
Abstract
Parahydrogen-induced polarization is a hyperpolarization method for enhancing nuclear magnetic resonance signals by chemical reactions/interactions involving the para spin isomer of hydrogen gas. This method has allowed for biomolecules to be hyperpolarized to such a level that they can be used for real time in vivo metabolic imaging. One particularly promising example is fumarate, which can be rapidly and efficiently hyperpolarized at low cost by hydrogenating an acetylene dicarboxylate precursor molecule using parahydrogen. The reaction is relatively slow compared to the timescale on which the hyperpolarization relaxes back to thermal equilibrium, and an undesirable 2nd hydrogenation step can convert the fumarate into succinate. To date, the hydrogenation chemistry has not been thoroughly investigated, so previous work has been inconsistent in the chosen reaction conditions in the search for ever-higher reaction rate and yield. In this work we investigate the solution preparation protocols and the reaction conditions on the rate and yield of fumarate formation. We report conditions to reproducibly yield over 100 mM fumarate on a short timescale, and discuss aspects of the protocol that hinder the formation of fumarate or lead to irreproducible results. We also provide experimental procedures and recommendations for performing reproducible kinetics experiments in which hydrogen gas is repeatedly bubbled into an aqueous solution, overcoming challenges related to the viscosity and surface tension of the water.
Similar content being viewed by others


1 Introduction
Nuclear magnetic resonance (NMR) and magnetic resonance imaging (MRI) are among the most widely applied and versatile analytical and diagnostic techniques in chemistry, biophysics, medicine and pharmacology. The major challenge for NMR or MRI is their low sensitivity, caused by the tiny interaction between nuclear spins and magnetic fields and the resulting small polarization of spin states in thermal equilibrium, which is typically on the order of 10–5 and below. To solve this sensitivity problem, hyperpolarization techniques, which transfer spin-polarization from a highly polarized spin-reservoir to the nuclear spins, were developed. Dynamic nuclear polarization [1,2,3] (DNP) employs for example stable radicals. Chemically induced dynamic nuclear polarization (CIDNP) [4, 5], optical nuclear polarization (ONP) [6, 7] and its time resolved variants, stimulated nuclear polarization (SNP) [8,9,10] or spin exchange by optical pumping (SEOP) [11, 12] use optically created transient triplet states of noble-gasses. Despite the high signal enhancements obtainable by these techniques, they generally require sophisticated and expensive apparatus to achieve the hyperpolarization.
An alternative, less expensive hyperpolarization technique known as parahydrogen-induced polarization (PHIP) [13,14,15] employs hydrogenation reactions of an unsaturated precursor molecule with parahydrogen (p-H2) gas as the source of the hyperpolarization. A more recent variation of this technique, signal amplification by reversible exchange (SABRE) [16] employs a reversible coordination of p-H2 and a target molecule to an organometallic complex. Para-enriched hydrogen gas can be easily produced by cooling hydrogen gas to cryogenic temperatures (liquid nitrogen or below) in the presence of a catalyst (typically charcoal or iron oxide), which accelerates the spin-conversion process by which an H2 molecule can transition from an ortho to a para state. Without those catalysts the back-reaction to orthohydrogen (o-H2) is slow and p-H2 can be easily stored for several days at ambient temperature [17]. PHIP can lead to NMR signals enhanced by factors of 10,000 and more, compared to the thermal polarization at high field. The resulting NMR signal pattern of PHIP depends strongly on the experimental protocol. In PASADENA [15], the hydrogenation is performed inside—and in ALTADENA [18] experiments, outside—the NMR magnet. PHIP experiments were successfully employed for the detailed investigation of mechanistic problems in organic and inorganic synthesis [19,20,21], for monitoring chemical processes in microreactors, [22,23,24] in time-domain detected experiments [25, 26], or in field-cycling [27, 28] and ultra-low-field NMR experiments [29,30,31]. They were employed for hyperpolarization of amino acids and peptides [32,33,34,35,36,37,38] and in MRI [39,40,41,42,43,44,45,46,47]. To prolong the application window of PHIP, which is limited by the lifetime of the hyperpolarization, it is advantageous to transfer the hyperpolarization from the hyperpolarized protons to spin-1/2 X-nuclei such as 13C. Glöggler et al. [48, 49] reported a pulsed experiment, which can nearly completely transfer the hyperpolarization from the protons to the 13C. The Side Arm Hydrogenation (PHIP-SAH)50,51 protocol greatly expanded the range of PHIP-polarizable molecules, which allowed for the hyperpolarization and subsequent in vivo detection of 13C-labeled pyruvate and its metabolic product lactate in a 1T MRI scanner [52].
Fumaric acid is an organic compound present in all aerobic organisms, and the analysis of its behavior and interactions in the body are of great scientific interest [53]. It can be found in beverages and food products [54], in animal feed, in the cosmetic industry, and in medicines [55]. In the latter case it is used for the clinical treatment of psoriasis and its methyl ester (dimethyl fumarate) is used in multiple sclerosis therapy since it was approved in 2013. Its salts, the fumarates, are a side product of the urea cycle in which ammonia is converted into urea via fumarate [56]. Fumarate is also a key component of the citric acid cycle, through which all aerobic organisms produce energy and biologically important intermediates by the oxidation of organic substances in a series of chemical reaction steps. One step in this cycle is the dehydrogenation of succinate to fumarate by the enzyme succinate dehydrogenase, which in turn is converted to malate by the enzyme fumarase [57]. This last step is of particular interest for hyperpolarized in vivo imaging, and has been used to probe the efficacy of cancer therapies [58, 59]. Employing 13C-DNP hyperpolarization, Gallagher et al. [60] showed that malate production from fumarate is increased in treated lymphoma cells and tumors by tumor cell necrosis. This seminal discovery initiated intense research into the applications of hyperpolarized fumarate as a real-time metabolic contrast agent for MRI [59, 61,62,63,64,65,66]. Eills and coworkers [67,68,69,70,71] realized that PHIP is a convenient method to efficiently hyperpolarize fumarate and that it can be employed for MRI, which has since been demonstrated by Stewart et al. [72] Some advantages of the PHIP-based approach are that the production of the hyperpolarized substrates is faster, at least an order of magnitude less expensive than the DNP approach, scalable with respect to the amounts of hyperpolarized molecules that can be produced, and that DNP apparatus is not required.
Until now papers in which PHIP-polarized fumarate is developed as an MRI contrast agent report different reaction conditions for the hydrogenation step. To further develop this technique into a clinically applicable one it is important to gain further understanding in and optimize the interplay of chemical kinetics, spin-dynamics and relaxation in this system. In the present study the formation of fumarate by hydrogenation and the subsequent reaction to succinate are investigated by means of nuclear magnetic resonance (NMR), and in particular we investigate the reaction dependence on conditions and the sample preparation protocol. By working with normal H2 gas (i.e. not para-enriched), or by observing hyperpolarized of molecules containing a symmetry carbon-13 nucleus to generate observable signals.
2 Materials and Methods
2.1 Sample Preparation
As the final perspective of these studies are biophysical or medical applications, all reactions were performed in aqueous solution. The monopotassium salt of acetylene dicarboxylic acid (K-ADC) was used as a water-soluble precursor.
The production and detection of the hydrogenation is done in two steps (Fig. 1). The first step (Fig. 1a) involves the reduction of a carbon–carbon triple bond (C≡C) in ADC to a double bond (C=C) to form fumarate, and the second step is the reduction of the double bond to a carbon–carbon single bond (C–C), succinate (Fig. 1b). The hydrogenations are performed in a deuterated water solvent, with a ruthenium catalyst (Pentamethylcyclopentadienyltris (acetonitrile)ruthenium(II) hexafluorophosphate (Ru-Cat)) and sodium sulphite (Na2SO3). K-ADC was purchased from TCI with a purification of > 95%. All other chemicals were obtained from Sigma-Aldrich (Darmstadt, Germany) and employed without further purification.

Reaction scheme of the two-step hydrogenation of ADC (M=Na, K or H) via fumarate (a) to succinate (b)
2.2 NMR and Hydrogenation Protocol
NMR experiments were performed in 5 mm O.D. 528-TR-7 NMR tubes from Rototec Spintec on a Bruker Avance III HD NMR spectrometer at 11.7 T (proton frequency 500 MHz). For hydrogenation reactions, hydrogen gas was bubbled through a glass capillary into the samples at a pressure of 7 bar. To generate para-enriched hydrogen gas for PHIP experiments, we employed a home-built PHIP setup, described in detail in Ref. [73].
For all NMR experiments a 600 μL sample was used. To quantify the concentrations of species from the NMR signals, an external DSS (sodium trimethylsilyl propanesulfonate) standard was used. The standard sample concentrations are given in Table 1, and the standard sample pH was 7.8 and temperature was 88 °C. These values were used for each sample, unless otherwise stated. The temperature was calibrated via the procedure described in Sect. 2.8.
In the kinetics plots, each data point represents concentration of the species (fumarate or succinate) as determined from the peak integrals from the 1H NMR spectra. The peak integral, which indicates the proton concentration, was divided by a factor of 2 to give the fumarate concentration, and 4 to give the succinate concentration, and these are the values that are plotted. For succinate this is inaccurate since a large proportion of the succinate is partially deuterated (i.e. most succinate molecules do not have 4 protons, see Supporting Information), and we discuss this more in the Discussion.
2.3 Experiments to Determine the Reaction Temperature Dependence
All substances were weighed together and 3 mL D2O was added. To dissolve the sample at a neutral pH, NaOH dissolved in D2O (2 M) was added to a concentration of 250 mM to compensate for the acidic starting material. The sample was sonicated at 60 °C until it had completely dissolved. The concentrations of each substance are listed in Table 1. For the evaluation of the correct calculation of the concentrations the total volume (VH2O + VNaOH) was taken into account. After dissolving, the pH value was measured using a digital glass-electrode pH meter HI 221 from Hanna Instruments, and adjusted to pH 7.8 with aqueous deuterium chloride and sodium deuteroxide solution. Afterwards, the sample was filtered twice through a 0.22 µm, 13 mm syringe filter from Carl Roth to remove possible sediments inside the sample and to reduce the foaming during the bubbling.
2.4 Experiments to Determine the Optimal Sodium Sulphite Concentration
In these experiments the sample preparation was performed as described in Sect. 2.3, but here the concentration of sodium sulphite was varied. The concentrations used were: 0, 50, 150, 250 and 350 mM, while the concentrations of K-ADC and Ru-Cat were fixed at 250 and 7 mM, respectively. The pH was adjusted to 7.8 and the sample temperature was 88 °C.
2.5 Determination of the pH Influence on the Reaction Behavior
For these experiments, the sample preparation was slightly modified to ensure the pH at which the catalyst was dissolved, which helped to improve experimental reproducibility (see Discussion for further details). The K-ADC and Na2SO3 were dissolved in D2O, the pH was adjusted to a given value, and the catalyst was added only after the other substances had completely dissolved. In some experiments, where stated, the pH was then readjusted to the desired value after the catalyst had dissolved.
2.6 Investigation of the Sample Stability
For the analysis of the sample stability, the sample was prepared as described in Sect. 2.5.
2.7 PHIP Experiment
For the PHIP experiment a sample was prepared as described in Sect. 2.5. As opposed to the kinetics experiments, the PHIP experiment only used a single hydrogen bubbling interval, followed by a helium bubbling interval to stop the ongoing reaction. After a delay of 20 s, a Singlet-to-Magnetization (S2M) pulse sequence was used to generate proton magnetization from singlet order, and the signal was acquired. After a 30 min delay for relaxation, a thermal equilibrium spectrum was acquired following a 90° excitation pulse.
2.8 Temperature Calibration of the NMR Probe
The sample temperature inside the probe differs from the nominal temperature, set at the spectrometer. To determine the real temperature the chemical shift difference between DSS and water was employed as a temperature standard. The instrument was set to temperatures from 25 to 90 °C and the chemical shift difference between the DSS and the water signal was measured. Figure 2 shows the calibration curve. There is a direct linear correlation between the nominal and the real temperature. All temperatures stated henceforth refer to the sample temperature.

Temperature calibration curve showing the instrument temperature read by a sensor directly below the sample tube plotted against the temperature of the sample which was determined by the chemical shift difference of the DSS and water
3 Results and Discussion
The aim of this work was to optimize the production of fumarate by hydrogenation of ADC salts, which includes suppressing the subsequent over-reduction to succinate. To achieve the highest possible concentration of fumarate the kinetics of this reaction are investigated and optimized. Parameters that influence the reaction are the sample preparation, sample pH, the reaction temperature, the concentration of the reactants, the gas stream of hydrogen (i.e. the shape, size and speed of the bubbles passing through the solution), and the pressure of hydrogen. While investigating one reaction parameter, it was important to implement experimental controls to keep the other parameters constant. In the first part of the results Sect. (3.1–3.4), we describe the methods developed to give reproducibility in the experiments and in the second part (3.5–3.10) we describe the experimental results and discuss them.
3.1 Setup Optimization for Kinetic Measurements in Aqueous Solution
Bubbling hydrogen into aqueous solutions is far more challenging than experiments in typical organic solvents like methanol, owing to the higher viscosity and surface tension of water leading to a tendency to form foams and persistent bubbles, as well as for the solution to stick to the sides of the NMR tube. These challenges cause problems both in the field-homogeneity and in the measured signal intensities because the amount of solution inside the RF-coil varies. While the foaming problem has been solved previously using a defoamer [74], which suppresses or at least reduces the foaming, the other problems needed more attention. In the following, we describe the setup optimizations that made measurements in aqueous solutions easier and more reproducible.
3.2 Cleaning of the NMR Tubes
The cleaning of the tubes is an important step preceding the sample preparation since the smallest amounts of impurities results in water drops as well as gas bubbles sticking to the wall of the NMR tubes and preventing reaction solution from flowing down the tube properly. To avoid this problem, we adopted the following cleaning practice: The used tubes were rinsed with deionized water twice and then filled with aqua regia. The tubes were then heated in a heating block at 60 °C for a minimum of 1 h. We note that it is important not to overfill the tubes, because the impurities can cause the formation of gases and the aqua regia might spill over. Afterwards, the tubes were rinsed again with deionized water and with acetone twice to make sure that all the aqua regia was removed. Finally, the tubes were dried in air.
3.3 Insertion of Centering Guides
In our initial setup we employed a PTFE capillary for hydrogen gas bubbling. However, due to its flexibility, it was not possible to keep it straight in the NMR tube. Moreover, the capillary moved inside the tube while bubbling, so that centering of the PTFE capillary could not be guaranteed. The PTFE capillary was replaced by a sintered glass capillary, and while this solved the deformation problem, a new problem arose: the capillary stuck to the wall of the NMR tube due to adhesion, leading to small and irreproducible bubbles in the water, independent of the gas type. Additionally, some of these small bubbles stuck to the capillary or between the capillary and the wall of the tube and influenced the shim of the instrument and hence the spectral resolution. To solve this new problem, we fixed the capillary in the center of the NMR tube by means of centering guides. Those guides have to be chemically stable and should interfere as little as possible with the sample, the gas flow, and the shim of the instrument. Silicon inserts fulfill those requirements and we tried various spacer geometries (Fig. 3a). In a first attempt, 4 mm diameter silicon discs were punched out of a silicon mat with a thickness of 2 mm. A small hole was made in the discs for the capillary. In our test experiments we found that these simple discs interfered too much with the gas flow and caused a pressure rise in the lower part of the tube. For this reason, we removed parts of the discs and created the threefold shape, to make sure that the gas could leave the NMR tube and to let rising water flow back down again. Two of these shaped discs were threaded onto the top of the capillary above the solvent level. This optimization improved the bubbling behavior, the reproducibility, and the shim of the instrument. After several kinetic measurements it turned out that water may be stuck above these centering guides due to some gas bubbles under the guides, leading to a loss of sample after some time. In a third design, silicon strips of 2 mm × 2 mm × 4 mm (HxWxL) were used instead of discs with cut-outs. Three of these strips were arranged in a star-like pattern when looked at from above. A schematic illustration of the arrangement of the strips threaded onto the capillary and the resulting bubbling is depicted in Fig. 3a, b photo is shown in Fig. 3c. After this modification, the problems emerging from the bubbling in water were solved and the setup was ready for the optimization of the pulse program used for the kinetic studies in water.

Centering of the capillary in the NMR tube: a Different spacer designs for centering the capillary; b Illustration of the sample tube with a centered capillary; c Photo of the tube with spacers and capillary inside
3.4 Optimization of the Pulse Program
For the measurements, a pseudo 2D experiment was designed with a pulse program containing seven building blocks (see Fig. 4). These building blocks can be arranged individually for each experiment. This leads to a high flexibility of these experiments and a wide range of application possibilities. The building blocks have different functions. The whole timing of the program is controlled by the spectrometer, employing our previously described automized setup [73].

Schematic illustration of the building blocks used in the pulse program. For kinetics experiments, the whole sequence was run once, with the hydrogen bubbling and signal acquisition block repeated many times
The red block is used as a vacuum step to remove residual gases before gas bubbling, which guarantees fast and pure bubbling of the desired gas. A similar step is placed before the H2 / p-H2 bubbling block shown in purple which is the main part of the experiment. Here the possible repetition of the measurement needed for kinetic studies can be performed. To make sure that no other gases in the sample interfere with the hydrogen, a He bubble block (green) is applied after the initial vacuum period to degas the sample. The He bubble block is repeated at the end of the experiment to get rid of the remaining hydrogen in the sample to stop possible ongoing reactions.
The first purple block is the Dummy Scan block and is used to bubble helium through the sample to avoid changes in the temperature due to non-tempered gas and to lead to a stable temperature during the bubbling and the actual measurement.
Owing to its modular design, the constructed pulse program is versatile and can be used for a single 1H measurement, or for kinetic studies. In this work to collect the kinetics data for each sample the entire sequence was run once, but the 2nd purple block was repeated many times. The bubbling time was 5 s and the acquisition time was 6.5 s. The time shown in the kinetics plots in the following sections represents just the H2 bubbling time; the acquisition time of 6.5 s is not taken into account.
3.5 Influence of the Temperature on the Reaction
Samples were prepared (see Materials and Methods) with a pH value of 7.8, and hydrogenation reactions were carried out at different temperatures: 60, 70, 80, 88 and 95 °C. The resulting concentration courses are depicted in Fig. 5.

Temperature dependence of the formation of fumarate (upper) and succinate (lower); the inset plot shows the maximal concentration reached and the time it took against the temperature. The NMR spectra were acquired with an excitation pulse of 9° after 5 s bubbling with H2
It is clear that the reaction rate depends strongly on temperature. The maxima were reached after 150 s (95 °C), 165 s (88 °C), 380 s (80 °C), 890 s (70 °C) and 2090s (60 °C). The optimum temperature was found to be 88 °C, for which 123 mM fumarate was reached, which corresponds to 53.4% yield. Above 88 °C the catalyst starts to degrade and only a maximum of 90 mM of fumarate was achieved. A suggested reaction mechanism modified from [67] can be found in the Supporting Information (Fig. S1).
3.6 Influence of the Sodium Sulphite Concentration on the Reaction
A sample at pH 7.8 was prepared as described in the Materials and Methods, and experiments were run at 88 °C. The resulting kinetic curves are depicted in Fig. 6 and show clearly the influence of the sodium sulphite on the reaction rate. The reaction without any Na2SO3 is very slow and doesn’t reach a maximum in the measured time frame. Also, many side products like the cis-configuration (maleate) can be detected when Na2SO3 is not included. A spectrum is given in the Supporting Information (Fig. S2). An interesting side product from this reaction is one in which the protons end up in geminal positions, which can lead to geminal-PHIP, and this is discussed in a paper by L. Dagys et al. [75]. The other curves show less significant differences. Using 50 mM of Na2SO3 leads to a concentration of 123 mM fumarate which correspond to a yield of 54.4%. A slightly higher turnover of 55.4% and faster reaction kinetics 205 s vs. 345 s to maximum concentrations could be reached with 150 mM Na2SO3. These results indicate that both the reaction rate and yield depend on Na2SO3 concentration. The reaction with 350 mM reaches its maximum already after 145 s but shows a lower turnover to fumarate with just 114 mM (49.2%) formed. The reaction with 250 mM Na2SO3 is only 20 s slower compared to 350 mM Na2SO3 but achieves a turnover of 123 mM (53.4%). The reaction with 150 mM showed the best yield. A compromise must be found between the yield and the reaction time, since in experiments involving para-enriched hydrogen the hyperpolarized signals would be relaxing during the hydrogenation. As a compromise between reaction speed and yield 250 mM of Na2SO3 was used in subsequent experiments.

Influence of the sodium sulphite concentration on the formation of fumarate (upper) and succinate (lower); the inset plot shows the maximal concentration reached and the time it took against the sodium sulphite concentration. The NMR spectra were acquired with an excitation pulse of 9° after 5 s bubbling with H2
3.7 Analysis of the Optimal pH Value
Samples of differing pH were prepared as described in the Materials and Methods, and experiments were carried out at 88 °C. The pH values before and after dissolving the catalyst in the solution are shown in Fig. 7 in the legend, along with the kinetics results. The kinetic curve for pH 5 shows just a small formation of fumarate and no formation of succinate (see Fig. 7). When the pH is close to neutral, the reaction speeds up which is visible in the kinetics measurement of a reaction solution at pH 7. Here, a maximum is reached after 1075 s with a yield of already 91 mM fumarate (39.8%). After adjusting the pH to 7.2 the reaction speeds up even more and a maximum of 108 mM fumarate (47.6%) was reached after only 155 s. By increasing the pH even further to 7.7 again a change can be observed. Here, the conversion leads to a concentration of 125 mM fumarate which correspond to a turnover of 54.3%. Looking at the succinate formation it is interesting that despite the difference in the fumarate reaction, the formation of succinate is similar for both a pH of 7.2 and 7.7. Increasing the pH further to pH 8 and 10 led to similar fumarate and succinate reaction kinetics between the two. Concentrations of 96 mM (41.6%) and 91 mM (39.5%) fumarate were achieved in nearly the same times (180/175 s). Additionally, the formation of succinate is also nearly equal and reaches a maximum concentration of 70 mM.

Influence of the pH on the formation of fumarate (left) and succinate (right); the inset plot shows the maximal concentration reached and the time it took against the pH. The NMR spectra were acquired with an excitation pulse of 9° after 5 s bubbling with H2
It is evident that the pH of the sample is of fundamental importance for fumarate formation. It is also important is to consider the pH change of the sample by adding the catalyst which also has a significant impact on the final pH value. Here, it was observed that under basic conditions the pH change can be drastic when dissolving the catalyst. For example, when the prepared pH of the K-ADC/ Na2SO3 solution was 8.1, after dissolving the catalyst the pH was 11.4.
Seeing that small changes in the pH have a large effect on the reaction, and noting that the pH changes while all of the chemicals are dissolving, we modified the sample preparation for all subsequent experiments to be more robust. We dissolved the K-ADC and Na2SO3, adjusted the pH, and then added the catalyst as a final step, since we suspect it is only the catalyst that is sensitive to pH.
To understand how pH changes during sample preparation affect the results, we prepared samples, including catalyst, at a certain pH and then adjusted the pH to 7.8 since this value showed the best results. The kinetics results are shown in Fig. 8. It is visible that dissolving the sample at a lower pH and adjusting it upwards after dissolution produces good results, while dissolving the sample at a high pH and adjusting it downwards leads to poor reaction rate and yield. This leads to the conclusion that the catalyst degrades at high pH which is not reversible, while dissolving the sample at reduced pH does not significantly change the catalyst.

Kinetic curves acquired on samples for which the pH was adjusted after all chemicals were dissolved. Formation of fumarate (upper) and succinate (lower). The NMR spectra were acquired with an excitation pulse of 9° after 5 s bubbling with H2
We also performed a kinetics experiment using the optimal solution parameters but including 500 mM phosphate buffer in the reaction solution to maintain pH 7.8 during the reaction. This reduced the rate of fumarate formation by a factor of about 2, and the yield was just 106 mM.
3.8 Stability Experiments
To determine how stable the precursor solutions are, a stock sample solution was prepared at pH 7.8. The first sample was measured directly after preparation, another one was left at room temperature for 1.5 h before measuring, and the last one was stored for 1 h in the magnet at a temperature of 88 °C before starting the measurement. Figure 9 shows the results of the stability tests. At room temperature, the sample is stable after 1.5 h as can be deduced from the unchanged reaction kinetics in Fig. 9. Storage at 88 °C for 1 h lead to a turnover of just 73 mM fumarate (29.1%) compared to the original sample with 125 mM fumarate (49.8%) turnover and the sample stored 1.5 h at room temperature which resulted in 126 mM fumarate (50.3%). It seems that storage at elevated temperature affects the formation of fumarate but not succinate.

Kinetic curves acquired on a sample at pH 7.8 and 88 °C under different storage conditions prior starting the experiment. Formation of fumarate (upper) and succinate (lower);
Finally we observed (not shown) that freezing the precursor solutions for several days and then thawing for use also has no measurable effect on reaction kinetics.
The NMR spectra were acquired with an excitation pulse of 9° after 5 s bubbling with H2.
3.9 PHIP Experiment
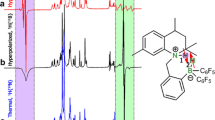
Having optimized reaction conditions using n-H2 gas, we demonstrated the results in an experiment using p-H2 to generate hyperpolarized signals. It is challenging to observe hyperpolarized signals of fumarate since the protons remain chemically equivalent, and so the spin order remains mostly locked in the nonmagnetic singlet state. To liberate the singlet order as hyperpolarized magnetic signals, a Singlet-to-Magnetization (S2M) pulse sequence was used. The sequence takes advantage of the weak magnetic inequivalence between the two protons due to their asymmetric J-couplings to a 13C nucleus to yield inphase magnetization for the 2.2% of molecules containing a 13C isotope at one of the carboxylic groups. In Fig. 10 the hyperpolarized and the thermal equilibrium spectra of three separate measurements are depicted. We measured enhancement factors of 3511, 3538 and 3487 for the 2.2% of molecules contributing to the signal. With these results, we can show the good reproducibility of the investigated setup and sample preparation. The reduced hyperpolarization efficacy at high field, compared to previous results at low-field is mainly attributed to a loss of singlet order, as discussed in ref. [76].

Reaction scheme of the hydrogenation of ADC to fumarate with para-enriched hydrogen. An S2M pulse sequence is applied to the protons to excite a hyperpolarized NMR signal. The red, yellow and green spectrum are the hyperpolarized spectrum after 90 s bubbling with p-H2., and the turquoise, pink and blue spectra are the thermal equilibrium spectra acquired after the hyperpolarized signals had fully relaxed
4 Further Discussion
A closer look at the succinate resonance in the 1H spectra of a hydrogenated sample reveals a three-peak structure that indicates partially deuterated succinate is present in higher concentration than fully protonated succinate. Figure S3 in the Supporting Information shows the three peaks from a reaction sample, and two more spectra of the same sample after pure succinic acid was added. Only one peak increased after adding succinic acid, which suggests that the two other peaks are from partially deuterated succinate molecules and the protons are exhibiting an isotope shift. This was confirmed by deuterium spectra which show a multiplet peak corresponding to partially deuterated succinate. The deuterium must come from molecules of D2O, although the mechanism is not known. This however has an important implication: the concentrations of succinate reported in the Results come from taking integral of all three succinate peaks and assuming only fully protonated succinate is present. Since much of the succinate contains less than 4 protons, the true succinate concentrations are higher than reported. We have not investigated this further to provide more quantitative results because the succinate is an undesirable side product of the reaction, although we note that this catalytic route would likely not be suitable for generating PHIP-polarized succinate due to the degree of deuteration.
In many of the results the overall concentration of reaction products (fumarate + succinate) appears to decrease after a time. This is partially because the true succinate concentration is being underestimated. However, we do observe that during the reaction a polymerization takes place and after some time a solid black precipitate can be seen in the solutions (see figure S4). We do not know the origin of this solid material, but it is possible it comes from further reaction of the fumarate and/or succinate.
Our results show that the catalyst degrades at high pH, and we do not have data below pH 4.8, but it is likely the catalyst would also degrade if subjected to very low pH. By dissolving all chemicals except the catalyst, balancing the pH of the solution, and adding the catalyst as a final step, the results were more reproducible, since the catalyst was not subjected to significant pH changes as the powders dissolved.
We performed reactions using the disodium salt of ADC as opposed to the monopotassium salt, and saw no difference in the reaction kinetics as long as the pH was adjusted to be the same. However, we note an important and surprising discovery: when experiments were repeated using sodium sulphite obtained from a different supplier, the reaction kinetics were dramatically altered, giving less than 4% fumarate yield. The reason for this is not currently understood, but is not a pH effect since the solution pH was set using the described procedure. We postulate that the seemingly low level of impurities in the sodium sulphite is an important consideration, since the sodium sulphite is at such high concentration (35 times higher concentration than the catalyst).
In some of the experiments the reaction rate was limited by the rate at which hydrogen dissolved into solution. This was unavoidable in our experiments without going to significantly higher hydrogen gas pressures or changing the reaction vessel. The rate of hydrogen dissolution into solution strongly depends on the precise details of the experimental setup, and so the time required to reach maximum fumarate yield is a parameter that must be optimized for a given experimental setup.
The 2nd reaction step in which fumarate is hydrogenated to succinate limits the fumarate yield, and if this step could be suppressed this should further improve the reaction. This offers scope for future investigations into this important chemical process.
5 Conclusion
In conclusion, we have investigated the formation of fumarate via trans-hydrogenation, an important route to produce fumarate for applications in hyperpolarized metabolic MRI. We have optimized the reaction conditions to yield high (> 100 mM) fumarate concentration on a short timescale, which is important given the transient nature of the hyperpolarization. We also investigated a number of important questions surrounding this reaction such as how to reliably prepare the solutions, and how stable the solutions are before the reaction. This work required bubbling gases into aqueous solutions, which is notoriously difficult in small inner-diameter tubes because the high viscosity and surface tension of the water lead to adhesion to the walls and bubbles forming. We discuss experimental considerations such as effective tube cleaning, and made use of an anti-foaming agent to negate these effects.
We determined that for precursor solutions of 7 mM ruthenium catalyst and 250 mM acetylene dicarboxylate in D2O, the best results were obtained when 250 mM sodium sulphite was included and the reaction was carried out at 88 °C and pH 7.8. Using a phosphate buffer to stabilize the pH at 7.8 was detrimental. The results were identical for the disodium and monopotassium forms of the ADC precursor as long as the pH was adjusted to be the same, but the reaction was dramatically worse when sodium sulphite from a different supplier was used. For more reproducible results, all chemicals except the catalyst should be dissolved, the pH adjusted, and finally the catalyst dissolved, so that the catalyst is not subjected to significant pH changes during sample preparation. The precursor solution can be frozen for days, or stored at room temperature for hours, without jeopardizing the reaction. The optimal reactions shown here are limited in reaction rate by the rate at which hydrogen dissolves into solution. This should not be a limiting factor if the reaction vessel is carefully designed and higher hydrogen pressure is used. The rate of reaction might be further increased by increasing the catalyst concentration, something that was not investigated in this work. In this work we used 250 mM starting material and the optimal fumarate yields were just over 50%—there is certainly scope to increase the concentration of fumarate by further reaction optimization, or by increasing the starting material concentration.
The results of this work are particularly pertinent as they follow a demonstration that hyperpolarized fumarate can be formed via trans-hydrogenative PHIP with 13C polarization of > 30%, and then purified of contaminants by precipitation as a pure solid, and redissolved in a biocompatible solution for biological applications. [71] We hope that the findings presented here allow others to more easily perform the reactions to generate PHIP-polarized fumarate.
References
A.W. Overhauser, Phys. Rev. 92, 411 (1953)
T.P. Carver, C.P. Slichter, Phys. Rev. 92, 212 (1953)
K.H. Hausser, D. Stehlik, Adv. Mag. Res. 3, 79 (1968)
H.R. Ward, Acc. Chem. Res. 5(1), 18 (1972)
K.M. Salikhov, R.Z. Sagdeev, A.L. Buchachenko, Spin Polarization and Magnetic Effects in Radical Reactions (Elsevier, Netherlands, 1984)
G. Maier, U. Haeberlen, H.C. Wolf, K.H. Hausser, Phys. Lett. 25a, 384 (1967)
G. Buntkowsky, D. Stehlik, H.M. Vieth, K.M. Salikhov, J. Phys. Condens. Mat. 3(32), 6093 (1991)
S.A. Mikhailov, K.M. Salikhov, M. Plato, Chem. Phys. 117(2), 197 (1987)
R.Z. Sagdeev, E.G. Bagryanskaya, Pure. App. Chem. 62(8), 1547 (1990)
S.V. Dvinskikh, G. Buntkowsky, K.M. Salikhov, H.M. Vieth, Chem. Phys. Lett. 268(5–6), 401 (1997)
W. Happer, E. Miron, S. Schaefer, W.A. Schreiber, W.A.V. Wijngaarden, X. Zeng, Phys. Rev. A 29, 3092 (1984)
A. Bifone, Y.-Q. Song, R. Seydoux, R.E. Taylor, B.M. Goodson, T. Pietrass, T.F. Budinger, G. Navon, A. Pines, Proc. Natl. Acad. Sci 93, 12932 (1996)
C.R. Bowers, D.P. Weitekamp, Phys. Rev. Lett. 57, 2645 (1986)
T.C. Eisenschmidt, R.U. Kirss, P.P. Deutsch, S.I. Hommeltoft, R. Eisenberg, J. Bargon, J. Am. Chem. Soc. 109, 8089 (1987)
C.R. Bowers, D.P. Weitekamp, J. Am. Chem. Soc. 109(18), 5541 (1987)
R.W. Adams, J.A. Aguilar, K.D. Atkinson, M.J. Cowley, P.I.P. Elliott, S.B. Duckett, G.G.R. Green, I.G. Khazal, J. Lopez-Serrano, D.C. Williamson, Science 323(5922), 1708 (2009)
S. Wagner, MAGMA 27(3), 195 (2014)
M.G. Pravica, D.P. Weitekamp, Chem. Phys. Lett. 145(4), 255 (1988)
S.B. Duckett, C.J. Sleigh, Prog. Nucl. Magn. Reson. Spectrosc. 34, 71 (1999)
J. Bargon, in Handbook of homogeneous hydrogenation (Wiley-VCH, Eds.: J.G. de Vries C.J. Elsevier, 2007). https://doi.org/10.1002/9783527619382
A.S. Kiryutin, G. Sauer, A.V. Yurkovskaya, H.H. Limbach, K.L. Ivanov, G. Buntkowsky, J. Phys. Chem. C 121(18), 9879 (2017)
L.S. Bouchard, S.R. Burt, M.S. Anwar, K.V. Kovtunov, I.V. Koptyug, A. Pines, Science 319(5862), 442 (2008)
L.S. Bouchard, K.V. Kovtunov, S.R. Burt, M.S. Anwar, I.V. Koptyug, R.Z. Sagdeev, A. Pines, Angew. Chem. Int. Edit. 46(22), 4064 (2007)
A.A. Lysova, I.V. Koptyug, Chem. Soc. Rev. 39(12), 4585 (2010)
J.F. Dechent, L. Buljubasich, L.M. Schreiber, H.W. Spiess, K. Münnemann, Phys Chem Chem Phys 14(7), 2346 (2012)
T. Ratajczyk, T. Gutmann, S. Dillenberger, S. Abdulhussaein, J. Frydel, H. Breitzke, U. Bommerich, T. Trantzschel, J. Bernarding, P.C. Magusin, G. Buntkowsky, Solid State Nucl. Mag. Res. 43–44, 14 (2012)
K.L. Ivanov, A.V. Yurkovskaya, H.M. Vieth, J. Chem. Phys. 128, 154701 (2008)
S.E. Korchak, K.L. Ivanov, A.V. Yurkovskaya, H.M. Vieth, Phys. Chem. Chem. Phys. 11(47), 11146 (2009)
Q. Gong, A. Gordji-Nejad, B. Blümich, S. Appelt, Anal. Chem. 82(17), 7078–7082 (2010)
S. Appelt, S. Glöggler, F.W. Häsing, U. Sieling, A.G. Nejad, B. Blümich, Chem. Phys. Lett. 485(1–3), 217 (2010)
I.V. Zhukov, A.S. Kiryutin, F. Ferrage, G. Buntkowsky, A.V. Yurkovskaya, K.L. Ivanov, J. Phys. Chem. Lett. 11(17), 7291 (2020)
S. Glöggler, M. Emondts, J. Colell, R. Müller, B. Blümich, S. Appelt, Analyst 136(8), 1566 (2011)
T. Trantzschel, J. Bernarding, M. Plaumann, D. Lego, T. Gutmann, T. Ratajczyk, S. Dillenberger, G. Buntkowsky, J. Bargon, U. Bommerich, Phys. Chem. Chem. Phys. 14(16), 5601–5604 (2012)
T. Ratajczyk, G. Buntkowsky, T. Gutmann, B. Fedorczyk, A. Mames, M. Pietrzak, P. Szkudlarek, Z. Puzio, ChemBioChem 22, 855–860 (2021)
M. Koerner, G. Sauer, A. Heil, D. Nasu, M. Empting, D. Tietze, S. Voigt, H. Weidler, T. Gutmann, O. Avrutina, H. Kolmar, T. Ratajczyk, G. Buntkowsky, Chem. Comm. 49(71), 7839 (2013)
G. Sauer, D. Nasu, D. Tietze, T. Gutmann, S. Englert, O. Avrutina, H. Kolmar, G. Buntkowsky, Angew. Chem. Int. Edit. 53(47), 12941 (2014)
A.S. Kiryutin, G. Sauer, D. Tietze, M. Brodrecht, S. Knecht, A.V. Yurkovskaya, K.L. Ivanov, O. Avrutina, H. Kolmar, G. Buntkowsky, Chem-Eur. J. 25(16), 4025 (2019)
A.N. Pravdivtsev, G. Buntkowsky, S.B. Duckett, I.V. Koptyug, J.-B. Hövener, Angew. Chem. Int. Edit. (2021). https://doi.org/10.1002/anie.202100109
K. Golman, O. Axelsson, H. Johannesson, S. Mansson, C. Olofsson, J.S. Petersson, Magn. Reson. Med. 46(1), 1 (2001)
H. Jóhannesson, O. Axelsson, M. Karlsson, C. R. Phys. 5(3), 315 (2004)
L.E. Olsson, C.M. Chai, O. Axelsson, M. Karlsson, K. Golman, J.S. Petersson, Magn. Reson. Med. 55(4), 731 (2006)
E.Y. Chekmenev, J. Hövener, V.A. Norton, K. Harris, L.S. Batchelder, P. Bhattacharya, B.D. Ross, D.P. Weitekamp, J. Am. Chem. Soc. 130(13), 4212 (2008)
S. Aime, W. Dastru, R. Gobetto, D. Santelia, A. Viale, Mol. Imaging (2008). https://doi.org/10.1007/978-3-540-72718-7_12
E. Terreno, D.D. Castelli, A. Viale, S. Aime, Chem. Rev 110(5), 3019 (2010)
U. Bommerich, T. Trantzschel, S. Mulla-Osman, G. Buntkowsky, J. Bargon, J. Bernarding, Phys. Chem. Chem. Phys. 12, 10309 (2010)
K. Münnemann, M. Kölzer, I. Blakey, A.K. Whittaker, K.J. Thurecht, Chem. Comm. 48, 1583 (2012)
A.B. Schmidt, S. Berner, M. Braig, M. Zimmermann, J. Hennig, D. von Elverfeldt, J.-B. Hövener, PLoS ONE 13(7), e0200141 (2018)
S. Korchak, S. Yang, S. Mamone, S. Glöggler, ChemistryOpen 7(5), 344 (2018)
S. Korchak, S. Mamone, S. Glöggler, ChemistryOpen 7(9), 672 (2018)
F. Reineri, T. Boi, S. Aime, Nat. Comm. 6, 5858 (2015)
N.V. Chukanov, O.G. Salnikov, R.V. Shchepin, K.V. Kovtunov, I.V. Koptyug, E.Y. Chekmenev, ACS Omega 3(6), 6673 (2018)
E. Cavallari, C. Carrera, M. Sorge, G. Bonne, A. Muchir, S. Aime, F. Reineri, Sci. Rep. 8(1), 8366 (2018)
L. Shan, Hyperpolarized [1, 4–13C2] fumarate as an imaging agent of tumor cell death in vivo. In: Molecular Imaging and Contrast Agent Database (MICAD) [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2004–2013.
C.A. Roa Engel, A.J.J. Straathof, T.W. Zijlmans, W.M. van Gulik, L.A.M. van der Wielen, Appl. Microbiol. Biotechnol. 78, 379 (2008)
M. Moo-Young, Comprehensive Biotechnology (Elsevier, 2019)
J. Adam, M. Yang, C. Bauerschmidt, M. Kitagawa, L. O’Flaherty, P. Maheswaran, G. Özkan, N. Sahgal, D. Baban, K. Kato, K. Saito, K. Iino, K. Igarashi, M. Stratford, C. Pugh, D.A. Tennant, C. Ludwig, B. Davies, P.J. Ratcliffe, M. El-Bahrawy, H. Ashrafian, T. Soga, P.J. Pollard, Cell Rep. 3(5), 1440 (2013)
M. Akram, Cell Biochem. Biophys. 68(3), 475 (2014)
S.E. Bohndiek, M.I. Kettunen, D.-E. Hu, K.M. Brindle, Cancer Res. 72(4), 854 (2012)
L. Mignion, P. Dutta, G.V. Martinez, P. Foroutan, R.J. Gillies, B.F. Jordan, Cancer Res 74(3), 686 (2014)
F.A. Gallagher, M.I. Kettunen, D.E. Hu, P.R. Jensen, R. in’t Zandt, M. Karlsson, A. Gisselsson, S.K. Nelson, T.H. Witney, S.E. Bohndiek, G. Hansson, T. Peitersen, M.H. Lerche, K.M. Brindle, Proc. Natl. Acad. Sci 106(47), 19801 (2009)
M.R. Clatworthy, M.I. Kettunen, D.E. Hu, R.J. Mathews, T.H. Witney, B.W.C. Kennedy, S.E. Bohndiek, F.A. Gallagher, L.B. Jarvis, K.G.C. Smith, K.M. Brindle, Proc. Natl. Acad. Sci 109(33), 13374 (2012)
A. Eldirdiri, A. Clemmensen, S. Bowen, A. Kjaer, J.H. Ardenkjaer-Larsen, NMR Biomed. 30(12), e3803 (2017)
B. Feuerecker, M. Durst, M. Michalik, G. Schneider, D. Saur, M. Menzel, M. Schwaiger, F. Schilling, J. Cancer 8(15), 3078 (2017)
J.J. Miller, A.Z. Lau, P.M. Nielsen, G. McMullen-Klein, A.J. Lewis, N.R. Jespersen, V. Ball, F.A. Gallagher, C.A. Carr, C. Laustsen, H.E. Botker, D.J. Tyler, M.A. Schroeder, JACC-Cardiovasc. Imag. 11(11), 1594 (2018)
E. Coleman, Z. Wang, M. He, E. Bier, J. Nouls, S. Womack, J. Mammarappallil, B. Driehuys, Y.T. Huang, Am. J. Resp. Crit. Care 199 (2019).
C. Laustsen, P.M. Nielsen, H.Y. Qi, M.H. Lobner, J. Palmfeldt, L.B. Bertelsen, Sci. Rep. (2020). https://doi.org/10.1038/s41598-020-66265-6
B. Ripka, J. Eills, H. Kourilova, M. Leutzsch, M.H. Levitt, K. Münnemann, Chem. Comm. 54(86), 12246 (2018)
J. Eills, G. Stevanato, C. Bengs, S. Glöggler, S.J. Elliott, J. Alonso-Valdesueiro, G. Pileio, M.H. Levitt, J. Magn. Reson. 274, 163 (2017)
J. Eills, E. Cavallari, C. Carrera, D. Budker, S. Aime, F. Reineri, J. Am. Chem. Soc. 141(51), 20209 (2019)
J. Eills, E. Cavallari, R. Kirchner, G. Di Matteo, C. Carrera, L. Dagys, M.H. Levitt, K. Ivanov, S. Aime, F. Reineri, K. Münnemann, D. Budker, G. Buntkowsky, S. Knecht, Angew. Chem. Int. Edit. 60, 6791–6798 (2021)
S. Knecht, J.W. Blanchard, D. Barskiy, E. Cavallari, L. Dagys, E. van Dyke, M. Tsukanov, B. Bliemel, K. Münnemann, S. Aime, F. Reineri, M. Levitt, G. Buntkowsky, A. Pines, P. Blümler, D. Budker, J. Eills, Proc. Natl. Acad. Sci 118, 1 (2021)
N.J. Stewart, H. Nakano, S. Sugai, M. Tomohiro, Y. Kase, Y. Uchio, T. Yamaguchi, Y. Matsuo, T. Naganuma, N. Takeda, I. Nishimura, H. Hirata, T. Hashimoto, S. Matsumoto, ChemPhysChem (2020). https://doi.org/10.1002/cphc.202001038
A.S. Kiryutin, G. Sauer, S. Hadjiali, A.V. Yurkovskaya, H. Breitzke, G. Buntkowsky, J. Magn. Reson. 285, 26 (2017)
G. Sauer, NMR sensitivity enhancement of functional biomolecules: design and development of PHIP-based hyperpolarization approaches. PhD Thesis, Technical University Darmstadt (2016)
L. Dagys, B. Ripka, M. Leutzsch, G.A.I. Moustafa, J. Eills, J.F.P. Colell, M.H. Levitt, Magn. Reson. 1, 175 (2020)
B.A. Rodin, V.P. Kozinenko, A.S. Kiryutin, A.V. Yurkovskaya, J. Eills, K.L. Ivanov, J. Magn. Reson. 327, 106978 (2021)
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft under contract Bu-911/29-1 is gratefully acknowledged. J.E. acknowledges funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 766402.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Dedicated to Prof. Klaus Möbius and Prof. Kev Salikhov on the occasion of their 85th birthday.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wienands, L., Theiß, F., Eills, J. et al. Optimizing the Reaction Conditions for the Formation of Fumarate via Trans-Hydrogenation. Appl Magn Reson 53, 615–634 (2022). https://doi.org/10.1007/s00723-021-01371-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-021-01371-w