
Abstract
Propionibacterium acnes (P. acnes) has been known to produce various exogenous proteases, however, their role in acne pathogenesis remains largely unknown. Proteases elicit cellular responses, at least in part, via proteinase-activated receptor-2 (PAR-2), which is known to mediate inflammation and immune response. In this study, we investigated whether proteases from P. acnes could activate PAR-2 on keratinocytes and induce pro-inflammatory cytokines, antimicrobial peptides (AMPs), and matrix metalloproteinases (MMPs) via PAR-2 signaling. We examined PAR-2 expression and protease activity in acne lesions using immunofluorescence staining and in situ zymography. The effect of the culture supernatant of P. acnes on Ca2+ signaling in immortalized keratinocytes (HaCaT) was measured using a fluorescence method. HaCaT cells were treated with P. acnes strain ATCC 6919 culture supernatant, with or without pretreatment with serine protease inhibitor or selective PAR-2 antagonist and the gene expression of pro-inflammatory cytokines, AMPs, and MMPs was detected using real-time reverse transcription-polymerase chain reaction. We found that the protease activity and PAR-2 expression were increased in acne lesions. The P. acnes culture supernatant induced calcium signaling in keratinocytes via PAR-2 and stimulated the mRNA expression of interleukin (IL)-1α, -8, tumor necrosis factor (TNF)-α, human beta defensin (hBD)-2, LL-37, MMP-1, -2, -3, -9, and -13 in keratinocytes, which was significantly inhibited by serine protease inhibitor as well as selective PAR-2 specific antagonist. These results indicate that PAR-2 plays an important role in the pathogenesis of acne by inducing inflammatory mediators in response to proteases secreted from P. acnes.
Similar content being viewed by others


Introduction
Acne vulgaris is a chronic inflammatory disorder of pilosebaceous unit characterized by increased sebum production, altered keratinization, Propionibacterium acnes (P. acnes) colonization, and inflammation [35]. Propionibacterium acnes, which is a facultative anaerobic gram-positive bacterium, is believed to play a critical role in the induction and maintenance of inflammation in acne by activating inflammatory cells, keratinocytes, and sebocytes to induce various inflammatory mediators [3, 23]. It is now accepted that P. acnes contributes to inflammation via activation of toll-like receptors (TLRs) [17, 18]. It is also known that, besides TLRs activation, P. acnes triggers acne inflammation by releasing various enzymes which lead to rupture of follicular walls and tissue injury such as lipases, proteases and hyaluronidases [8, 10, 28]. A previous study showed that P. acnes, laboratory strain P-37, produced extracellular proteinase, which was a heterogeneous mixture of three molecular species of enzyme including a neutral proteinase with a serine group and two kinds of alkaline proteinases [10]. In addition, the genome sequence of P. acnes demonstrated various extracellular peptidases, including homologs of an O-sialoglycoprotein endopeptidase, an extracellular subtilisin-like protease and a tripeptidyl aminopeptidase [4]. Extracellular proteases secreted by P. acnes may contribute to inflammation of acne by matrix breakdown and proteolytic detachment of follicular keratinocytes, thereby releasing inflammatory mediators, however, their precise role in the development of acne has not been fully elucidated.
Proteases play an important role in skin homeostasis and various disease conditions [7]. A number of biological activities of proteases are mediated, at least in part, via the activation of its receptor, protease-activated receptors (PARs) [7]. In the human skin, PAR-2 is abundantly expressed by keratinocytes and seems to regulate permeability barrier homeostasis, inflammation, pruritus, pigmentation, and wound healing in response to various endogenous and exogenous serine proteases [7]. Functional PAR-2 is also expressed by keratinocytes of hair follicles and sebaceous glands, fibroblasts, endothelium, afferent neuron, as well as inflammatory cells [29]. During cutaneous inflammation, PAR-2 is activated by endogenous activators such as leukocyte elastase and mast cell tryptase, thereby amplifying inflammation via upregulation of inflammatory mediators [30]. PAR-2 is also known to be activated by various pathogenic organisms with protease activity such as house dust mites, cockroaches, certain bacteria, or parasites [12, 32].
As P. acnes has been reported to produce various proteases, exogenous proteases from P. acnes also can react with PAR-2 on keratinocytes to induce and amplify inflammation in acne.
In this study, we investigated whether the culture supernatant of P. acnes can activate PAR-2 on keratinocytes and then induce the gene expression for pro-inflammatory cytokines, antimicrobial peptides (AMPs), and matrix metalloproteinases (MMPs). In addition, we tested whether protease activity of P. acnes supernatant and PAR-2 are involved in the induction of these inflammatory mediators.
Materials and methods
Samples from patients
A total of six patients were enrolled in this study, with four patients diagnosed with acne vulgaris and two patients with nevus comedonicus. Skin of comedonal lesions were obtained from the face of patients with acne vulgaris and nevus comedonicus by 3-mm punch biopsy. The tissues from acne patients were divided into two groups; one group was fixed in 10% buffered formalin for paraffin embedding, and the other was embedded in optimal cutting temperature compound and immediately frozen in liquid nitrogen, and stored at −80°C for in situ zymography. All biopsies were taken after obtaining informed consents from the patients, and this study was approved by the institutional review board of Severance Hospital, Yonsei University College of Medicine, Seoul, Korea.
Measurement of protease activity by in situ zymography
Frozen sections (5 μm thickness) were rinsed with washing solution (1% Tween 20 in deionized water) and incubated at 37°C for 1 h with 250 μl of BODIPY-Fl-casein substrate (1 μg/μl; EnZCheck® Protease Assay Kits, Molecular Probes, Eugene, OR, USA) in deionized water (2 μl/ml). After removal of excess of substrate solution, nuclei were stained with propidium iodide (PI; Sigma, MO, USA) and the sections were washed with 1% Tween 20 in deionized water. After incubation, the sections were rinsed with washing solution and visualized immediately under confocal microscope C1 Plus (Nikon, Japan).
Immunofluorescence study
Paraffin-embedded tissue sections were deparaffinized in xylene and rehydrated in an ethanol series. Sections were then incubated for 30 min in blocking buffer [1% BSA, 0.1% cold-water fish gelatin in phosphate-buffered saline (PBS)]. For PAR-2 staining, sections were incubated with a 1:250 dilution of a rabbit polyclonal antibody against PAR-2 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) diluted in blocking buffer for 2 h at room temperature. After washing with PBS, the sections were incubated with secondary antibodies consisting of 1:100 dilution of FITC-labeled goat anti-rabbit antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 1 h at room temperature and diluted in blocking buffer. Tissue sections then were washed with PBS, and visualized under a confocal microscope C1 Plus (Nikon, Japan).
Preparation of culture supernatant of P. acnes
Propionibacterium acnes (ATCC 6919, Manassas, VA, USA) was grown in brain heart infusion broth (Difco, Sparks, MD, USA) at 37°C for 24 h under anaerobic condition. The log phase culture was centrifuged at 5,000×g for 15 min. The supernatants were harvested, filtered through a 0.2 μm-pore size filter and then stored at −20°C until used.
Protease activity measurement
To detect the secreted protease activity of P. acnes, the protease activity of the P. acnes culture supernatant was assayed using an EnzChek Protease Assay Kit (Molecular Probes Inc., Eugene, OR, USA). The pH-insensitive green fluorescent BODIPY-FL-conjugated casein was used as a substrate. Trypsin type IX-S (Sigma Co., St Louis, MO, USA), was used as a positive control. BODIPY-FL-conjugated casein solution (10 μg/ml) was prepared with 10 mM Tris–HCl buffer solution (pH 7.8) containing 0.1 mM sodium azide. The exponential phase culture supernatant of P. acnes strain ATCC 6919 was incubated with the substrate in a 96-well plate (OptiPlate 96F; Perkin Elmer, Boston, MA, USA) according to the manufacturer’s protocol for 1 h at 37°C and the fluorescence was measured by the HTS Multilabel Reader (Perkin Elmer, Boston, MA, USA) with an excitation wavelength of 485 nm and an emission wavelength of 530 nm.
Reagents
PAR-2 agonist peptide (AP) (SLIGKV-NH2) was purchased from Peptron (Daejeon, Korea) and dissolved in PBS at a concentration ranged from 0.3125 to 2.5 μM. Serine protease inhibitor, PMSF (phenylmethyl sulfonyl fluoride) was purchased from Sigma Chemicals (St Louis, MO, USA) and dissolved in ethanol, to a final concentration of 1.5 mM. A novel and selective PAR2 antagonist, ENMD-1068, was dissolved in PBS at a concentration ranged from 0.2 to 1.5 mM. The selectivity for PAR-2 in vivo in mice and in vitro in both murine and human cell lines of this PAR-2 antagonist was confirmed previously [16].
Culture and stimulation of keratinocytes
The human keratinocyte cell line HaCaT grown to 80% confluency were maintained in EPILIFE Medium (Gibco, Invitrogen Ltd., Paisley, UK) with human keratinocyte growth supplement (Gibco, Invitrogen Ltd., Paisley, UK) for 24 h before stimulation. HaCaT cells were treated with the culture supernatant of P. acnes (2.5%) or AP (2.5 μM) at the optimal concentration to induce the maximal calcium influx for 3, 6, 12, 18 h at 37°C/5% CO2 in culture medium. In the inhibition study, cells were pretreated with PMSF (1.5 mM) or ENMD-1068 (1.5 mM) for 30 min and 6 h, respectively, and then treated with P. acnes culture supernatant (2.5%) or AP (2.5 μM) under the same condition.
Measurement of Ca2+ mobilization
We assessed PAR-2-mediated intracellular calcium mobilization in keratinocytes using a fluorometric imaging plate reader (FLIPR) calcium assay kit (Molecular Devices, Sunnyvale, CA, USA). Keratinocytes were plated in 96-well plates 24 h prior to the assay and grown to reach confluence at 37°C, 5% CO2. A calcium indicator dye (100 μl per well; FLIPR Calcium 4 assay kit; Molecular Devices, Sunnyvale, CA, USA) was added, and then plates were incubated for 1 h at 37°C. Test compound solutions were prepared in vehicle, consisting of 20 mM HEPES in Hank’s balanced salt solution (HBSS) with 0.1% BSA at pH 7.4. Test solutions were added at 25 μl to each well in duplicate for each experiment. Test compounds were added 30 min prior to the agonist stimulation. Then cells were stimulated with various concentrations of AP or P. acnes supernatant and the fluorescence change measured at 25°C (excitation 485 nm and emission 525 nm). ENMD-1068 was added 30 min prior to the stimulation of cells. Inhibitory effects of ENMD-1068 on calcium responses were expressed as percentage of the reference calcium response induced by either AP (2.5 μM) or P. acnes supernatant (2.5%) alone.
Real-time reverse transcription-polymerase chain reaction (RT-PCR)
After treatment, the cells were harvested using 0.25% trypsin/ethylene diamine tetra acetic acid (EDTA) and resuspended in PBS. Total RNA was extracted from the cells using Trizol reagent (Invitrogen). The complementary DNA was synthesized with 500 ng of total RNA using a TaKaRa RNA PCR kit (AMV) Ver. 3.0 (Takara, Shuzo Shiga, Japan). The primer sets were designed using NCBI Primer-Blast. Real-time PCR was performed using the SYBR Green PCR master mix and the ABI 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using the following parameters: 50°C for 2 min, 95°C for 10 min; 40 cycles of denaturation at 95°C for 15 s; and primer extension at 60°C for 1 min. The expression of β-actin was used as reference. The PCR conditions for human β-actin were denaturation at 94°C for 1 min, annealing at 55°C for 1 min, and elongation at 72°C for 1 min for 30 cycles. All reactions were done in triplicate. Melting-curve analysis of the PCR products was performed at the end of each assay to confirm the specificity of the amplification and absence of primer dimers. The relative amount of all mRNAs was calculated using the comparative threshold (CT) method. Primers in the present study are shown in Table 1.
Statistical analysis
All experiments were carried out in triplicate, and results are expressed as mean ± standard deviation. Comparisons between tests were done by the Student’s t test.
Results
Protease activity was increased in the lesional epithelium of acne vulgaris patients
Propionibacterium acnes has been shown to produce several extracellular virulence factors including proteases which may contribute to the pathogenesis of acne vulgaris. To determine the presence of protease activity in vivo, we performed in situ zymography analysis on the lesions of acne patients. As shown by in situ zymography, protease activity was increased in the lesional epithelium of acne vulgaris patients (Fig. 1b) in comparison with that seen in perilesional epidermis (Fig. 1a). Although the accurate quantitative analysis of specific proteases were not possible by zymography using BODIPY FL casein as a substrate, the increased protease activity in acne lesions compared with perilesional skin is thought to be due, in part, to the P. acnes-derived proteases.

In situ zymography analysis. Total protease activities of the lesions and perilesional normal-appearing skin obtained from acne patients were detected by the degradation of the BODIPY FL casein substrate. Protease activity was observed as the intensity of green fluorescence. Lesions of acne showed higher protease activity compared to the perilesional normal-appearing skin. SC stratum corneum, SG stratum granulosum, SB stratum basale
PAR-2 expression was upregulated in the comedone of acne vulgaris
Next, we assessed the expression pattern of PAR-2 in comedones obtained from patients with acne vulgaris. Comedonal lesion of nevus comedonicus was used as a negative control. As shown in Fig. 2, PAR-2 was highly expressed in the suprabasal layers of follicular epithelium lining the comedone obtained from acne patients. In contrast, the fluorescence intensity of PAR-2 was barely detected in the comedone obtained from patients with nevus comedonicus.

PAR-2 expression in the comedonal lesions of patients with acne vulgaris and nevus comedonicus. PAR-2 immunostaining was performed using rabbit polyclonal anti-PAR-2 (H99, Santa Cruz Biotechnology, CA, USA) antibody and FITC-labeled goat anti-rabbit antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Immunoreactivity of PAR-2 was highly expressed in comedone of patients with acne vulgaris (a) in comparison to that of patients with nevus comedonicus (b). (c) and (d) Enlarged views in the white box in a and b, respectively
Assay of protease activity
To estimate the secreted protease activity of P. acnes, the exponential phase culture supernatant of P. acnes strain ATCC 6919 was obtained and tested with a commercially available protease assay, EnzChek (Molecular Probes) on a casein substrate. Trypsin type IX-S (Sigma Co., St Louis, MO, USA), was used as a positive control. The P. acnes culture supernatant was observed to have 202.5 units/mL protease activity (data not shown).
Proteases released by P. acnes activate PAR-2 in cultured keratinocytes
PAR-2 activation is known to stimulate mobilization of intracellular calcium concentrations ([Ca2+] i ) in keratinocytes [14]. To investigate that proteases from P. acnes are potent activators of PAR-2, we assessed the effects of P. acnes supernatant on [Ca2+] i of keratinocytes. [Ca2+] i was measured by calcium mobilization assay at different concentrations of P. acnes supernatant ranging from 0 to 2.5%. AP induced a significant increase in relative fluorescence intensity with the peak relative fluorescence units (RFU) around 40 s (Fig. 3a). The amplitude of the spike was dependent on the concentration of AP. Similarly, P. acnes supernatant induced an increase of [Ca2+] i , in a dose-dependent manner comparable with AP, as a positive control (Fig. 3b). ENMD-1068 dose-dependently inhibited the calcium influx in response to P. acnes supernatant (Fig. 3c). To clarify whether the effect of P. acnes supernatant on [Ca2+] i increases was mediated by PAR-2, desensitization protocol was performed as described in previous studies [17, 18]. Receptor desensitization by repeated treatment with 2.5% P. acnes supernatant abolished the intracellular calcium signaling in response to AP, suggesting that P. acnes supernatant targets the same receptor with AP (Fig. 3d). These results indicate that P. acnes supernatant-induced intracellular calcium signaling in keratinocytes is mediated by PAR-2 activation.

PAR-2 activation induced by the culture supernatant of P. acnes ATCC6919. HaCaT cells were stimulated with SLIGKV (a) or P. acnes supernatant (b), and intracellular Ca2+ mobilization was measured using a fluorescence method as described in the materals and methods section. Fluorescence was measured over 120 s. ENMD-1068 was incubated with cells 6 h prior to addition of agonist stimulation (c). Desensitization protocol showed that these calcium responses are induced via PAR-2 signaling (d). RFU relative fluorescence units
Propionibacterium acnes culture supernatant stimulates mRNA expression of inflammatory cytokines via protease/PAR signaling in keratinocytes
Next, we investigated whether the activation of PAR-2 by P. acnes culture supernatant is involved in the regulation of inflammatory cytokine gene expression. Keratinocytes were incubated for 18 h with P. acnes culture supernatant (2.5%) at the concentration of maximum response in intracellular calcium levels. Then the transcript level of IL-1α, IL-6, IL-8, and TNF-α in the culture medium was measured by RT-PCR. Propionibacterium acnes supernatant significantly increased the mRNA expression of IL-1α, IL-6, IL-8, and TNF-α in keratinocytes to a comparable level to that of AP-treated keratinocytes, as positive controls (Fig. 4).

Effects of serine protease inhibitor or PAR-2 specific antagonist on P. acnes supernatant-induced mRNA expression of pro-inflammatory cytokines in HaCaT cells. The mRNA expression levels of IL-1α, IL-6, IL-8, and TNF-α in HaCaT cells treated with the culture supernatant of P. acnes for 18 h with or without pretreatment with serine protease inhibitor or selective PAR-2 antagonist were determined using RT-PCR. The mRNA levels were normalized to human β-actin and are presented as fold increases over the control (untreated cells) values. Data are mean ± standard deviation. Asterisks indicate statistically significant differences (*P < 0.05, **P < 0.01) versus the control. Double crosses indicate statistically significant differences (# P ≤ 0.05, ## P ≤ 0.01) between the P. acnes sup-treated and inhibitor-pretreated group
To determine whether this upregulation was due to the protease activity, we assessed the effect of serine protease inhibitor. Pretreatment with a serine protease inhibitor (PMSF) significantly abolished the P. acnes culture supernatant-induced upregulation of IL-1α, IL-8, and TNF-α mRNA expression (Fig. 4). Next we showed that pretreatment with a selective PAR-2 antagonist, ENMD-1068 significantly inhibited the induction of IL-1α, IL-8, and TNF-α mRNA expression in response to P. acnes culture supernatant (Fig. 4). These findings suggest that serine protease activity of P. acnes supernatant induces IL-1α, IL-8, and TNF-α gene expression in keratinocytes partially via PAR-2 activation.
Propionibacterium acnes culture supernatant stimulates mRNA expression of hBD-2 and LL-37 via protease/PAR signaling in keratinocytes
Keratinocytes treated with P. acnes culture supernatant for 12 h showed increased mRNA level of hBD-2 and LL-37 by about 7- and 4.5-fold, respectively, compared to that of untreated cells (Fig. 5a). Since the peak expression of hBD-2 and LL-37 mRNA occurred after 12 h of treatment in our preliminary study (data not shown), this time point was chosen for the next assay to investigate the ability of serine protease inhibitor and PAR-2 antagonist to inhibit AMP gene expression. Pretreatment with a PMSF significantly suppressed the P. acnes supernatant-induced upregulation of hBD-2 and LL-37 mRNA level, suggesting that serine protease activity is involved in this induction (Fig. 5a). To assess the role of PAR-2 in P. acnes culture supernatant-induced AMP gene expression, cells were pretreated with ENMD-1068 and then stimulated with P. acnes culture supernatant for 12 h. The P. acnes supernatant-induced upregulation of hBD-2 gene expression was significantly suppressed by ENMD-1068, whereas, expression of LL-37 gene was suppressed slightly by ENMD-1068, however, the suppression was not statistically significant (Fig. 5a).

Effects of serine protease inhibitor or selective PAR-2 antagonist on P. acnes culture supernatant-induced mRNA expression of antimicrobial peptides in HaCaT cells. The mRNA expression levels of hBD-2 and LL-37 in HaCaT cells treated with the culture supernatant of P. acnes ATCC6919 for 12 h (a) or AP for 3, 6, and 12 h (b) were determined using RT-PCR. The mRNA levels were shown as a fold increase versus 0 h untreated samples (b). Cells were pretreated with serine protease inhibitor or selective PAR-2 antagonist and then stimulated with P. acnes supernatant for 12 h (a). The mRNA levels were normalized to human β-actin and are presented as fold increases over the control (untreated cells) values. Data are mean ± standard deviation. Asterisks indicate statistically significant differences (*P < 0.05, **P < 0.01) vs the control. Double crosses indicate statistically significant differences (# P ≤ 0.05, ## P ≤ 0.01) between the P. acnes sup-treated and inhibitor-pretreated group
Conversely, treatment of keratinocytes with AP significantly increased hBD-2 and LL-37 expression. As shown in Fig. 5b, hBD-2 mRNA level was increased by about 5.5-fold at 3 h and about tenfold at 6 h after the treatment with AP compared to that of untreated cells. LL-37 mRNA level was maximally increased by about eightfold after 3 h treatment with AP. These results suggest that protease activity of P. acnes supernatant stimulates hBD-2 and LL-37 release from keratinocytes partially via activation of PAR-2.
Propionibacterium acnes culture supernatant increases mRNA expression of MMP-1, -2, -3, -9, and -13, but not tissue inhibitor of metalloproteinases (TIMP)-1 and -2
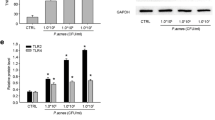
The effect of P. acnes supernatant on the gene expression of MMPs and TIMPs in keratinocytes was investigated by RT-PCR analysis. As shown in Fig. 6a, HaCaT cells treated with P. acnes supernatant for 18 h demonstrated a significant increase in the mRNA level of MMP-1, -2, -3, -9, and -13 by about 9-, 5-, 14-, 17-, and 5.5-fold, respectively, compared to that of untreated cells. However, the gene expression of TIMP-1 and -2 was not affected by P. acnes supernatant after 12 h and 18 h treatment (Fig. 6b).

Effects of serine protease inhibitor or selective PAR-2 antagonist on P. acnes culture supernatant-induced mRNA expression of matrix metalloproteinases in HaCaT cells. The mRNA expression levels of MMP-1, -2, -3, -9, -13, TIMP-1, and -2 in HaCaT cells treated with the culture supernatant of P. acnes ATCC6919 for 12 and 18 h (a, b) or AP for 3, 6, 12, and 18 h (c, d) were determined using RT-PCR. The mRNA levels were shown as a fold increase versus 0 h untreated samples (a–d). Cells were pretreated with serine protease inhibitor or selective PAR-2 antagonist and then stimulated with P. acnes supernatant for 18 h and assessed the mRNA expression levels of MMP-1, -2, -3, -9, and -13 (e). The mRNA levels were normalized to human β-actin and are presented as fold increases over the control (untreated cells) values. Data are mean ± standard deviation. Asterisks indicate statistically significant differences (*P < 0.05, **P < 0.01) versus the control. Double crosses indicate statistically significant differences (# P ≤ 0.05, ## P ≤ 0.01) between the P. acnes sup-treated and inhibitor-pretreated group
Protease activity of P. acnes culture supernatant stimulates mRNA expression of MMP-1, -2, -3, -9, and -13 via PAR-2 activation in keratinocytes
Then we assessed the effect of PMSF or ENMD-1068 on this upregulation (Fig. 6e). The peak expression of MMPs mRNA occurred after 18 h of treatment with P. acnes supernatant (Fig. 6a), this time point was chosen for the next assay to investigate the ability of serine protease inhibitor and PAR-2 antagonist to inhibit MMPs gene expression. Pretreatment with a PMSF significantly inhibited the P. acnes supernatant-induced upregulation of MMP-1, -2, -3, -9, and -13 mRNA level, suggesting that protease activity of P. acnes supernatant is involved in the stimulation of MMPs gene expression. ENMD-1068 also significantly inhibited the upregulation of MMP-1, -2, -3, -9, and -13 mRNA level by P. acnes supernatant. To further support the role of PAR-2 in regulating P. acnes supernatant-induced gene expression of MMPs, we directly stimulated keratinocytes with AP (Fig. 6c). AP significantly increased the mRNA level of MMP-1, -3, -9, and -13 by about 5-, 20-, 18-, and 5-fold, respectively, compared to that of untreated cells after 6 h co-culture. The peaks of these mRNA expressions were at 6 h. For MMP-2, AP significantly increased its mRNA level by about 4.5-fold at 6 h, and its level continued to increase by about 17.5-fold at 18 h. These results suggest that P. acnes supernatant-induced gene expression of MMP-1, -2, -3, -9, and -13 in keratinocytes is mediated the activation of PAR-2.
Discussion
PAR-2 is known to be expressed in the adnexal structures of the skin. As a sensor for exogenous or endogenous danger molecules, PAR-2 activation is tightly regulated by endogenous serine proteases as well as by secreted proteases from various microbes [29, 30]. In previous studies, P. acnes was found to produce various proteases and contribute to tissue injury [8, 10, 28]. In addition to this direct proteolytic effect, proteases derived from P. acnes could be potential activators of PAR-2 and activate innate immune response and inflammation. However, the possible role of PAR-2 in the pathogenesis of acne has not been investigated. Consistent with previous studies, we demonstrated that the culture supernatant of P. acnes strain ATCC 6919 in the exponential phase of growth had proteolytic activity with a casein substrate (data not shown). We also showed that the protease activity was increased in follicular epithelium of acne lesion compared to that of non-lesional normal epidermis by in situ zymography, suggesting that the proteases secreted by P. acnes might be attributed to the higher protease activity in acne lesions. Then we demonstrated that the culture supernatant of P. acnes induced intracellular calcium influx and desensitized PAR-2 activation, suggesting that the proteases derived from P. acnes can proteolytically activate PAR-2 on keratinocytes. To further confirm the involvement of P. acnes proteases in the PAR-2 expression in comedonal lesions of acne, we compared the expression of PAR-2 immunoreactivity in the comedo of acne vulgaris and nevus comedonicus. As a negative control, we used the lesion of nevus comedonicus, because it is characterized by the abnormal keratinization and comedo formation similar to acne vulgaris without colonization of P. acnes [19]. PAR-2 immunoreactivity was upregulated in the follicular wall of comedones of acne vulgaris in compared with that of nevus comedonicus. Taken together we hypothesized that protease/PAR-2 signaling might have an important role in acne pathogenesis. Based on this hypothesis, we investigated whether P. acnes-derived protease activity is able to stimulate keratinocytes to induce inflammatory mediators via PAR-2 activation.
The culture supernatant of P. acnes increased the gene expression of IL-1α, IL-6, IL-8, and TNF-α in keratinocytes. The induction of IL-1α, IL-8, and TNF-α mRNA expression was significantly inhibited by incubating P. acnes culture supernatants with serine protease inhibitor or selective PAR-2 antagonist, suggesting that P. acnes proteases contribute to the upregulation of IL-1α, IL-8, and TNF-α via PAR-2 activation, thereby attributes to the comedo formation [9], neutrophil infiltration [1], and sustained inflammation in the acne pathogenesis. Viable P. acnes in the exponential and stationary phase of growth was found to stimulate the production of TNF-α and granulocyte macrophage-colony stimulating factor (GM-CSF) by human keratinocytes [6]. Viable P. acnes in the stationary phase of growth also reported to contribute to IL-1α production in keratinocytes [6]. In addition, it is well established that P. acnes induces keratinocyte IL-8 production through a TLR2-dependent pathway [24]. In the present study we demonstrated that P. acnes proteases induce the gene expression of proinflammatory cytokines in keratinocytes via activation of PAR-2. These results are agreement with previous studies which reported that several pathogen-derived serine proteases are able to activate PAR-2, thereby inducing inflammation [14, 31]. Protease derived from Aggregatibacter actinomycetemcomitans extracts was demonstrated to upregulate IL-8 and intercellular adhesion molecule-1 expression in human gingival cells via PAR-2 activation [31]. Recent study also showed that serine proteases in mite culture extracts activated PAR-2 on keratinocytes leading to upregulation of IL-8 and GM-CSF [14].
Next, we demonstrated that the P. acnes supernatant increased the gene expression of hBD-2 and LL-37 in keratinocytes, which was blocked by serine protease inhibitor as well as PAR-2 specific inhibitor, suggesting that P. acnes-derived protease contributes to these AMPs induction via PAR-2.
Our findings are consistent with previous studies which demonstrated that PAR-2 stimulation on gingival epithelial cells by Porphyromonas gingivalis-secreted proteases led to an upregulation of hBD-2 expression [5]. In addition, immunoglobulin A in human milk with protease activity has been demonstrated to activate PAR-2 on intestinal epithelium, inducing hBD-2 expression via PAR-2 signaling [2]. Previous study showed that hBD-2 expression is upregulated in acne lesions, suggesting the role of hBD-2 in acne pathogenesis [27]. Nagy et al. found that P. acnes induced hBD-2 in keratinocytes through TLR-2 and -4 and reported that hBD-2 does not have any bacteriostatic or bactericidal effect against any of the four P. acnes strains they used, suggesting that the primary contribution of hBD2 to acne pathogenesis is the regulation of adaptive immunity [24]. In contrast, Nakatsuji et al. demonstrated that high concentrations of synthetic hBD-2 (5–20 μM) had bactericidal effect on P. acnes (ATCC 6919), dose dependently [25]. These observations suggest that hBD-2 has a dual role in the pathogenesis of acne: the protective role against P. acnes colonization when its local concentration rises above the bactericidal level and the role of innate immune responses and cutaneous inflammation. Nakatsuji et al. demonstrated that sebum free fatty acids (FFAs) enhanced hBD-2 mRNA levels up to 45,260-fold in sebocytes and that the FFA-induced sebocyte culture supernatant showed antimicrobial activity against P. acnes [25]. Our results showed that P. acnes culture supernatant or AP treatment of keratinocytes significantly induced the gene expression of hBD-2 by sevenfold or tenfold, respectively, but to a lesser extent than in a previous report [25]. These observations may require further study to investigate the clinical significance of PAR-2-mediated hBD-2 induction in response to P. acnes.
The regulation and role of cathelicidins, which are another group of AMPs, in acne are largely unknown. Cathelicidins are reported to be controlled at transcriptional and post-transcriptional level by infection, inflammation, and 1,25-dihydroxyvitamin D3 [20]. Our results demonstrated that P. acnes-derived protease and PAR-2 signaling induced the upregulation of cathelicidin expression in keratinocytes. The importance of serine protease-mediated proteolysis in the regulation of the antimicrobial effects of cathelicidin has been well known. Recent study observed increased protease activity and cathelicidin expression in the lesions of rosacea patients and suggested that increased protease activity leads to abnormal cathelicidin processing, thereby promotes inflammation in rosacea [34]. However, this protease-induced activation of cathelicidin was post-transcriptional regulation. Taken together, it remains to be determined whether the increased gene expression of LL-37 in response to P. acnes supernatant in our study is a nonspecific reaction to inflammatory cytokines or a more specific response to protease/PAR-2 signaling.
Recently, an active role of MMPs in acne has become an interesting topic of research in acne pathogenesis [11, 26]. Papakonstantinou et al. reported that facial sebum from acne patients contained MMP-1, MMP-13, TIMP-1, and TIMP-2, which are thought to originate in keratinocytes and sebocytes [26]. A recent in vitro study by Jalian et al. demonstrated that P. acnes induced MMP-9, MMP-1, and TIMP-1 transcript in human monocytes [11]. We demonstrated here that keratinocytes are important source of MMPs in acne and that protease activity of P. acnes could induce the expression of several MMPs, including MMP-1, -2, -3, -9, and -13 via PAR-2 activation. We also observed that MMP-1, -2, -3, and -9 were significantly upregulated in acne lesions by immunohistochemistry (data not shown) as previous study which showed that MMP-1, -3, and -9 were markedly elevated in inflammatory acne lesions [15]. Although the mechanism underlying the elevation of MMPs in acne lesions has not been fully elucidated, one report suggested that the enhanced expression of activator protein-1 (AP-1) regulated MMPs in inflammatory acne lesions by demonstrating that AP-1 was activated in acne lesions [15]. Retinoids, which are known to modulate AP-1 expression, have been demonstrated to reduce MMP-9 and -13 in sebum of acne patients [26], whereas, upregulate TIMP-1 in vitro [11], thereby inducing clinical improvement of acne. It was reported that PAR-2 induced c-Jun/AP-1 activation via Jun activation domain-binding protein 1 (Jab1)-mediated signaling pathway [21]. These data led us to suggest that PAR-2-induced AP-1 activation may stimulate MMPs in keratinocytes when exposed to P. acnes with protease activity. The upregulated MMPs might contribute to the acne pathogenesis by inducing inflammation and tissue destruction.
It has been known that there are two distinct types of P. acnes (types I and II) based on serological agglutination tests and cell-wall sugar analysis [13]. Recent studies have profiled these two phenotypes of P. acnes using nucleotide sequencing of the recA housekeeping gene, demonstrating that types I and II represent phylogenetically distinct lineages and the type I strains could be split into two further clusters, types IA and IB [22]. Because of the antigenic and biochemical differences between these phylogenetic types of P. acnes, including the expression of putative Christie–Atkins–Munch–Peterson (CAMP) factor [33], a number of recent studies have attempted to correlate the different phylogenetic types of P. acnes with their different role in acne pathogenesis [23, 24]. The P. acnes strain ATCC 6919, which we used in our study, belongs to the P. acnes type IA strain [33]. A previous microbiological study has found that P. acnes isolates from patients with acne were predominantly type IA [22] and type IA isolates have been demonstrated to have a greater effect on AMPs and proinflammatory cytokines production by keratinocytes [24] and sebocytes [23]. However, the lesions of acne patients are likely to harbor different skin microbes and P. acnes strains other than type IA. From these findings, our study has a limitation that should be taken into account in interpreting the results. It should be noted that our data obtained with only one isolate type of P. acnes, do not necessarily translate into other phylogenetic types of P. acnes and their role in acne pathogenesis. Moreover, in future study it might be worthwhile to investigate the possible involvement of PAR-2 in the pathogenesis of acne using the different other phylogenetic types of P. acnes.
In conclusion, our results indicate that PAR-2 is an important sensor for exogenous danger molecules, such as exogenous proteases from P. acnes, and plays an important role in acne pathogenesis by inducing inflammation, innate immune responses, and acne scar formation. Together, these finding also suggest that specific protease activity or PAR-2 might be a future target for therapeutic intervention for the treatment of acne vulgaris.
References
Baggiolini M, Loetscher P (2000) Chemokines in inflammation and immunity. Immunol Today 21:418–420
Barrera GJ, Portillo R, Mijares A et al (2009) Immunoglobulin A with protease activity secreted in human milk activates PAR-2 receptors, of intestinal epithelial cells HT-29, and promotes beta-defensin-2 expression. Immunol Lett 123:52–59
Bojar RA, Holland KT (2004) Acne and Propionibacterium acnes. Clin Dermatol 22:375–379
Bruggemann H (2005) Insights in the pathogenic potential of Propionibacterium acnes from its complete genome. Semin Cutan Med Surg 24:67–72
Dommisch H, Chung WO, Rohani MG et al (2007) Protease-activated receptor 2 mediates human beta-defensin 2 and CC chemokine ligand 20 mRNA expression in response to proteases secreted by Porphyromonas gingivalis. Infect Immun 75:4326–4333
Graham GM, Farrar MD, Cruse-Sawyer JE et al (2004) Proinflammatory cytokine production by human keratinocytes stimulated with Propionibacterium acnes and P. acnes GroEL. Br J Dermatol 150:421–428
Hansen KK, Oikonomopoulou K, Baruch A et al (2008) Proteinases as hormones: targets and mechanisms for proteolytic signaling. Biol Chem 389:971–982
Hoeffler U (1977) Enzymatic and hemolytic properties of Propionibacterium acnes and related bacteria. J Clin Microbiol 6:555–558
Ingham E, Eady EA, Goodwin CE et al (1992) Proinflammatory levels of interleukin 1 alpha-like bioactivity are present in the majority of open comedones in acne vulgalis. J Invest Dermatol 98:895–901
Ingram E, Holland KT, Gowland G et al (1983) Studies of the extracellular proteolytic activity produced by Propionibacterium acnes. J Appl Bacteriol 54:263–271
Jalian HR, Liu PT, Kanchanapoomi M et al (2008) All-trans retinoic acid shifts Propionibacterium acnes-induced matrix degradation expression profile toward matrix preservation in human monocytes. J Invest Dermatol 128:2777–2782
Jeong SK, Kim HJ, Youm JK et al (2008) Mite and cockroach allergens activate protease-activated receptor 2 and delay epidermal permeability barrier recovery. J Invest Dermatol 128:1930–1939
Johnson JL, Cummins CS (1972) Cell wall composition and deoxyribonucleic acid similarities among the anaerobic coryneforms, classical propionibacteria, and strains of Arachnia propionica. J Bacteriol 109:1047–1066
Kato T, Takai T, Fujimura T et al (2009) Mite serine protease activates protease-activated receptor-2 and induces cytokine release in human keratinocytes. Allergy 64:1366–1374
Kang S, Cho S, Chung JH et al (2005) Inflammation and extracellular matrix degradation mediated by activated transcription factors nuclear factor-kappaB and activator protein-1 in inflammatory acne lesions in vivo. Am J Pathol 166:1691–1699
Kelso EB, Lockhart JC, Hembrough T et al (2006) Therapeutic promise of proteinase-activated receptor-2 antagonism in joint inflammation. J Pharmacol Exp Ther 316:1017–1024
Kim J (2005) Review of the innate immune response in acne vulgaris: activation of Toll-like receptor 2 in acne triggers inflammatory cytokine responses. Dermatology 211:193–198
Kim J, Ochoa MT, Krutzik SR et al (2002) Activation of toll-like receptor 2 in acne triggers inflammatory cytokine responses. J Immunol 169:1535–1541
Kurokawa I, Nakai Y, Nishimura K et al (2007) Cytokeratin and filaggrin expression in nevus comedonicus. J Cutan Pathol 34:338–341
Lai Y, Gallo RL (2009) AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol 30:131–141
Luo W, Wang Y, Hanck T et al (2006) Jab1, a novel protease-activated receptor-2 (PAR-2)-interacting protein, is involved in PAR-2-induced activation of activator protein-1. J Biol Chem 281:7927–7936
McDowell A, Valanne S, Ramage G et al (2005) Propionibacterium acnes types I and II represent phylogenetically distinct groups. J Clin Microbiol 43:326–334
Nagy I, Pivarcsi A, Kis K et al (2006) Propionibacterium acnes and lipopolysaccharide induce the expression of antimicrobial peptides and proinflammatory cytokines/chemokines in human sebocytes. Microbes Infect 8:2195–2205
Nagy I, Pivarcsi A, Koreck A et al (2005) Distinct strains of Propionibacterium acnes induce selective human beta-defensin-2 and interleukin-8 expression in human keratinocytes through toll-like receptors. J Invest Dermatol 124:931–938
Nakatsuji T, Kao MC, Zhang L et al (2010) Sebum free fatty acids enhance the innate immune defense of human sebocytes by upregulating beta-defensin-2 expression. J Invest Dermatol 130:985–994
Papakonstantinou E, Aletras AJ, Glass E et al (2005) Matrix metalloproteinases of epithelial origin in facial sebum of patients with acne and their regulation by isotretinoin. J Invest Dermatol 125:673–684
Philpott MP (2003) Defensins and acne. Mol Immunol 40:457–462
Puhvel SM, Reisner RM (1972) The production of hyaluronidase (hyaluronate lyase) by Corynebacterium acnes. J Invest Dermatol 58:66–70
Rattenholl A, Steinhoff M (2003) Role of proteinase-activated receptors in cutaneous biology and disease. Drug Dev Res 59:408–416
Rattenholl A, Steinhoff M (2008) Proteinase-activated receptor-2 in the skin: receptor expression, activation and function during health and disease. Drug News Perspect 21:369–381
Shimada T, Sugano N, Ikeda K et al (2009) Protease-activated receptor 2 mediates interleukin-8 and intercellular adhesion molecule-1 expression in response to Aggregatibacter actinomycetemcomitans. Oral Microbiol Immunol 24:285–291
Shpacovitch V, Feld M, Bunnett NW et al (2007) Protease-activated receptors: novel PARtners in innate immunity. Trends Immunol 28:541–550
Valanne S, McDowell A, Ramage G et al (2005) CAMP factor homologues in Propionibacterium acnes: a new protein family differentially expressed by types I and II. Microbiology 151:1369–1379
Yamasaki K, Di Nardo A, Bardan A et al (2007) Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med 13:975–980
Zouboulis CC, Eady A, Philpott M et al (2005) What is the pathogenesis of acne? Exp Dermatol 14:143–152
Conflicts of interest
None declared.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lee, S.E., Kim, JM., Jeong, S.K. et al. Protease-activated receptor-2 mediates the expression of inflammatory cytokines, antimicrobial peptides, and matrix metalloproteinases in keratinocytes in response to Propionibacterium acnes . Arch Dermatol Res 302, 745–756 (2010). https://doi.org/10.1007/s00403-010-1074-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00403-010-1074-z