca2+ transfer in conditions when SR Ca2+ leak is chronically increased. Indeed, as shown in Fig. 8e, f, CPVT is accompanied by a significant increase in expression levels of an inhibitory variant of MCU (MCUb; [41, 58]), and consequently a ~ 200% increase in the MCUb/MCU ratio. Taken together, our data suggest that hereditary enhancement in RyR2-complex activity affects mito-ROS production, exacerbating RyR2 dysfunction. Furthermore, although compensatory program to reduce SR-mitochondria Ca2+ transfer is activated in CPVT, this protection is clearly incomplete. Under β-adrenergic stimulation, upregulation of inhibitory MCUb does not prevent a surge in mito-ROS and the subsequent oxidation of RyR2.
Discussion
Parallel changes in RyR2 activity and mito-ROS production are hallmark features in cardiac pathology such as HF, MI, aging, or diabetic-induced cardiomyopathy [4, 18, 22, 33, 34, 36, 64, 70]. We recently demonstrated that modulation of mito-Ca2+ efficiently regulates RyR2 activity via mito-ROS [32]. In the present study, we demonstrate that pharmacologically induced or genetic enhancement of RyR2 activity promotes mito-ROS production, and that SR-mitochondria Ca2+ transfer is essential for this process. We show that increased diastolic SR Ca2+ leak via hyperactive RyR2 channels drives further SR Ca2+ leak via mito-ROS, in an indirect, positive feedback process that is detrimental to intracellular Ca2+ handling in cardiac VMs.
Leak begets leak
Robust control of RyR2-mediated SR Ca2+ release is vital for cardiac function. The amplitude of intracellular Ca2+ release increases to achieve greater contraction in conditions when the metabolic demands of the body are increased, i.e., stress [7]. This is largely attributed to the enhanced activity of SR Ca2+ ATPase and thereby higher intra-SR Ca2+ available during release. However, there is ample evidence suggesting that parallel changes in RyR2 activity play a significant role in this process. For example, it has been proposed that β-adrenergic stimulation increases “synchronization” of SR Ca2+ release from individual RyR2 clusters [62, 67] and the maximal rate of [Ca2+]i rise [28]. Enhanced activity of RyR2 under β-adrenergic stimulation has been attributed to the direct effects of PKA phosphorylation [11, 53] or indirect effects via activation of CaMKII [19]. Importantly, prolonged exposure to β-adrenergic agonists was shown to evoke deleterious effects on Ca2+ cycling associated with enhanced RyR2 oxidation by mito-ROS and thereby its activity, resulting in th generation of pro-arrhythmic spontaneous Ca2+ waves [12, 13]. We previously showed that in chronic model of HF, enhanced phosphorylation levels of RyR2 are readily detectable relatively early during the course of pathological remodeling, while RyR2 oxidation becomes evident at much later stages [6]. Importantly, the combination of PKA/CaMKII-mediated phosphorylation and oxidation modifications of RyR2 led to more pronounced changes in RyR2-mediated SR Ca2+ leak and intracellular Ca2+ cycling than PKA/CaMKII-mediated phosphorylation alone [6, 40]. These studies strongly suggest that the effects of modulators of RyR2 activity can summate, and that primary modification of RyR2 can evoke secondary modification via systems of posttranslational control of RyR2 activity. To test this theory, we performed experiments in rat VMs with RyR2 activity directly enhanced by the pharmacological agent caffeine in the presence of β-adrenergic agonist ISO (Fig. 1). Parallel experiments with ROS biosensors targeted to mitochondria and the SR showed significant increases in ROS in the presence of caffeine (Figs. 2 and 3). This led to enhanced oxidation of RyR2 (Fig. 3). Application of mitochondria-specific ROS scavenger mitoTEMPO attenuated the effects of caffeine on RyR2-mediated SR Ca2+ leak (Fig. 1) and Ca2+ transient amplitude (Fig. 5), suggesting that initial insult produces a secondary effect on RyR2 function via a ROS-generating pathway involving mitochondria.
These results were further corroborated using calsequestrin2-null mouse model of RyR2-complex gain of function [37]. Western blot analysis using samples prepared from mouse hearts exposed to ISO shows significant increase in RyR2 oxidation in CPVT (Fig. 8c, d), while measurements of mito-ROS with mitoSOX in acutely isolated VMs demonstrate an eightfold increase in the rate of ROS production by mitochondria (Fig. 8a, b). Existing evidence of secondary posttranslational modulation of RyR2 activity in CPVT VMs includes increased phosphorylation of the channel by PKA [49] and CaMKII [45], while RyR2 oxidation was not previously shown. However, our findings are in line with a recent report demonstrating enhanced RyR2 oxidation and mito-ROS emission in a mouse model with chronic SR Ca2+ leak induced by phosphomimetic RyR2 mutation [60]. Incubation of ISO-challenged CPVT VMs with mitoTEMPO restored SR Ca2+ content and reduced spontaneous Ca2+ release (Fig. 7), which implies that oxidation of RyR2 secondary to genetically evoked gain of function of SR Ca2+ release plays a key role in Ca2+-dependent arrhythmia.
Mitochondrial [Ca2+] and ROS
The close proximity of mitochondria and the SR allows for bidirectional communication between the organelles, whereby production of ATP by mitochondria can be closely coupled to RyR2-mediated Ca2+ release and the energy demand of the myocyte [20, 25]. Activity of several mitochondrial enzymes involved in oxidative phosphorylation depends on [Ca2+], thereby enhanced mitochondrial Ca2+ influx is thought to promote electron transport and ATP production [52]. However, increased activity of electron transport chain results in enhanced mito-ROS production, which can overcome intracellular antioxidant defenses and produce cell-wide deleterious effects [9, 29]. This is most evident during ischemia–reperfusion when mito-Ca2+ overload was directly linked to a massive surge in ROS [39, 51]. However, in other pathological conditions accompanied by enhanced catecholaminergic drive such as HF, the link between mito-[Ca2+] and rate of mito-ROS production remains a subject of controversy. Indeed, in guinea pig model of HF and rat model of hypertrophy, the levels of mitochondria ROS were shown to be increased, while levels of [Ca2+]m dramatically reduced [38, 46, 48]. These data strongly suggest that the [Ca2+]/ROS relationship in mitochondria is not linear. It appears that in the absence of a catastrophic event such as ischemia–reperfusion, excessive Ca2+ accumulation can be effectively limited by robust control mechanisms, which clearly fail to stop the generation of mito-ROS. In the present work, we for the first time directly tested how acute enhancement of RyR2 activity affects intra-mitochondrial [Ca2+]. Experiments using intra-mitochondrial Ca2+ biosensor mtRCamp1h showed that periodic pacing in the presence of ISO induces mito-Ca2+ accumulation (Fig. 4). Pharmacological facilitation of RyR2 activity with low-dose caffeine to mimic HF phenotype produces a decrease in peak [Ca2+]m, an increase in the maximum rate of mito-Ca2+ loading during pacing, and an accelerated loss of [Ca2+] upon pacing cessation. Phenomenologically, these results are consistent with a previously reported loss of mito-Ca2+ in disease models with enhanced RyR2 activity, i.e., guinea pig HF and rat model of cardiac hypertrophy induced by pressure overload [32, 48]. Notably, the net reduction in [Ca2+]m could be explained by a decrease in systolic [Ca2+]. Moreover, the caffeine-induced RyR2 leak appears to facilitate mito-Ca2+ loss, as shown in Fig. 4. Our experiments using voltage-sensitive indicator showed ΔΨ m depolarization in the presence of low-dose caffeine (Fig. 5c, d), which is expected to reduce the driving force for mito-Ca2+ uptake and can indicate mPTP opening, promoting rapid Ca2+ extrusion [15, 26, 35, 50, 69]. This can at least partially explain the reduction in net [Ca2+]m in conditions with enhanced RyR2-mediated SR Ca2+ leak. Increased Ca2+ extrusion by the mitochondrial Na+/Ca2+/Li+ exchanger (NCLX) may also play a role in reducing net [Ca2+]m during enhanced SR Ca2+ leak, as seen in conditions of cytosolic Na+ overload [38, 48].
Importantly, our results contradict the findings obtained in several models with hyperactive RyR2 channels. The Ca2+ content of isolated mitochondria was shown to be increased in mice with phosphomimetic RyR2 mutation [60] and in aging human hearts [59]. An explanation for this discrepancy could be that our measurements of free [Ca2+]m with a genetically targeted biosensor in live VMs do not reflect total mito-Ca2+ accumulation, because we are not tracking Ca2+ complexed with phosphates. However, according to Chalmers and Nicholls [17, 54], free mito-Ca2+ must first be increased to promote the formation of Ca2+-phosphate complexes. A more likely scenario is that differences in cellular [Na+] between control and diseased VMs, and thereby changes in NCLX-mediated mito-Ca2+ efflux, are omitted upon mitochondria isolation. Furthermore, the reduction of cytosolic Ca2+ transient amplitude and [Ca2+] available for uptake observed in live VMs are not directly mimicked in in vitro studies. At the same time, our data demonstrating an increase in maximal mito-Ca2+ uptake rate (Fig. 4h) supports the hypothesis that MCU activity is increased, likely due to oxidation of the complex [23]. This may contribute to an increased [Ca2+] in isolated mitochondria from samples with leaky RyR2s. Future studies using protocols that match mito-Ca2+ fluxes in live cells and isolated preparations are needed to reconcile this controversy.
The role of SR-mitochondria Ca2+ transfer in mito-ROS production
It is well established that increase of oxygen consumption by mitochondria under β-adrenergic stimulation is a Ca2+-dependent process [55]. Increased Ca2+ release in the presence of β-adrenergic agonists stimulates contraction and accelerates expenditure of ATP, decreasing the ATP/ADP ratio and thus promoting generation of ATP by mitochondria. The by-product of this process will be mito-ROS, which can lead to RyR2 oxidation and consequently generation of spontaneous Ca2+ waves. Indeed, Bovo et al. [13] recently showed that the presence of blebbistatin to specifically inhibit contraction substantially reduced oxidative stress in VMs under prolonged exposure to β-adrenergic agonist. However, this data does not fully exclude the potential role of direct SR-mitochondria Ca2+ transfer in modulation of mitochondria function and production of mito-ROS. Our recent study revealed that facilitation of mito-Ca2+ influx using pharmacological activators of MCU in the presence of ISO fails to increase mitochondria Ca2+ loading, and, similar to the effects of low-dose caffeine, depolarizes ΔΨm [32]. Likewise, both caffeine to increase RyR2 activity (Figs. 2 and 3) and MCU agonists accelerate production of mito-ROS. Previously, we also showed that inhibition of MCU was sufficient to reduce mito-ROS in VMs from hypertrophic rats [32]. Using the dominant-negative MCU variant, we obtained additional evidence that in the presence of caffeine, SR-mitochondria Ca2+ transfer is necessary for the mito-ROS surge (Fig. 5). Further indirect evidence that SR-mitochondria Ca2+ transfer plays a key role in disease-related mitochondria dysfunction was obtained by measuring the expression levels of MCU and its inhibitory variant MCUb in the hearts of CPVT mice (Fig. 8e, f). Importantly, recent electron microscopy studies in a mouse model of RyR2 loss of function demonstrated a substantial increase in tunneling between mitochondria, highlighting the responsiveness of the organelle to disturbances in SR Ca2+ release in an attempt to preserve mitochondrial function and energy supply [43]. A significant increase in MCUb/MCU ratio in CPVT suggests that RyR2 hyperactivity facilitates adaptive remodeling directed toward the reduction of mito-Ca2+ uptake.
Limitations
Sarcoplasmic reticulum Ca2+ leak-induced changes in intracellular ROS, [Ca2+]m and ΔΨm may produce secondary effects on other key components of intracellular Ca2+ handling machinery including SERCa2a, Na+/Ca2+ exchanger and L-type Ca2+ channels [8, 33, 68]. In the present study, we did not measure changes in activities of these complexes in caffeine-treated rat VMs or CPVT mouse VMs, nor did we measure intracellular [ATP]. Notably, our previous data demonstrates lack of changes and/or effects of ROS scavenging on activities of these complexes in aged rabbit VMs and canine HF and MI VMs with leaky RyR2 channels [4,5,6, 18]. This supports the hypothesis that mito-ROS-mediated, self-imposed enhancement of SR Ca2+ leak is the major contributor to defective Ca2+ homeostasis in conditions with hyperactive RyR2s.
Conclusion
Taken together, our results suggest that both acute and inherited increase in activity of RyR2 evoke multiple mechanisms that protect mitochondria from Ca2+ overload. However, under conditions mimicking stress, these mechanisms fail to fully alleviate mito-Ca2+-dependent increase in mito-ROS, resulting in oxidation of RyR2. This further increases channel activity and exacerbates defective intracellular Ca2+ homeostasis.
Abbreviations
- ΔΨm :
-
Mitochondrial membrane potential
- [Ca2+]i :
-
Intracellular calcium concentration
- [Ca2+]m :
-
Mitochondrial calcium concentration
- [Ca2+]SR :
-
Sarcoplasmic reticulum calcium concentration
- CAFF:
-
Caffeine
- CPVT:
-
Catecholaminergic polymorphic ventricular tachycardia
- HF:
-
Heart failure
- ISO:
-
Isoproterenol
- MCU:
-
Mitochondrial Ca2+ uniporter
- MCU-DN:
-
Dominant negative subunit of mitochondrial Ca2+ uniporter
- Mito-Ca2+ :
-
Mitochondrial Ca2+
- Mito-ROS:
-
Mitochondria-derived reactive oxygen species
- NCLX:
-
Na+/Ca2++/Li+ exchanger
- OMM:
-
Outer mitochondrial membrane
- ROS:
-
Reactive oxygen species
- RyR2:
-
Ryanodine receptor
- SCW:
-
Spontaneous Ca2+ wave
- SERCa2a:
-
Sarco/endoplasmic reticulum Ca2+-ATPase
- SR:
-
Sarcoplasmic reticulum
- VM:
-
Ventricular myocyte
- VT/VF:
-
Ventricular tachycardia/ventricular fibrillation
- WT:
-
Wild type
References
Akerboom J, Carreras Calderón N, Tian L, Wabnig S, Prigge M, Tolö J, Gordus A, Orger MB, Severi KE, Macklin JJ, Patel R, Pulver SR, Wardill TJ, Fischer E, Schüler C, Chen TW, Sarkisyan KS, Marvin JS, Bargmann CI, Kim DS, Kügler S, Lagnado L, Hegemann P, Gottschalk A, Schreiter ER, Looger LL (2013) Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front Mol Neurosci 4:2. https://doi.org/10.3389/fnmol.2013.00002
Avezov E, Cross BC, Kaminski Schierle GS, Winters M, Harding HP, Melo EP, Kaminski CF, Ron D (2013) Lifetime imaging of a fluorescent protein sensor reveals surprising stability of ER thiol redox. J Cell Biol 201:337–349. https://doi.org/10.1083/jcb.201211155
Banyasz T, Lozinskiy I, Payne CE, Edelmann S, Norton B, Chen B, Chen-Izu Y, Izu LT, Balke CW (2008) Transformation of adult rat cardiac myocytes in primary culture. Exp Physiol 93:370–382. https://doi.org/10.1113/expphysiol.2007.040659
Belevych AE, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA, Györke S (2007) Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys J 93:4083–4092. https://doi.org/10.1529/biophysj.107.114546
Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, Carnes CA, Billman GE, Györke S (2012) Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res 110:569–577. https://doi.org/10.1161/CIRCRESAHA.111.260455
Belevych AE, Terentyev D, Terentyeva R, Nishijima Y, Sridhar A, Hamlin RL, Carnes CA, Györke S (2011) The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovasc Res 90:493–502. https://doi.org/10.1093/cvr/cvr025
Bers DM (2002) Cardiac excitation-contraction coupling. Nature 415:198–205. https://doi.org/10.1038/415198a
Bers DM (2014) Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol 76:107–127. https://doi.org/10.1146/annurev-physiol-020911-153308
Bertero E, Maack C (2018) Calcium signaling and reactive oxygen species in mitochondria. Circ Res 122:1460–1478. https://doi.org/10.1161/CIRCRESAHA.118.310082
Bilan DS, Pase L, Joosen L, Gorokhovatsky AY, Ermakova YG, Gadella TW, Grabher C, Schultz C, Lukyanov S, Belousov VV (2013) HyPer-3: a genetically encoded H(2)O(2) probe with improved performance for ratiometric and fluorescence lifetime imaging. ACS Chem Biol 8:535–542. https://doi.org/10.1021/cb300625g
Bovo E, Huke S, Blatter LA, Zima AV (2017) The effect of PKA-mediated phosphorylation of ryanodine receptor on SR Ca2+ leak in ventricular myocytes. J Mol Cell Cardiol 104:9–16. https://doi.org/10.1016/j.yjmcc.2017.01.015
Bovo E, Lipsius SL, Zima AV (2012) Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during β-adrenergic receptor stimulation in rabbit cardiomyocytes. J Physiol 590:3291–3304. https://doi.org/10.1113/jphysiol.2012.230748
Bovo E, Mazurek SR, de Tombe PP, Zima AV (2015) Increased energy demand during adrenergic receptor stimulation contributes to Ca(2+) wave generation. Biophys J 109:1583–1591. https://doi.org/10.1016/j.bpj.2015.09.002
Bovo E, Mazurek SR, Zima AV (2018) Oxidation of ryanodine receptor following ischemia/reperfusion increases propensity of Ca2+ waves during β-adrenergic receptor stimulation. Am J Physiol Heart Circ Physiol 315:H1032–H1040. https://doi.org/10.1152/ajpheart.00334.2018
Broekemeier KM, Klocek CK, Pfeiffer DR (1998) Proton selective substate of the mitochondrial permeability transition pore: regulation by the redox state of the electron transport chain. Biochemistry 37:13059–13065. https://doi.org/10.1021/bi980820c
Brunello L, Slabaugh JL, Radwanski PB, Ho HT, Belevych AE, Lou Q, Chen H, Napolitano C, Lodola F, Priori SG, Fedorov VV, Volpe P, Fill M, Janssen PM, Györke S (2013) Decreased RyR2 refractoriness determines myocardial synchronization of aberrant Ca2+ release in a genetic model of arrhythmia. Proc Natl Acad Sci U S A 110:10312–10317. https://doi.org/10.1073/pnas.1300052110
Chalmers S, Nicholls DG (2003) The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J Biol Chem 278:19062–19070. https://doi.org/10.1074/jbc.M212661200
Cooper LL, Li W, Lu Y, Centracchio J, Terentyeva R, Koren G, Terentyev D (2013) Redox modification of ryanodine receptors by mitochondria-derived reactive oxygen species contributes to aberrant Ca2+ handling in ageing rabbit hearts. J Physiol 591:5895–5911. https://doi.org/10.1113/jphysiol.2013.260521
Curran J, Hinton MJ, Ríos E, Bers DM, Shannon TR (2007) Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res 100:391–398. https://doi.org/10.1161/01.RES.0000258172.74570.e6
De la Fuente S, Sheu SS (2019) SR-mitochondria communication in adult cardiomyocytes: A close relationship where the Ca2+ has a lot to say. Arch Biochem Biophys 663:259–268. https://doi.org/10.1016/j.abb.2019.01.026
De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476:336–340. https://doi.org/10.1038/nature10230
Dedkova EN, Seidlmayer LK, Blatter LA (2013) Mitochondria-mediated cardioprotection by trimetazidine in rabbit heart failure. J Mol Cell Cardiol 59:41–54. https://doi.org/10.1016/j.yjmcc.2013.01.016
Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, Breves SL, Zhang X, Tripathi A, Palaniappan P, Riitano MF, Worth AM, Seelam A, Carvalho E, Subbiah R, Jaña F, Soboloff J, Peng Y, Cheung JY, Joseph SK, Caplan J, Rajan S, Stathopulos PB, Madesh M (2017) Mitochondrial Ca2+ uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol Cell 65:1014–1028. https://doi.org/10.1016/j.molcel.2017.01.032
Eisner DA, Caldwell JL, Kistamás K, Trafford AW (2017) Calcium and excitation-contraction coupling in the heart. Circ Res 121:181–195. https://doi.org/10.1161/CIRCRESAHA.117.310230
Eisner V, Csordás G, Hajnóczky G (2013) Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle - pivotal roles in Ca2+ and reactive oxygen species signaling. J Cell Sci 126:2965–2978. https://doi.org/10.1242/jcs.093609
Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, Karch J, Gabel S, Farber J, Force T, Brown JH, Murphy E, Molkentin JD (2010) Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest 120:3680–3687. https://doi.org/10.1172/JCI43171
García-Rivas Gde J, Carvajal K, Correa F, Zazueta C (2006) Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br J Pharmacol 149:829–837. https://doi.org/10.1038/sj.bjp.0706932
Ginsburg KS, Bers DM (2004) Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J Physiol 556:463–480. https://doi.org/10.1113/jphysiol.2003.055384
Giordano FJ (2005) Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 115:500–508. https://doi.org/10.1172/JCI24408
Györke S (2009) Molecular basis of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 6:123–129. https://doi.org/10.1016/j.hrthm.2008.09.013
Györke S, Lukyanenko V, Györke I (1997) Dual effects of tetracaine on spontaneous calcium release in rat ventricular myocytes. J Physiol 500:297–309. https://doi.org/10.1113/jphysiol.1997.sp022021
Hamilton S, Terentyeva R, Kim TY, Bronk P, Clements RT, O-Uchi J, Csordás G, Choi BR, Terentyev D (2018) Pharmacological modulation of mitochondrial Ca2+ content regulates sarcoplasmic reticulum Ca2+ release via oxidation of the ryanodine receptor by mitochondria-derived reactive oxygen species. Front Physiol 9:1831. https://doi.org/10.3389/fphys.2018.01831
Hamilton S, Terentyev D (2018) Proarrhythmic remodeling of calcium homeostasis in cardiac disease; implications for diabetes and obesity. Front Physiol 9:1517. https://doi.org/10.3389/fphys.2018.01517
Hamilton S, Terentyev D (2019) Altered intracellular calcium homeostasis and arrhythmogenesis in the aged heart. Int J Mol Sci 20:2386. https://doi.org/10.3390/ijms20102386
Hüser J, Blatter LA (1999) Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem J 343:311–317
Kim TY, Terentyeva R, Roder KH, Li W, Liu M, Greener I, Hamilton S, Polina I, Murphy KR, Clements RT, Dudley SC Jr, Koren G, Choi BR, Terentyev D (2017) SK channel enhancers attenuate Ca2+-dependent arrhythmia in hypertrophic hearts by regulating mito-ROS-dependent oxidation and activity of RyR. Cardiovasc Res 113:343–353. https://doi.org/10.1093/cvr/cvx005
Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K (2006) Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest 116:2510–2520. https://doi.org/10.1172/JCI29128
Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Böhm M, O'Rourke B, Maack C (2010) Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 121:1606–1613. https://doi.org/10.1161/CIRCULATIONAHA.109.914911
Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD (2015) The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile Stress In The Heart. Cell Rep 12:15–22. https://doi.org/10.1016/j.celrep.2015.06.002
Kyrychenko S, Poláková E, Kang C, Pocsai K, Ullrich ND, Niggli E, Shirokova N (2013) Hierarchical accumulation of RyR post-translational modifications drives disease progression in dystrophic cardiomyopathy. Cardiovasc Res 97:666–675. https://doi.org/10.1093/cvr/cvs425
Lambert JP, Luongo TS, Tomar D, Jadiya P, Gao E, Zhang X, Lucchese AM, Kolmetzky DW, Shah NS, Elrod JW (2019) MCUB regulates the molecular composition of the mitochondrial calcium uniporter channel to limit mitochondrial calcium overload during stress. Circulation 140:1720–1733. https://doi.org/10.1161/CIRCULATIONAHA.118.037968
Landstrom AP, Dobrev D, Wehrens XHT (2017) Calcium signaling and cardiac arrhythmias. Circ Res 120:1969–1993. https://doi.org/10.1161/CIRCRESAHA.117.310083
Lavorato M, Iyer VR, Dewight W, Cupo RR, Debattisti V, Gomez L, De la Fuente S, Zhao YT, Valdivia HH, Hajnóczky G, Franzini-Armstrong C (2017) Increased mitochondrial nanotunneling activity, induced by calcium imbalance, affects intermitochondrial matrix exchanges. Proc Natl Acad Sci U S A 114:E849–E858. https://doi.org/10.1073/pnas.1617788113
Liu B, Ho HT, Velez-Cortes F, Lou Q, Valdivia CR, Knollmann BC, Valdivia HH, Gyorke S (2014) Genetic ablation of ryanodine receptor 2 phosphorylation at Ser-2808 aggravates Ca(2+)-dependent cardiomyopathy by exacerbating diastolic Ca2+ release. J Physiol 592:1957–1973. https://doi.org/10.1113/jphysiol.2013.264689
Liu N, Ruan Y, Denegri M, Bachetti T, Li Y, Colombi B, Napolitano C, Coetzee WA, Priori SG (2011) Calmodulin kinase II inhibition prevents arrhythmias in RyR2(R4496C+/-) mice with catecholaminergic polymorphic ventricular tachycardia. J Mol Cell Cardiol 50:214–222. https://doi.org/10.1016/j.yjmcc.2010.10.001
Liu T, O’Rourke B (2008) Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res 103:279–288. https://doi.org/10.1161/CIRCRESAHA.108.175919
Liu T, O'Rourke B (2009) Regulation of mitochondrial Ca2+ and its effects on energetics and redox balance in normal and failing heart. J Bioenerg Biomembr 41:127–132. https://doi.org/10.1007/s10863-009-9216-8
Liu T, Takimoto E, Dimaano VL, DeMazumder D, Kettlewell S, Smith G, Sidor A, Abraham TP, O'Rourke B (2014) Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ Res 115:44–54. https://doi.org/10.1161/CIRCRESAHA.115.303062
Loaiza R, Benkusky NA, Powers PP, Hacker T, Noujaim S, Ackerman MJ, Jalife J, Valdivia HH (2013) Heterogeneity of ryanodine receptor dysfunction in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res 112:298–308. https://doi.org/10.1161/CIRCRESAHA.112.274803
Lu X, Kwong JQ, Molkentin JD, Bers DM (2016) Individual cardiac mitochondria undergo rare transient permeability transition pore openings. Circ Res 118:834–841. https://doi.org/10.1161/CIRCRESAHA.115.308093
Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, Tsai EJ, Molkentin JD, Chen X, Madesh M, Houser SR, Elrod JW (2017) The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 545:93–97. https://doi.org/10.1038/nature22082
Mammucari C, Raffaello A, Vecellio Reane D, Gherardi G, De Mario A, Rizzuto R (2018) Mitochondrial calcium uptake in organ physiology: from molecular mechanism to animal models. Pflugers Arch 470:1165–1179. https://doi.org/10.1007/s00424-018-2123-2
Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR (2000) PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101:365–376. https://doi.org/10.1016/s0092-8674(00)80847-8
Nicholls DG, Chalmers S (2004) The integration of mitochondrial calcium transport and storage. J Bioenerg Biomembr 36:277–281. https://doi.org/10.1023/B:JOBB.0000041753.52832.f3
Opie LH, Thandroyen FT, Muller C, Bricknell OL (1979) Adrenaline-induced "oxygen-wastage" and enzyme release from working rat heart. Effects of calcium antagonism, beta-blockade, nicotinic acid and coronary artery ligation. J Mol Cell Cardiol 11:1073–1094. https://doi.org/10.1016/0022-2828(79)90395-x
Page E, McCallister LP (1973) Quantitative electron microscopic description of heart muscle cells. Application to normal, hypertrophied and thyroxin-stimulated hearts. Am J Cardiol 31:172–181. https://doi.org/10.1016/0002-9149(73)91030-8
Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, DeSimone L, Coltorti F, Bloise R, Keegan R, Cruz Filho FE, Vignati G, Benatar A, DeLogu A (2002) Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 106:69–74. https://doi.org/10.1161/01.cir.0000020013.73106.d8
Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabò I, Rizzuto R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 32:2362–2376. 10.1038/emboj.2013.157.
Ruiz-Meana M, Minguet M, Bou-Teen D, Miro-Casas E, Castans C, Castellano J, Bonzon-Kulichenko E, Igual A, Rodriguez-Lecoq R, Vázquez J, Garcia-Dorado D (2019) Ryanodine receptor glycation favors mitochondrial damage in the senescent heart. Circulation 139:949–964. https://doi.org/10.1161/CIRCULATIONAHA.118.035869
Santulli G, Xie W, Reiken SR, Marks AR (2015) Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A 112:11389–11394. https://doi.org/10.1073/pnas.1513047112
Song LS, Sham JS, Stern MD, Lakatta EG, Cheng H (1998) Direct measurement of SR release flux by tracking 'Ca2+ spikes' in rat cardiac myocytes. J Physiol 512:677–691. https://doi.org/10.1111/j.1469-7793.1998.677bd.x
Song LS, Wang SQ, Xiao RP, Spurgeon H, Lakatta EG, Cheng H (2001) beta-Adrenergic stimulation synchronizes intracellular Ca(2+) release during excitation-contraction coupling in cardiac myocytes. Circ Res 88:794–801. https://doi.org/10.1161/hh0801.090461
Suzuki J, Kanemaru K, Ishii K, Ohkura M, Okubo Y, Iino M (2014) Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat Commun 5:4153. https://doi.org/10.1038/ncomms5153
Terentyev D, Györke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Györke S (2008) Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res 103:1466–1472. https://doi.org/10.1161/CIRCRESAHA.108.184457
Terentyev D, Rees CM, Li W, Cooper LL, Jindal HK, Peng X, Lu Y, Terentyeva R, Odening KE, Daley J, Bist K, Choi BR, Karma A, Koren G (2014) Hyperphosphorylation of RyRs underlies triggered activity in transgenic rabbit model of LQT2 syndrome. Circ Res 115:919–928. https://doi.org/10.1161/CIRCRESAHA.115.305146
Terentyev D, Viatchenko-Karpinski S, Valdivia HH, Escobar AL, Györke S (2002) Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ Res 6:414–420. https://doi.org/10.1161/01.RES.0000032490.04207.BD
Viatchenko-Karpinski S, Györke S (2001) Modulation of the Ca(2+)-induced Ca(2+) release cascade by beta-adrenergic stimulation in rat ventricular myocytes. J Physiol 533:837–848. https://doi.org/10.1111/j.1469-7793.2001.t01-1-00837.x
Venetucci LA, Trafford AW, O'Neill SC, Eisner DA (2008) The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc Res 77:285–292. https://doi.org/10.1093/cvr/cvm009
Zhao Z, Gordan R, Wen H, Fefelova N, Zang WJ, Xie LH (2013) Modulation of intracellular calcium waves and triggered activities by mitochondrial ca flux in mouse cardiomyocytes. PLoS ONE 8:e80574. https://doi.org/10.1371/journal.pone.0080574
Zima AV, Blatter LA (2006) Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res 71:310–321. https://doi.org/10.1016/j.cardiores.2006.02.019
Acknowledgements
We thank Dr. Jin O-Uchi, University of Minnesota Twin Cities, for obtaining the MCU-DN adenovirus.
Funding
This work was supported by American Heart Association Grant #18POST33960456 to SH, and National Heart, Lung, and Blood Institute at the National Institutes of Health (NIH) RO1 HL142588 and NIH R01HL121796 to DT.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted (The Ohio State University Institutional Animal Care and Use Committee (IACUC), permit number 2019A00000041 and 2010A00000117-R3). This article does not contain any studies with human participants performed by any of the authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hamilton, S., Terentyeva, R., Martin, B. et al. Increased RyR2 activity is exacerbated by calcium leak-induced mitochondrial ROS. Basic Res Cardiol 115, 38 (2020). https://doi.org/10.1007/s00395-020-0797-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-020-0797-z